
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visualization of long-term Mg2+ dynamics in apoptotic cells using a novel targetable fluorescent probe†
Yusuke
Matsui
 a,
Yosuke
Funato
b,
Hiromi
Imamura
c,
Hiroaki
Miki
b,
Shin
Mizukami
*d and
Kazuya
Kikuchi
*ae
a,
Yosuke
Funato
b,
Hiromi
Imamura
c,
Hiroaki
Miki
b,
Shin
Mizukami
*d and
Kazuya
Kikuchi
*ae
aDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: kkikuchi@mls.eng.osaka-u.ac.jp
bDepartment of Cellular Regulation, Research Institute for Microbial Diseases, Osaka University, Suita, Osaka 565-0871, Japan
cGraduate School of Biostudies, Kyoto University, Kyoto 606-8501, Japan
dInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Sendai, Miyagi 980-8577, Japan. E-mail: shin.mizukami@m.tohoku.ac.jp
eImmunology Frontier Research Center, Osaka University, Suita, Osaka 565-0871, Japan
First published on 20th October 2017
Abstract
Mg2+ plays important roles in many physiological processes. However, the underlying molecular mechanisms, especially in the apoptotic pathway, remain unclear due to the diffusion of Mg2+ probes, which hinders long-term imaging in specific organelles. We developed an immobilized Mg2+ probe, MGH, which is covalently conjugated with the HaloTag protein in various organelles. HaloTag-coupled MGH enabled long-term imaging of intracellular local Mg2+ dynamics for 24 h. To exploit this remarkable property, MGH was applied to the investigation of intracellular Mg2+ dynamics during apoptosis. Time-lapse imaging revealed an increase in the Mg2+ concentration after apoptotic cell shrinkage. Combined imaging analyses of intracellular Mg2+ and ATP concentrations strongly suggested that this Mg2+ concentration increase was caused by the dissociation of Mg2+ from ATP, along with a decrease in the intracellular ATP concentration. Thus, this protein-coupled Mg2+ probe could be a new chemical tool to elucidate intracellular Mg2+ dynamics with high spatiotemporal resolution.
Introduction
Mg2+ is an essential divalent cation in cells, and the overall intracellular Mg2+ concentration ranges between 17 and 20 mM. However, the intracellular free Mg2+ concentration ([Mg2+]i) is maintained between 0.5 and 1 mM, because most Mg2+ forms complexes with nucleotides, such as ATP or DNA.1,2 Mg2+ plays vital roles in a variety of physiological processes such as regulation of enzyme activities, stabilization of nucleotides, and cell proliferation.2–4 It is understood that intracellular Mg2+ homeostasis is tightly regulated by Mg2+ transporters and Mg2+ channels and buffered by intracellular ATP.1 In addition, Mg2+ plays a role as a secondary messenger in T cells.5 Abnormal Mg2+ homeostasis is involved in several disorders including diabetes, hypertension, Parkinson’s disease, and cancer.6,7Despite Mg2+ being obviously important for regulating cellular functions, regulation of the underlying molecular mechanisms of [Mg2+]i still remains unclear.8 One phenomenon for which Mg2+ dynamics are unclear is apoptosis. Apoptosis is a programmed cell death process occurring over several hours to eliminate damaged cells. While Ca2+ plays very important roles in regulating the apoptotic process,9 changes in [Mg2+]i have also been observed in apoptotic cells, for example hepatocytes treated with glycodeoxycholate10 and B lymphocytes treated with Fas ligand.11 However, the intracellular Mg2+ dynamics during apoptosis have not been continuously visualized due to the lack of suitable analytical tools. To date, commercially available Mg2+ fluorescent probes, such as magnesium green and Mag-Fura-2, have often been utilized to detect free Mg2+ in cells. However, these probes tend to quickly diffuse over the entire cytoplasm and leak out of the cells via anion transporters.12,13 These unfavorable properties have made it impossible to image local Mg2+ concentration changes over long time periods, resulting in insufficient information regarding the intracellular Mg2+ dynamics in apoptotic cells.
To improve the spatial and temporal resolution of Mg2+ imaging, various Mg2+ probes have been developed. Recently, genetically encoded fluorescent protein-based Mg2+ sensors, MagFRET14 and MagIC,15 have been reported. These probes were easily targeted to various intracellular compartments by adding a localization signal peptide, and are likely to be capable of long-term detection. However, the application of these probes is limited, probably due to their low sensitivity in living cells14 and/or pH sensitivity under physiological conditions.15 Therefore, ideal protein-based Mg2+ sensors have not yet been developed.
Therefore, small molecule-based Mg2+ probes with improved fluorescence properties and intracellular behaviors have been developed for the exploration of intracellular Mg2+ dynamics. A chemical Mg2+ sensor functionalized with a lipophilic cationic alkylphosphonium group, Mag-mito, showed targetability to mitochondria.16 Mag-mito detected changes in the free Mg2+ concentration in mitochondria by calculating the ratio of fluorescence signals obtained using two different excitation wavelengths. Mag-mito successfully detected an increase in free Mg2+ in mitochondria in the early stages of staurosporine-induced apoptosis. However, this probe diffused from the mitochondria to the cytosol after 40 min of the compound treatment due to the depolarization of the mitochondrial membrane during apoptosis.16 It is likely that such noncovalent targetable probes diffuse away from the target site during long-term experiments. Furthermore, for monitoring Mg2+ concentration in various other organelles, novel fluorescent probes with different targeting moieties must be developed.
Therefore, a versatile method is to use a protein labeling system, with a pairing of a genetically encoded tag and its specific ligand. So far, several targetable fluorescent sensors have been developed on the basis of protein labeling systems, such as SNAP-tag and HaloTag.17–25 However, genetically encoded tag-based targeting of Mg2+ probes has rarely been reported. One example is KMG-104-AsH,26 which was developed based on the highly selective Mg2+ sensors in the KMG series.27–29 KMG-104-AsH can be anchored to a tetracysteine peptide tag (TC-tag), which is genetically expressed in specific organelles, enabling the detection of local Mg2+ concentration changes. However, long-term imaging of more than 4 h has not been carried out, probably owing to the cellular toxicity of an additive (ethanedithiol) that is necessary to reduce nonspecific binding.30 For the above-mentioned reasons, novel targetable fluorescent probes for long-term monitoring of local Mg2+ dynamics in living cells have been pursued.
To address these issues, we developed a tag protein-conjugatable Mg2+ probe that can be localized in specific organelles for imaging local Mg2+ dynamics in living cells. Through covalent anchoring of the Mg2+ probe to a tag protein expressed in various cellular compartments, intracellular diffusion and extracellular leakage of the Mg2+ probe were suppressed. This enabled not only the localization of the Mg2+ probe, but also long-term imaging of intracellular Mg2+ dynamics. This approach provided the first long-term imaging of [Mg2+]i during apoptosis.
Results
Design and synthesis of the Mg2+ probe with the HaloTag ligand
In order to visualize local Mg2+ dynamics in living cells for a long time, we designed and synthesized a new Mg2+ sensor, MGH, which covalently labels HaloTag (Fig. 1a). HaloTag is a modified haloalkane dehalogenase derived from Rhodococcus, and it quickly forms a covalent bond with specific ligands, including a chloroalkyl group.31 HaloTag can be expressed as a fusion protein with a localization signal peptide in mammalian cells. Thus, MGH, which involves a HaloTag ligand, was expected to be localized in specific target organelles, since similar strategies have previously been exploited to localize various small-molecule metal ion sensors in specific organelles.22–24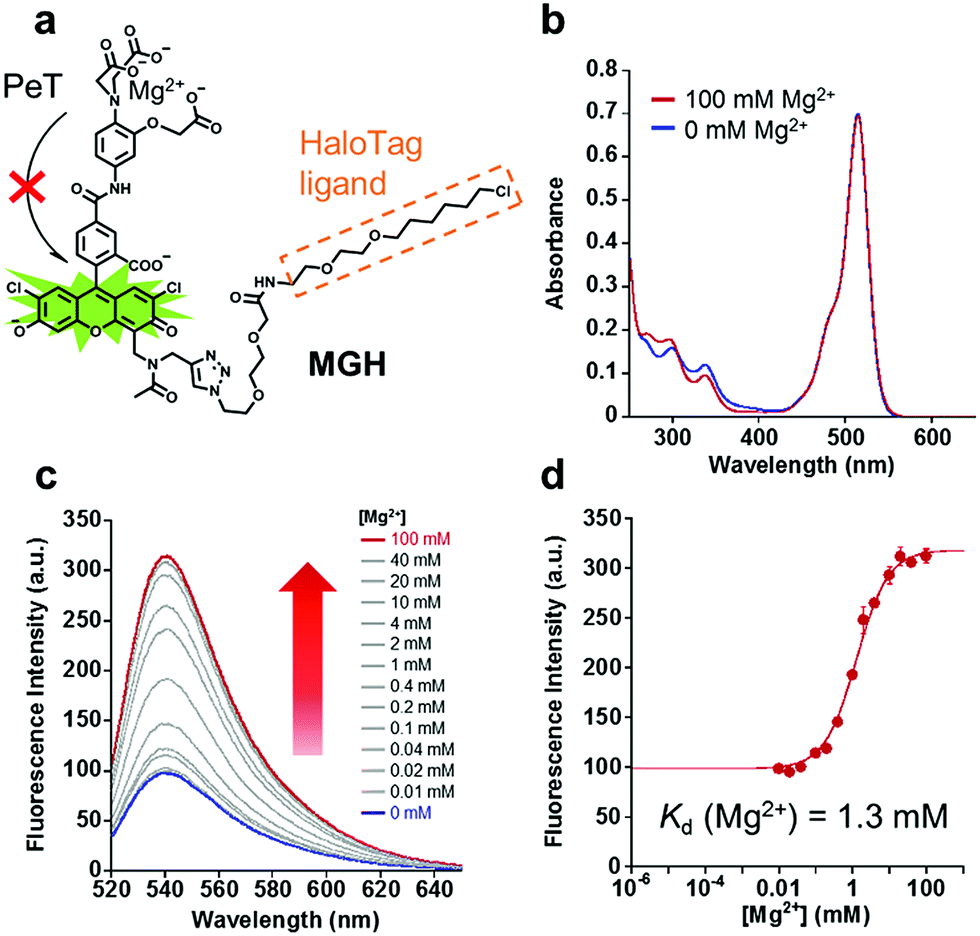 | ||
| Fig. 1 Structure and spectroscopic properties of MGH. (a) An overview of MGH, an Mg2+ probe modified with a HaloTag ligand. (b) Absorption spectra of 10 μM MGH in the presence or absence of 100 mM Mg2+ (100 mM HEPES buffer, 115 mM KCl, 20 mM NaCl, pH 7.4, 37 °C). (c) The emission spectra of 1 μM MGH in the presence of Mg2+ (100 mM HEPES, 115 mM KCl, 20 mM NaCl, pH 7.4, 37 °C). [Mg2+] = 0, 0.01, 0.02, 0.04, 0.1, 0.2, 0.4, 1, 2, 4, 10, 20, 40, and 100 mM. λex = 515 nm. (d) Mg2+-titration curve of MGH emission at 538 nm (λex = 515 nm). The error bars show the SD (n = 3). | ||
Here, magnesium green was adopted for Mg2+ sensing due to its suitable dissociation constant for Mg2+ (Kd = 1 mM), making it able to detect [Mg2+]i changes.32 The probe was designed so that the fluorescence intensity of its Mg2+-unbound state is suppressed by photo-induced electron transfer (PeT),33–35 and it emits strong fluorescence via binding to Mg2+.
MGH is composed of three building blocks for synthesis: the Mg2+ chelator part, the fluorophore part, and the HaloTag ligand part. These components were individually synthesized, and were then conjugated to facilitate the synthetic process and optimization of each part. The Mg2+ chelator part (compound 4) was synthesized from o-aminophenol via SN2 reaction with chloroacetic acid, Fischer esterification, nitration, and alkaline hydrolysis (Scheme S1†).36 To improve the cell membrane permeability, compound 4 was then derivatized to tris-acetoxymethyl (AM) ester, which is quickly deprotected by endogenous intracellular esterases.37 In the fluorophore part, an alkynyl moiety of compound 10 was introduced at the 4-position of the xanthene ring of 5-carboxyfluorescein via the Mannich reaction (Scheme S2†).38 Then, the secondary amine, as well as the phenolic hydroxy groups, was modified with an acetyl group to avoid PeT from the free amino group to the fluorophore. Compound 15 was afforded via the amide condensation reaction of the chelator moiety (compound 6) and chromophore (compound 11), and then it was conjugated with the HaloTag ligand (compound 14) (Scheme S3†) via Cu(I)-catalyzed azide–alkyne cycloaddition to produce MGH(AM) (Scheme S4†). MGH(AM) was used for live cell imaging due to its sufficient cell membrane permeability. For the various in vitro spectral measurements, the deprotected probe MGH was synthesized via the alkaline hydrolysis of MGH(AM).
Spectroscopic properties of MGH and HaloTag-MGH
The absorption and emission spectra of MGH were measured in 100 mM HEPES buffer (pH 7.4) containing 115 mM KCl and 20 mM NaCl. Although the absorption spectra of MGH did not change significantly with 100 mM Mg2+ (Fig. 1b), the fluorescence intensity of MGH considerably increased as the Mg2+ concentration was increased (Φfree = 0.19, Φbound = 0.56, Fig. 1c). From the fluorescence intensity change, the dissociation constant of MGH for Mg2+ was calculated to be 1.3 mM (Fig. 1d), which is a suitable value for imaging [Mg2+]i changes. The metal ion selectivity of MGH, shown in Fig. S1c,† was similar to that of other o-amino-phenol-N,N,O-triacetic acid (APTRA)-based fluorescent probes.39 For live-cell Mg2+ imaging, the ability of the probe to discriminate changes in intracellular Ca2+ concentration ([Ca2+]i) is quite important. MGH bound to Ca2+ more strongly (Kd (Ca2+) = 12 μM) than to Mg2+ (Fig. S1a and b†), although this had already been predicted at the stage of the probe design. Since [Ca2+]i is roughly 100 nM and rises 10- to 100-fold during various cellular events,40 simultaneous monitoring of [Ca2+]i with a Ca2+-specific probe is necessary to correctly evaluate intracellular Mg2+ dynamics with MGH. In addition, the pH sensitivity of live-cell imaging probes is another important factor for practical use. Concerning the pH effect, MGH scarcely showed a fluorescence intensity change in a physiological pH range (pH 7–8) (Fig. S1d†). These spectroscopic properties of MGH mostly corresponded with those of magnesium green, indicating that the attachment of the HaloTag ligand scarcely affected the metal ion-detecting properties of the Mg2+ sensor (Table 1 and Fig. S2†).| λ abs [nm] | λ em [nm] | ε [cm−1 M−1] | Φ free (Φbound) | K d (Mg2+) [mM] | K d (Ca2+) [μM] | |
|---|---|---|---|---|---|---|
| a Measured at 37 °C in 100 mM HEPES buffer, 115 mM KCl, 20 mM NaCl, pH 7.4. b Relative fluorescence quantum yield determined using fluorescein (Φ = 0.85 in 0.1 M NaOH aq.) as a standard. Φfree and Φbound denote the relative fluorescence quantum yield in the absence and presence of 100 mM Mg2+, respectively. | ||||||
| MGH | 515 | 538 | 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0.19 (0.56) | 1.3 | 12 |
| HaloTag-MGH | 517 | 540 | 78000 |
0.21 (0.61) | 0.67 | 7.5 |
| Magnesium green | 509 | 534 | 77000 |
0.20 (0.56) | 0.88 | 12 |
It is likely that spectroscopic properties of fluorescent probes are affected by environmental changes such as binding on the protein surface. Hence, we measured the spectroscopic properties of MGH after incubation with a purified HaloTag protein for 1 h at 37 °C. SDS-PAGE analysis showed almost all MGH covalently bound to HaloTag (Fig. S3†). Overall, the fluorescence properties of HaloTag-MGH were similar to those of MGH and magnesium green (Table 1 and Fig. S4†). We infer that, in response to Mg2+, HaloTag-MGH has almost the same dynamic range of as that of free MGH, which was derived from the hydrophilic spacer between the Mg2+ sensor part and the HaloTag ligand part. The hydrophilic spacer prevented the interaction between the chromophore and the protein surface of the HaloTag.24 Although the Kd values of HaloTag-MGH for Mg2+ and Ca2+ were enhanced approximately 2-fold, the affinity of HaloTag-MGH for Mg2+ was still appropriate for visualizing intracellular Mg2+ dynamics.
Subcellular targeting of MGH and long-term imaging of Mg2+ in living cells
Here, we attempted to confirm the subcellular targeting of MGH to a variety of organelles. HEK293T cells were transfected with a plasmid encoding Halo-NLS, Lyn11-Halo, or HaloTag. NLS is a nuclear localization signal peptide41 and Lyn11 is a Lyn N-terminal sequence (GCIKSKGKDSA), which is used to target proteins to the inner leaflet of cell membrane.42,43 The transfected and non-transfected cells were incubated with 3 μM MGH(AM) for 1 h at 37 °C and were observed using a confocal spinning disk fluorescence microscope. In the case of the non-transfected cells, weak fluorescence was observed from the entire cell (Fig. 2a). However, in the transfected cells, strong fluorescence was observed from the target domains, such as nuclei, the cell membrane inner leaflet, or cytoplasm, without non-specific fluorescence signals (Fig. 2a).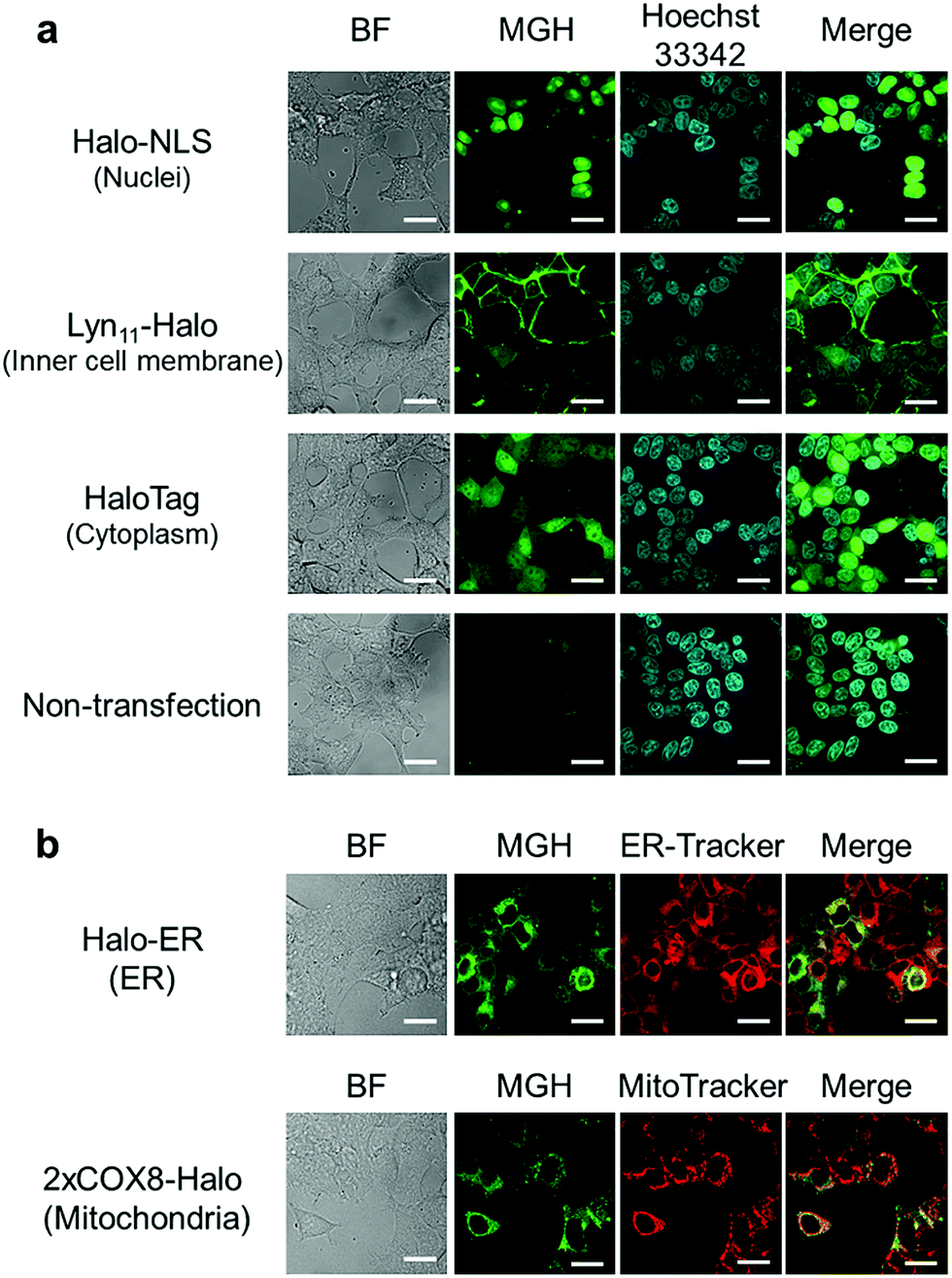 | ||
| Fig. 2 Subcellular localization of MGH in living cells. (a) Confocal fluorescence microscopic images of MGH localization in HEK293T cells transfected with a plasmid encoding Halo-NLS (nuclei), Lyn11-Halo (inner leaflet of cell membrane) or HaloTag (cytoplasm). Labeling reactions of fusion protein of Halo-NLS, Lyn11-Halo or HaloTag in HEK293T cells with 3 μM MGH(AM) and staining with 200 ng mL−1 Hoechst 33342 were performed for 1 h at 37 °C. (b) Confocal fluorescence microscopic images of MGH localization in ER or mitochondria matrix, which are surrounded by a lipid bilayer membrane. HEK293T cells transiently expressing Halo-ER or 2xCOX8-Halo were incubated with 3 μM MGH(AM) and either 200 nM ER-Tracker Red or 200 nM MitoTracker DeepRed for 1 h at 37 °C. Scale bar: 20 μm. | ||
Subsequently, MGH(AM) was added to HEK293T cells expressing HaloTag in the endoplasmic reticulum (ER) or the mitochondrial matrix. Since these organelles are covered by a lipid bilayer membrane, it was unclear whether fluorescent probes modified with AM esters could penetrate the second lipid bilayer membrane prior to the enzymatic hydrolysis of the AM esters by intracellular esterases. To express HaloTag in ER, the ER signal peptide and retention signal sequences (SEKDEL) were fused with HaloTag (Halo-ER). For mitochondrial matrix targeting, HaloTag was fused with the first 36 amino acids of subunit VIII of cytochrome c oxidase (COX8) in tandem to enhance the specificity of mitochondrial localization (2xCOX8-Halo).44 When the cells expressing Halo-ER or 2xCOX8-Halo were incubated with MGH(AM) for 1 h, the fluorescence of MGH was observed from the ER and mitochondria, respectively. The intended targeting of MGH to the ER and mitochondria was confirmed by the colocalization with the fluorescent signals of ER-Tracker Red and MitoTracker DeepRed, respectively (Fig. 2b). The results indicate that MGH(AM) diffused very quickly in living cells and accumulated inside of organelles surrounded by lipid bilayers, such as ER and the mitochondrial matrix, before enzymatic hydrolysis of the AM esters. However, it is likely that MGH mainly detects Ca2+ in the ER because the ER contains high concentrations of Ca2+ (hundreds of μM). Therefore, Mg2+-selective probes are necessary for the correct evaluation of changes in Mg2+ concentration in the ER.
Next, long-term Mg2+ imaging in living cells was attempted by using the nucleus-localized MGH. Commercially available small-molecule Mg2+ probes, such as magnesium green, are not suitable for long-term imaging owing to the extracellular leakage via anion transporters.12,13 We also confirmed that the fluorescence of magnesium green gradually decreased in a time-dependent manner, and completely disappeared after 24 h (Fig. 3b and c). In contrast, the fluorescence of nucleus-localized MGH was continuously detected for 24 h (Fig. 3a and c). The fluorescence signals of Hoechst 33342, which is a membrane-permeable cationic dye, gradually decreased, probably due to diffusion of the dye (Fig. 3a and c). These results indicated that MGH enables imaging of Mg2+ dynamics for more than 1 day.
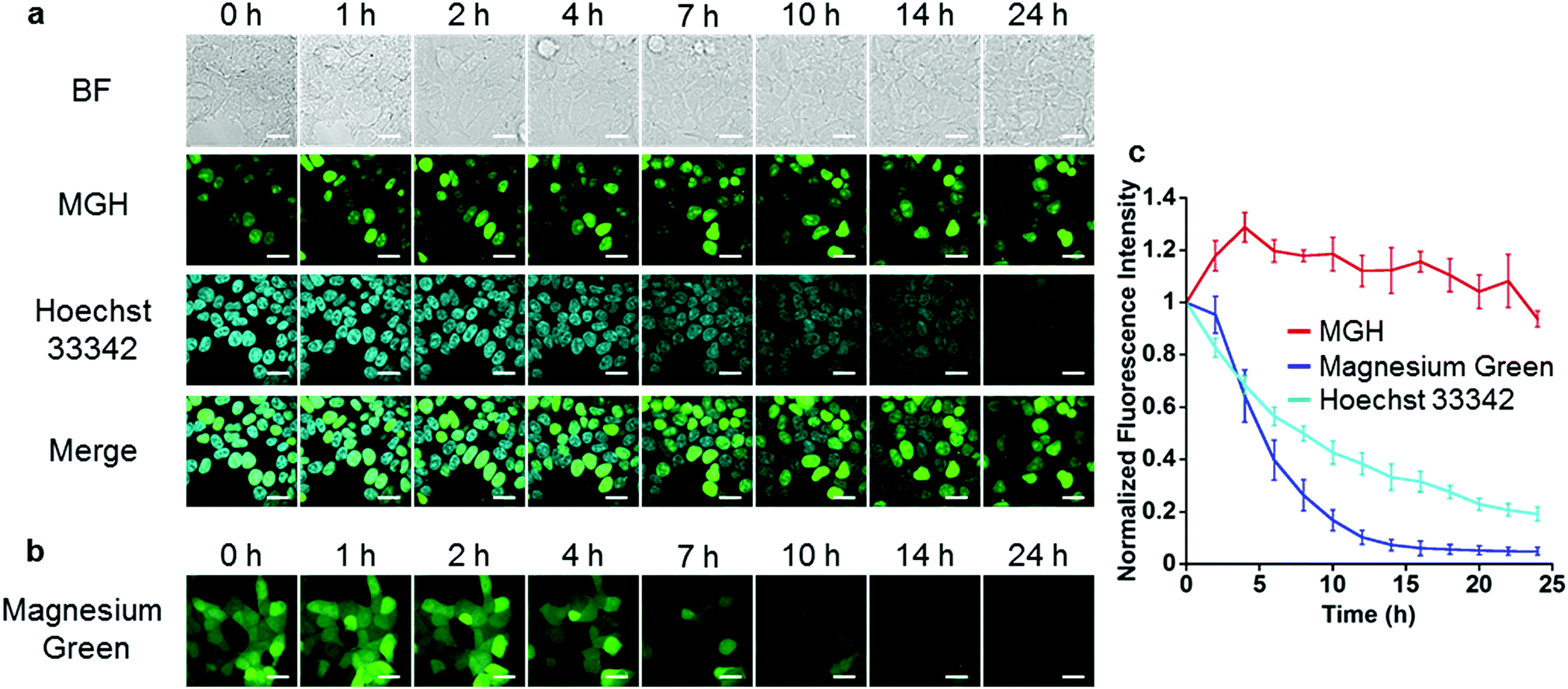 | ||
| Fig. 3 Comparison between MGH and magnesium green for long-term imaging. (a) Long-term Mg2+ imaging with MGH localized in nuclei for 24 h. HEK293T cells transiently expressing Halo-NLS were incubated with 3 μM MGH(AM) and 200 ng mL−1 Hoechst 33342 for 1 h at 37 °C. (b) Long-term Mg2+ imaging with magnesium green for 24 h. HEK293T cells were incubated with 1 μM magnesium green(AM) for 45 min at 37 °C. Scale bar: 20 μm. (c) Time-dependent changes of the relative fluorescence intensity of MGH, magnesium green or Hoechst 33342 in a single cell. The error bars denote SD (n = 4). | ||
Detection of Mg2+ extrusion through CNNM4
To examine the response of nucleus-localized MGH to Mg2+, we performed a Mg2+ extrusion experiment using a Mg2+ transporter, ancient conserved domain protein/cyclin M4 (CNNM4). CNNM4 is strongly expressed in the intestinal epithelium and can extrude Mg2+ by stimulating Na+/Mg2+ exchange.45 CNNM4-FLAG and Halo-NLS were co-expressed in HEK293 cells, in which the transient expression of CNNM4 was confirmed by western blot analysis (Fig. S5b†), and the cells were incubated with MGH(AM) in 40 mM Mg2+ buffer. After 1 h, the extracellular solution was exchanged with a Mg2+-free buffer to artificially promote Mg2+ extrusion via CNNM4, and time-lapse fluorescence microscopic images were captured. The fluorescence intensity of nucleus-localized MGH, as well as magnesium green, immediately decreased after Mg2+ depletion from the extracellular buffer (Fig. 4 and S5a†). The fluorescence of MGH and magnesium green in the control cells not overexpressing CNNM4 remained constant after the addition of the Mg2+-free buffer. Thus, it was confirmed that MGH maintained a quick fluorescence response to [Mg2+]i change. Since MGH showed promise for long-term imaging, the durability of the response to Mg2+ was investigated. The Mg2+ extrusion experiment was conducted after 24 h of MGH(AM) loading to the transfected HEK293 cells. As a result, the fluorescence of MGH quickly decreased after exchanging a buffer containing 40 mM Mg2+ with the Mg2+-free buffer (Fig. S5c and d†). This result showed that MGH retained the Mg2+ responsivity for 24 h in living cells.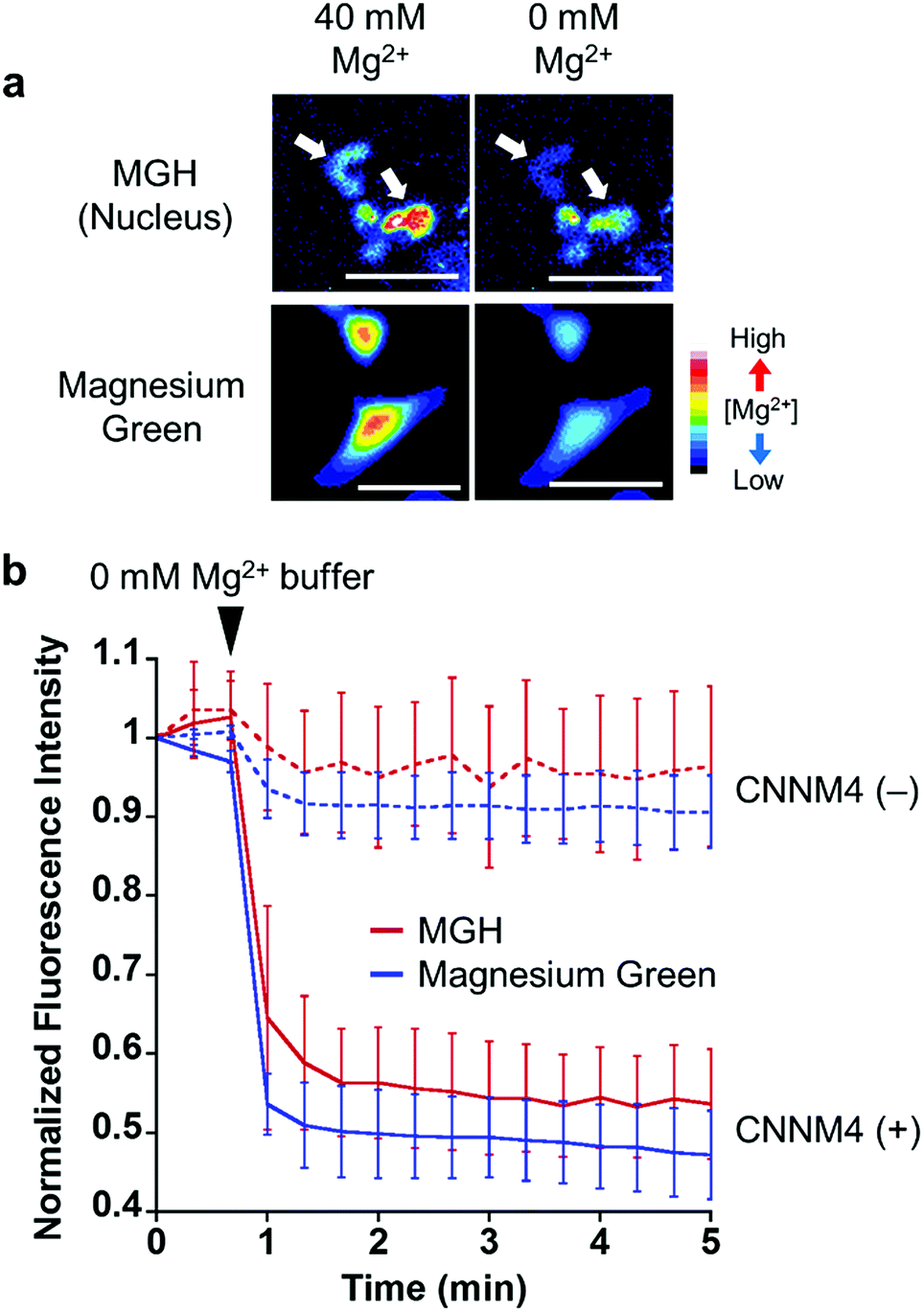 | ||
| Fig. 4 Visualization of Mg2+ extrusion via Mg2+ transporter, CNNM4, with MGH. (a) Epifluorescence microscopic images of Mg2+ extrusion with MGH or magnesium green. HEK293 cells transfected with CNNM4-FLAG and Halo-NLS were incubated with Mg2+-loading buffer (78.1 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 40 mM MgCl2, 5.5 mM glucose, 5.5 mM HEPES-KOH, pH 7.4), including 5 μM MGH(AM) or 2 μM magnesium green(AM), for 1 h at 37 °C. These cells were subjected to Mg2+ depletion 1 min after the imaging started. Scale bar: 40 μm. (b) The normalized fluorescence intensity of MGH or magnesium green in HEK293 cells subjected to Mg2+ depletion. The error bars denote SD (MGH: n = 6, magnesium green: n = 10). | ||
Long-term monitoring of intracellular Mg2+ dynamics during apoptosis
Taking advantage of the applicability to long-term imaging, MGH was applied to investigate intracellular Mg2+ dynamics during apoptosis induced by the anti-Fas antibody and cycloheximide (CHX). So far, anti-Fas antibody-mediated [Mg2+]i increase in apoptotic B cells was fragmentally detected for 16 h with a ratiometric Mg2+ probe, Mag-indo-1, by flow cytometric analysis.11 However, this method did not provide detailed information regarding the timing of Mg2+ mobilization along with the apoptotic process. On the other hand, use of MGH simultaneously enables real-time monitoring of [Mg2+]i and changes in cellular morphology over a long time period. However, MGH exhibits an increase in fluorescence intensity without a shift in wavelength. This means that the fluorescence intensity depends not only on Mg2+ concentration but also on other factors such as probe concentration and light intensity. Particularly, probe concentration must be changed during apoptosis, because apoptotic cells show shrinking. Therefore, to exclude the influence of such factors, an internal standard was adopted for calibration. As the internal standard, a red fluorescent HaloTag ligand, Halo-TMR, was used to label HaloTag, and the fluorescence intensity ratio of MGH and Halo-TMR was calculated.HeLa cells transiently expressing HaloTag in the cytoplasm were labeled with MGH(AM) for 30 min and subsequently Halo-TMR for 15 min. Then, the time-lapse imaging experiment was started, and anti-Fas antibody and CHX were added after 30 min. Alexa Fluor™ 350 annexin V conjugate, which binds phosphatidylserine-binding protein, was also added to identify apoptotic cells. As a result, the fluorescence signal ratio of MGH and Halo-TMR increased after the apoptotic cell shrinkage, and then plateaued (Fig. 5a). Quantitative analysis showed that [Mg2+]i increased from approximately 1 to 1.8 mM during apoptosis (Fig. S6†).
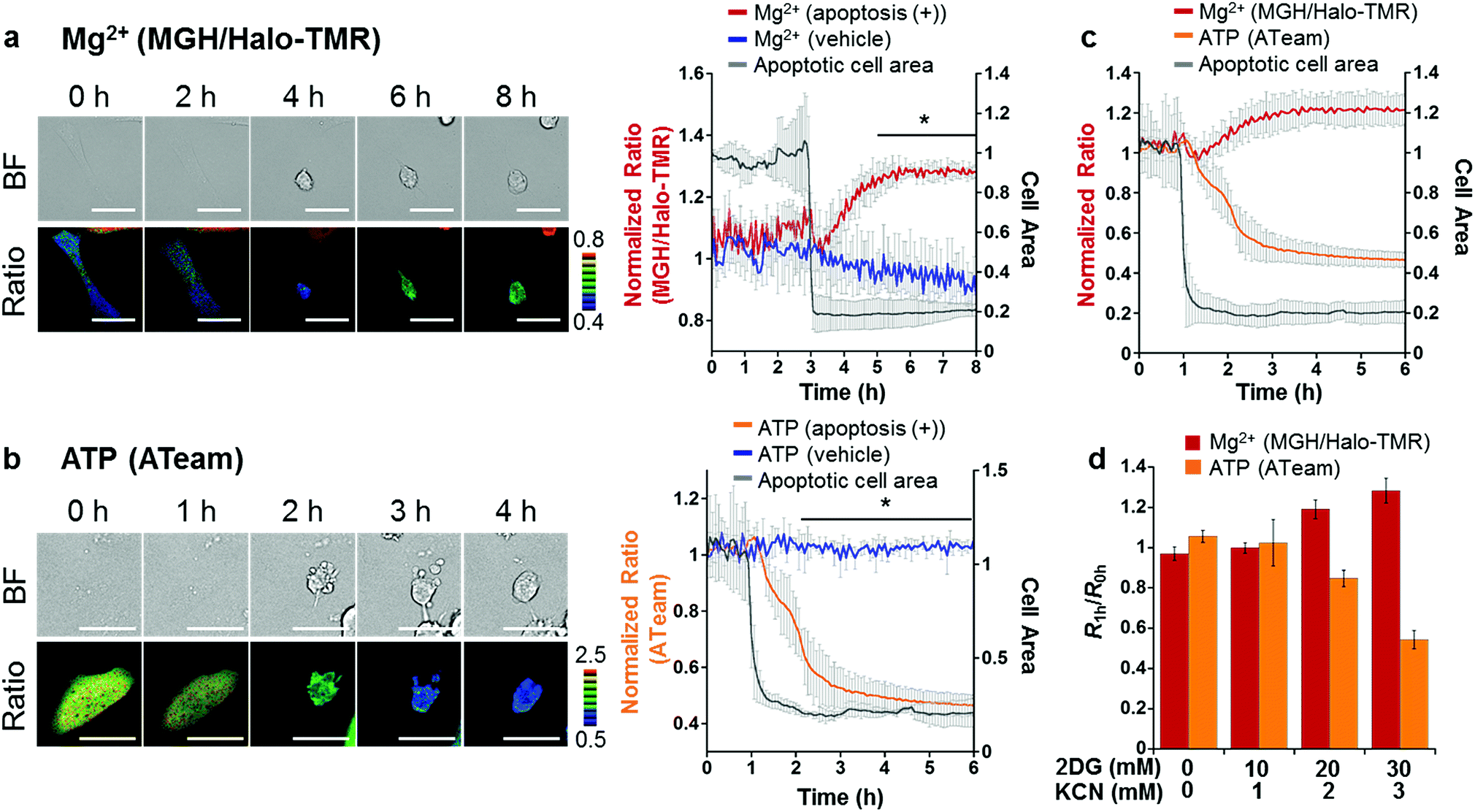 | ||
| Fig. 5 Confocal fluorescence imaging of (a) Mg2+ and (b) ATP dynamics with progression of apoptosis in HeLa cells. (a) HeLa cells were transfected with a plasmid encoding HaloTag. Fluorescence of 3 μM MGH(AM) was normalized with 50 nM Halo-TMR to exclude the influence of changes in probe concentration during apoptosis. (b) HeLa cells were transfected with a plasmid encoding ATeam, a FRET-based ATP sensor. Apoptosis inducers were anti-Fas antibody (250 ng mL−1) and cycloheximide (10 μg mL−1). Scale bar: 40 μm. Statistical analyses were performed using the Student’s t-test. *P < 0.05. The error bars denote SD (n = 3). (c) Correlation between [Mg2+] and [ATP] in apoptosis-induced HeLa cells. Each ratio change was individually recorded and plotted from 1 h before cell shrinkage. (Mg2+: n = 3, ATP: n = 3). (d) Dose-dependent effect of 2DG and KCN on [Mg2+]i and ATP concentration. Ratio changes for Mg2+ and ATP were individually recorded. The error bars denote SD (n = 5). | ||
We also checked the validity of our ratiometric imaging method with Halo-TMR as the internal standard. During long-term microscopic imaging experiments, fluorescent probes are likely to cause photobleaching. Since the photostability of fluorescent probes is dependent on the dye skeletons, we measured the photostability of MGH and Halo-TMR using a fluorometer under continuous irradiation at 490 and 550 nm, respectively (Fig. S7†). The fluorescence intensity of MGH decreased slightly during long-term irradiation as compared with that of Halo-TMR. Indeed, this result indicated that the fluorescence ratio of MGH/Halo-TMR may change without changes in [Mg2+]i during long-term imaging experiments. Actually, long-term microscopic imaging data demonstrated that the fluorescence ratio of MGH/Halo-TMR in HaloTag-expressing HeLa cells without the addition of apoptosis inducers continued to decrease slightly for several hours (Fig. 5a), and this small ratio change could be interpreted as the photobleaching of MGH. However, in apoptotic cells, the ratio tended to increase. Thus, we concluded that the fluorescence ratio change in the apoptotic cells properly reflected the changes in [Mg2+]i. Moreover, in another control experiment, the photostability of Halo-TMR during apoptosis was examined by calculating the ratio of Halo-OG to Halo-TMR (Fig. S8†). The fluorescence ratio of the two dyes remained constant during apoptosis. All of these results supported the adequacy of our ratiometric system.
As shown in Table 1, APTRA-based Mg2+ probes respond to high concentrations of Ca2+. It is suggested that Ca2+ release from ER through the IP3 receptor to the cytosol is one of the key apoptotic processes.46 Therefore, discrimination between the concentration changes of Ca2+ and Mg2+ is necessary. The change in [Ca2+]i during apoptosis was visualized with a genetically encoded Ca2+-specific fluorescent probe, R-GECO, which shows a Ca2+-dependent increase in red fluorescence (Kd (Ca2+) = 480 nM).47 R-GECO barely showed a fluorescence response to the Mg2+ introduced by the ionophore 4-bromo-A23187, in contrast to the increase in the fluorescence intensity of MGH, demonstrating sufficient specificity for Ca2+ (Fig. S9†). R-GECO and HaloTag were transiently expressed in HeLa cells, and the transfected cells were stained with green fluorescent Halo-OG to accurately evaluate Ca2+ dynamics by monitoring the fluorescence ratio of R-GECO/Halo-OG. A sharp spike was observed immediately after the addition of apoptosis inducers, which was followed by a slight increase during apoptosis (Fig. S10b†). This large spike was not observed by monitoring the MGH/Halo-TMR ratio, indicating that MGH has a sufficiently low affinity for Ca2+ in the physiological range of cytosolic [Ca2+]i during apoptosis (Fig. S10a†). Thus, the fluorescence response of MGH after the apoptotic cell shrinkage was ascribed to the increase of [Mg2+]i, not [Ca2+]i.
Another concern related to the increase in the fluorescence ratio of MGH/Halo-TMR is equilibration of intracellular and extracellular metal ions due to the loss of cell membrane integrity in the late stages of apoptosis.58 During apoptosis imaging, we observed further significant increases in the fluorescence ratios of both MGH/Halo-TMR and R-GECO/Halo-OG following increases in the ratios after apoptotic cell shrinkage (Fig. S10,† blue squares). This large fluorescence response during the late apoptotic stage is thought to reflect the loss of cell membrane integrity and the influx of extracellular metal ions, particularly Ca2+ (Ca2+ concentration in DMEM is approximately 1.8 mM).
To elucidate the mechanism of [Mg2+]i increase during apoptosis, we focused on the correlation between [Mg2+]i and intracellular ATP concentration; since most ATP forms a complex with Mg2+ in cells,1 [Mg2+]i is likely to be affected by ATP concentration. In fact, it has been reported that various stimulations, such as cyanide,48 fructose,49 and anoxia,50 trigger an increase in [Mg2+]i with a decrease in intracellular ATP or degradation of ATP to ADP or AMP. These reports suggested a close relationship between Mg2+ and ATP levels. Another previous study showed that intracellular ATP concentration considerably decreased at the late stage of apoptosis.51 Hence, the time course of ATP concentration change in apoptotic HeLa cells was visualized using a caspase-resistant version of ATeam,52 a genetically encoded fluorescence resonance energy transfer (FRET)-based ATP sensor.53 As a result, the ATP level significantly decreased in apoptotic cells after cell shrinkage (Fig. 5b).
Although the simultaneous imaging of [Mg2+]i and ATP concentration in apoptotic cells was desirable, it was not possible because the excitation and emission wavelengths of MGH and ATeam largely overlap. Therefore, the fluorescence ratio changes for [Mg2+]i and intracellular ATP concentration were plotted from 1 h before cell shrinkage (Fig. 5c). The time-course graphs of both concentrations indicated that [Mg2+]i and intracellular ATP concentration were almost inversely correlated.
Was this [Mg2+]i increase induced by the decrease in intracellular ATP concentration in apoptotic cells? We hypothesized that the increase in Mg2+ levels arose from disassociation of Mg2+ from ATP. To confirm this hypothesis, intracellular ATP concentration was lowered by adding 2-deoxyglucose (2DG) and potassium cyanide (KCN), which are inhibitors of glycolysis and oxidative phosphorylation, respectively.53 Incubation with the inhibitors for 1 h induced a decrease in ATP concentration depending on the inhibitor concentration, with a concomitant increase of [Mg2+]i (Fig. 5d). This relationship between Mg2+ and ATP concentrations suggested that the [Mg2+]i increase after apoptotic cell shrinkage was caused by the dissociation of Mg2+ from ATP. Our results indicated that Mg-ATP was a possible source of free Mg2+ in apoptotic cells. Importantly, a variety of endonucleases involved in DNA fragmentation during apoptosis requires Mg2+ to digest DNA.54 The increase in Mg2+ levels after the cell shrinkage may be relevant to endonuclease activities. Further studies will be necessary to investigate the role of Mg2+ following apoptotic cell shrinkage.
Discussion
Fluorescence imaging with higher temporal and spatial resolution is a crucial technique for the analysis of target molecules. To elucidate the intracellular dynamics and physiological roles of free Mg2+, this imaging system requires the long-term retention and localization of fluorescent Mg2+ probes in living cells. However, existing Mg2+ probes have not been suitable for such experiments, especially for long-term imaging. To overcome this problem, we developed a combined system of a novel small-molecule Mg2+ probe, MGH, and a HaloTag, which is genetically localized in a variety of organelles. This noteworthy feature improved the spatiotemporal resolution for detecting intracellular Mg2+ dynamics, enabling the investigation of unexplored phenomena related to Mg2+. Recently, Buccella and co-workers reported a similar imaging system combining a ratiometric Mg2+ probe, Mag-S-Tz, and HaloTag.25 This probe was also localized at the target subcellular compartments in living cells after the labeling of a strained bicyclononyne ligand to HaloTag and the subsequent fluorogenic click reaction between Mag-S-Tz and the bicyclononyne. However, this two-step strategy requires more than 4 h to start the imaging experiment, and long-term imaging has not been achieved. Therefore, MGH is the first imaging probe to have demonstrated long-term time-lapse imaging of local Mg2+ dynamics in living cells for 24 h.Apoptosis is a process that takes several hours. Therefore, long-term imaging is essential for the clarification of intracellular free Mg2+ dynamics in apoptotic cells. In the 1990s, some groups showed a [Mg2+]i increase during apoptosis.10,11 However, more detail on the mechanisms and timing of the [Mg2+]i increase have been unclear owing to the lack of long-term Mg2+ imaging methods. MGH had suitable properties for long-term monitoring of [Mg2+]i change during apoptosis. However, MGH showed a simple fluorescence in response to the change of [Mg2+] without an excitation or emission wavelength shift. This fluorescence spectral property was not ideal for the analysis of apoptotic cells because the fluorescence intensity is considerably affected by the probe concentration change during apoptosis. This problem was solved by introducing Halo-TMR as an internal standard, and thus calculating the signal ratio between MGH and Halo-TMR. Ratiometric two-fluorophore sensing systems have been employed in Ca2+ and Zn2+ imaging.5,55 In addition, extracellular leakage of the internal standard during long-term imaging was prevented by using Halo-TMR in HaloTag-expressing cells.
The data analysis of Mg2+ and ATP imaging with MGH and ATeam suggested that the [Mg2+]i increase after apoptotic cell shrinkage was caused by the dissociation of Mg2+ from ATP. The inverse correlation of [Mg2+]i and ATP concentration in the analysis of the dose-dependent effect of 2DG and KCN strongly supports the concept that Mg-ATP is the main resource for the Mg2+ increase after cell shrinkage during apoptosis. Although a close correlation between [Mg2+]i and ATP levels has been previously reported, further evidence was suggested by biochemical characterization after cell lysis without direct visualization of both molecular behaviors in cells.49 Therefore, our result provides strong evidence regarding the [Mg2+]i increase as a consequence of intracellular ATP depletion.
In HeLa cells, the intracellular ATP concentration was estimated at 3–4 mM,56,57 and it was reported that the intracellular ATP concentration in human T cells was almost fully depleted during apoptosis.51 If all of the discharged ATP released Mg2+ in cells, [Mg2+]i, which is maintained at the hundred-micromolar level in living cells, would increase to the several-millimolar level. However, [Mg2+]i increased by only approximately 0.8 mM during apoptosis (Fig. S6†). This result suggested that increased Mg2+ was buffered by Mg2+ transporters or channels. Recently, some Mg2+ transporters and channels were identified in the cell membrane and in the membrane of intracellular organelles (e.g. mitochondria and Golgi).1 These Mg2+ transport mechanisms may contribute to the regulation of intracellular Mg2+ homeostasis. Since MGH can be localized on a Mg2+ transporter by tandemly expressed HaloTag, Mg2+ influx via Mg2+ transporters would be visualized using such organelle-localized probes. Further studies using MGH should lead to the elucidation of Mg2+-buffering mechanisms in apoptotic cells.
Conclusions
In conclusion, we developed a novel Mg2+ probe, MGH, which covalently binds to HaloTag protein expressed in various cellular compartments. The conjugation of MGH to HaloTag dramatically suppressed the extracellular leakage of MGH, and the Mg2+ sensing ability of MGH was retained for 24 h. These noteworthy properties were successfully applied to long-term visualization of intracellular Mg2+ dynamics during apoptosis. The results demonstrated an increase in Mg2+ concentration after apoptotic cell shrinkage. Subsequent experiments utilizing a FRET-based ATP sensor showed that dissociation of Mg2+ from ATP in apoptotic cells was the source of the increase of Mg2+ concentration. Thus, long-term imaging with a HaloTag-coupled Mg2+ probe provided precise information regarding intracellular Mg2+ dynamics during apoptosis. This study includes the molecular design of a cell-functional probe that involves a synthetic organic molecule and a genetically encoded protein. In addition, the findings in this study should contribute to the understanding of Mg2+-related biology, which includes very wide areas in biology and medicine, and thus the results strongly suggest that this new methodology will be a robust tool for studying Mg2+ dynamics in living systems and diseases.Experimental section
Live-cell fluorescence imaging of MGH localization
HEK293T cells maintained in 10% FBS in DMEM at 37 °C in 5% CO2 were transfected with pcDNA-3.1-(+)-Halo-NLS, pcDNA-3.1-(+)-Lyn11-Halo, pcDNA-3.1-(+)-HaloTag, pKmc-2xCOX8-Halo or pmKate2-Halo-ER plasmids using Lipofectamine 3000, and the cells were incubated at 37 °C for 24 h. Then, the cells were washed three times with HBSS and incubated in FBS-free DMEM containing 3 μM MGH(AM) for 1 h in a CO2 incubator. After washing with HBSS, fluorescence images were captured in DMEM containing 10% FBS using a confocal fluorescence microscope at 37 °C.Mg2+ extrusion experiments
HEK293 cells were transfected with pCMV-CNNM4-FLAG and pcDNA-3.1-(+)-Halo-NLS using Lipofectamine 2000, and the cells were incubated at 37 °C for 24 h. Then, the cells were incubated with Mg2+-loading buffer (78.1 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 40 mM MgCl2, 5.5 mM glucose, 5.5 mM HEPES-KOH, pH 7.4), including 5 μM MGH(AM) or 2 μM magnesium green, for 1 h at 37 °C. The cells were rinsed once with loading buffer and viewed using an epifluorescence microscope (IX81 equipped with a DP30BW camera and a USH-1030L mercury lamp; Olympus). Fluorescence was measured every 20 s (excitation at 470–490 nm and emission at 505–545 nm) under the control of the MetaMorph software (Molecular Devices). Then, the buffer was changed to a Mg2+-free buffer (MgCl2 in the loading buffer was replaced with 60 mM NaCl). The Mg2+ extrusion experiment 24 h after loading MGH(AM) to HEK293 cells was performed as follows. HEK293 cells were firstly transfected with pcDNA-3.1-(+)-Halo-NLS, and the cells were incubated at 37 °C for 24 h. The cells were incubated with 5 μM MGH(AM) in DMEM (FBS free) for 1 h at 37 °C, and then the cells were transfected with pCMV-CNNM4-FLAG at 37 °C for 24 h. After incubation with Mg2+-loading buffer for 30 min, the cells were rinsed once with loading buffer and viewed using an epifluorescence microscope.Fluorescence imaging during apoptosis
| [Mg2+]i = KdQ(R − Rmin)/(Rmax − R), |
Dose-dependent effect of 2DG and KCN
HeLa cells were transfected with ATeam or HaloTag plasmids. The cells expressing HaloTag were washed twice with HBSS, incubated with 3 μM MGH(AM) for 30 min, and then treated with 50 nM Halo-TMR for 15 min at 37 °C. After washing with HBSS, fluorescence images were captured in DMEM at 37 °C for 1 h. The 2DG and KCN were added 10 min after the imaging started.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Grant-in-Aid for Scientific Research (Grant No. 24651259, 25220207, 15H03120, 15H00818, and 16K13102), and Innovative Areas “Molecular Robotics” (No. 24104004) and “Dynamic Ordering & Integrated Functions” (No. 16H00768) of The Ministry of Education, Culture, Sports, Science, and Technology, Japan; the Japanese Society for the Promotion of Science (JSPS) through its Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST Program), the Ministry of Health, Labour and Welfare Japan and CREST from the Japan Science and Technology Agency (JST), Suzuken Memorial Foundation; the Uehara Memorial Foundation, Takeda Science Foundation, and the Research Program of “Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials” in “Network Joint Research Center for Materials and Devices”.Notes and references
- A. M. Romani, Arch. Biochem. Biophys., 2011, 512, 1–23 CrossRef CAS PubMed.
- A. Hartwig, Mutat. Res., 2001, 475, 113–121 CrossRef CAS PubMed.
- F. I. Wolf and V. Trapani, Clin. Sci., 2008, 114, 27–35 CrossRef CAS PubMed.
- A. M. Romani and A. Scarpa, Arch. Biochem. Biophys., 1992, 298, 1–12 CrossRef CAS PubMed.
- F.-Y. Li, B. Chaigne-Delalande, C. Kanellopoulou, J. C. Davis, H. F. Matthews, D. C. Douek, J. I. Cohen, G. Uzel, H. C. Su and M. J. Lenardo, Nature, 2011, 475, 471–476 CrossRef CAS PubMed.
- J. H. de Baaij, J. G. Hoenderop and R. J. Bindels, Physiol. Rev., 2015, 95, 1–46 CrossRef PubMed.
- Y. Funato, D. Yamazaki, S. Mizukami, L. Du, K. Kikuchi and H. Miki, J. Clin. Invest., 2014, 124, 5398–5410 Search PubMed.
- V. Trapani, G. Farruggia, C. Marraccini, S. Iotti, A. Cittadini and F. I. Wolf, Analyst, 2010, 135, 1855–1866 RSC.
- S. Orrenius, B. Zhivotovsky and P. Nicotera, Nat. Rev. Mol. Cell Biol., 2003, 4, 552–565 CrossRef CAS PubMed.
- T. Patel, S. F. Bronk and G. J. Gores, J. Clin. Invest., 1994, 94, 2183–2192 CrossRef CAS PubMed.
- M. M. Chien, K. E. Zahradka, M. K. Newell and J. H. Freed, J. Biol. Chem., 1999, 274, 7059–7066 CrossRef CAS PubMed.
- F. Di Virgilio, T. H. Steinberg, J. A. Swanson and S. C. Silverstein, J. Immunol., 1988, 140, 915–920 CAS.
- M. Mitsui, A. Abe, M. Tajimi and H. Karaki, Jpn. J. Pharmacol., 1993, 61, 165–170 CrossRef CAS PubMed.
- L. H. Lindenburg, J. L. Vinkenborg, J. Oortwijn, S. J. Aper and M. Merkx, PLoS One, 2013, 8, e82009 Search PubMed.
- V. P. Koldenkova, T. Matsuda and T. Nagai, J. Biomed. Opt., 2015, 20, 101203 Search PubMed.
- G. Zhang, J. J. Gruskos, M. S. Afzal and D. Buccella, Chem. Sci., 2015, 6, 6841–6846 RSC.
- E. Tomat, E. M. Nolan, J. Jaworski and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 15776–15777 CrossRef CAS PubMed.
- M. Bannwarth, I. R. Corrêa Jr, M. Sztretye, S. Pouvreau, C. Fellay, A. Aebischer, L. Royer, E. Ríos and K. Johnsson, ACS Chem. Biol., 2009, 4, 179–190 CrossRef CAS PubMed.
- M. Kamiya and K. Johnsson, Anal. Chem., 2010, 82, 6472–6479 CrossRef CAS PubMed.
- D. Srikun, A. E. Albers, C. I. Nam, A. T. Iavarone and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 4455–4465 CrossRef CAS PubMed.
- M. Abo, R. Minakami, K. Miyano, M. Kamiya, T. Nagano, Y. Urano and H. Sumimoto, Anal. Chem., 2014, 86, 5983–5990 CrossRef CAS PubMed.
- D. Li, L. Liu and W.-H. Li, ACS Chem. Biol., 2015, 10, 1054–1063 CrossRef CAS PubMed.
- M. Best, I. Porth, S. Hauke, F. Braun, D.-P. Herten and R. Wombacher, Org. Biomol. Chem., 2016, 14, 5606–5611 CAS.
- T. Hirata, T. Terai, H. Yamamura, M. Shimonishi, T. Komatsu, K. Hanaoka, T. Ueno, Y. Imaizumi, T. Nagano and Y. Urano, Anal. Chem., 2016, 88, 2693–2700 CrossRef CAS PubMed.
- J. J. Gruskos, G. Zhang and D. Buccella, J. Am. Chem. Soc., 2016, 138, 14639–14649 CrossRef CAS PubMed.
- T. Fujii, Y. Shindo, K. Hotta, D. Citterio, S. Nishiyama, K. Suzuki and K. Oka, J. Am. Chem. Soc., 2014, 136, 2374–2381 CrossRef CAS PubMed.
- Y. Suzuki, H. Komatsu, T. Ikeda, N. Saito, S. Araki, D. Citterio, H. Hisamoto, Y. Kitamura, T. Kubota, J. Nakagawa, K. Oka and K. Suzuki, Anal. Chem., 2002, 74, 1423–1428 CrossRef CAS PubMed.
- H. Komatsu, N. Iwasawa, D. Citterio, Y. Suzuki, T. Kubota, K. Tokuno, Y. Kitamura, K. Oka and K. Suzuki, J. Am. Chem. Soc., 2004, 126, 16353–16360 CrossRef CAS PubMed.
- Y. Shindo, T. Fujii, H. Komatsu, D. Citterio, K. Hotta, K. Suzuki and K. Oka, PLoS One, 2011, 6, e23684 CAS.
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269–272 CrossRef CAS PubMed.
- G. V. Los, L. Encell, M. McDougall, D. Hartzell, N. Karassina, C. Zimprich, M. Wood, R. Learish, R. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. Klaubert, R. Bulleit and K. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- H. Szmacinski and J. R. Lakowicz, J. Fluoresc., 1996, 6, 83–95 CrossRef CAS PubMed.
- T. Ueno, Y. Urano, K. Setsukinai, H. Takakusa, H. Kojima, K. Kikuchi, K. Ohkubo, S. Fukuzumi and T. Nagano, J. Am. Chem. Soc., 2004, 126, 14079–14085 CrossRef CAS PubMed.
- T. Miura, Y. Urano, K. Tanaka, T. Nagano, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 8666–8671 CrossRef CAS PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- B. Metten, M. Smet, N. Boens and W. Dehaen, Synthesis, 2005, 11, 1838–1844 Search PubMed.
- R. Y. Tsien, Nature, 1981, 290, 527–528 CrossRef CAS PubMed.
- C. C. Woodroofe, R. Masalha, K. R. Barnes, C. J. Frederickson and S. J. Lippard, Chem. Biol., 2004, 11, 1659–1666 CrossRef CAS PubMed.
- R. P. Haugland, Handbook of Fluorescent Probes and Research Products, Molecular Probes Inc., Eugene, Oregon, 9th edn, 2002 Search PubMed.
- D. E. Clapham, Cell, 2007, 131, 1047–1058 CrossRef CAS PubMed.
- D. Kalderon, B. L. Roberts, W. D. Richardson and A. E. Smith, Cell, 1984, 39, 499–509 CrossRef CAS PubMed.
- M. D. Resh, Cell, 1994, 76, 411–413 CrossRef CAS PubMed.
- T. Inoue, W. D. Heo, J. S. Grimley, T. J. Wandless and T. Meyer, Nat. Methods, 2005, 2, 415–418 CrossRef CAS PubMed.
- L. Filippin, M. C. Abad, S. Gastaldello, P. J. Magalhães, D. Sandonà and T. Pozzan, Cell Calcium, 2005, 37, 129–136 CrossRef CAS PubMed.
- D. Yamazaki, Y. Funato, J. Miura, S. Sato, S. Toyosawa, K. Furutani, Y. Kurachi, Y. Omori, T. Furukawa, T. Tsuda, S. Kuwabata, S. Mizukami, K. Kikuchi and H. Miki, PLoS Genet., 2013, 9, e1003983 Search PubMed.
- D. Boehning, R. L. Patterson, L. Sedaghat, N. O. Glebova, T. Kurosaki and S. H. Snyder, Nat. Cell Biol., 2003, 5, 1051–1061 CrossRef CAS PubMed.
- Y. Zhao, S. Araki, J. Wu, T. Teramoto, Y. F. Chang, M. Nakano, A. S. Abdelfattah, M. Fujiwara, T. Ishihara, T. Nagai and R. E. Campbell, Science, 2011, 333, 1888–1891 CrossRef CAS PubMed.
- A. W. Harman, A. L. Nieminen, J. J. Lemasters and B. Herman, Biochem. Biophys. Res. Commun., 1990, 170, 477–483 CrossRef CAS PubMed.
- V. Gaussin, P. Gailly, J. M. Gillis and L. Hue, Biochem. J., 1997, 326, 823–827 CrossRef CAS PubMed.
- A. Gasbarrini, A. B. Borle, H. Farghali, C. Bender, A. Francavilla and D. Van Thiel, J. Biol. Chem., 1992, 267, 6654–6663 CAS.
- M. Leist, B. Single, A. F. Castoldi, S. Kühnle and P. Nicotera, J. Exp. Med., 1997, 185, 1481–1486 CrossRef CAS PubMed.
- H. Imamura, unpublished work.
- H. Imamura, K. P. Huynh Nhat, H. Togawa, K. Saito, R. Iino, Y. Kato-Yamada, T. Nagai and H. Noji, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15651–15656 CrossRef CAS PubMed.
- P. Widlak, P. Li, X. Wang and W. T. Garrard, J. Biol. Chem., 2000, 275, 8226–8232 CrossRef CAS PubMed.
- C. C. Woodroofe, A. C. Won and S. J. Lippard, Inorg. Chem., 2005, 44, 3112–3120 CrossRef CAS PubMed.
- T. W. Traut, Mol. Cell. Biochem., 1994, 140, 1–22 CrossRef CAS PubMed.
- T. Yoshida, A. Kakizuka and H. Imamura, Sci. Rep., 2016, 6, 39618 CrossRef CAS PubMed.
- V. A. Patel, A. Longacre, K. Hsiao, H. Fan, F. Meng, J. E. Mitchell, J. Rauch, D. S. Ucker and J. S. Levine, J. Biol. Chem., 2006, 281, 4663–4670 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc03954a |
| This journal is © The Royal Society of Chemistry 2017 |