
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanically induced pyrogallol[4]arene hexamer assembly in the solid state extends the scope of molecular encapsulation†
Sara N.
Journey
 ,
Kristine L.
Teppang
,
Cesar A.
Garcia
,
Shaylyn A.
Brim
,
David
Onofrei
,
J. Bennett
Addison
,
Gregory P.
Holland
and
Byron W.
Purse
*
,
Kristine L.
Teppang
,
Cesar A.
Garcia
,
Shaylyn A.
Brim
,
David
Onofrei
,
J. Bennett
Addison
,
Gregory P.
Holland
and
Byron W.
Purse
*
Department of Chemistry and Biochemistry, San Diego State University, San Diego, California 92182, USA. E-mail: bpurse@mail.sdsu.edu
First published on 25th September 2017
Abstract
Pyrogallol[4]arene hexamers are hydrogen-bonded molecular capsules of exceptional kinetic stability that can entrap small molecule guests indefinitely, without exchange, at ambient temperatures. Here, we report on the use of a ball mill to induce self-assembly of the capsule components and the guests in the solid state. Stoichiometric amounts of pyrogallol[4]arene and a guest, which can be an arene, alkane, amine, or carboxylic acid, were milled at 30 Hz for fixed durations, dissolved, and characterization by NMR. Most of the resulting encapsulation complexes were kinetically stable but thermodynamically unstable in solution, and the yield of their formation correlates with the duration of the milling and is related to the structures of guest and host. This method extends the scope of molecular encapsulation, as demonstrated by the preparation of kinetically trapped encapsulation complexes of [2.2]paracyclophane, for which we could find no other method of preparation. To gain mechanistic insights into the solid-state assembly process, we characterized the milled powders using 13C CP-MAS NMR, we studied the effects of changing the alkane domain of the host, and we examined how dissolution conditions impact on the distribution of observed encapsulation complexes once in solution. The results support a mechanism comprising mechanically induced solid-state reorganization to produce a mixture rich in nearly or fully assembled guest-filled capsules.
Introduction
Mechanochemistry—the use of mechanical forces to alter the free energy landscape of a system, thereby facilitating chemical change—has shown exciting applications both in the controlled degradation of polymers and in the activation of synthetic pathways that are otherwise inaccessible.1–4 Following its origin as a method for material homogenization and polymer degradation typically by ball milling,5 the advent of sonochemistry in solution has led to the discovery of reaction pathways that are inaccessible by other means. One quintessential example is the electrocyclic ring opening of benzocyclobutenes in a rotatory direction selected for polymer elongation and in contravention to the Woodward–Hoffmann rules.6 More recently, mechanochemical activation in the solid state by ball milling has been extended to applications in constructive bond formation. Swager et al. have used the method for polymer synthesis rather than degradation.7 Liquid-assisted grinding has been shown to offer advantages for metal-mediated coupling reactions.8,9Ball milling as a mechanochemical method has also gained attention in supramolecular chemistry, where it has been used to facilitate noncovalent self-assembly in the solid state.10 Most examples have involved the formation of relatively simple dimeric complexes driven by hydrogen bonding, salt bridge formation, or metal coordination. The first example of milling induced guest encapsulation, appearing in 2016, is Szumna's use of ball milling to facilitate the encapsulation of C60 or C70 in peptidic, dimeric molecular capsules based on a resorcin[4]arene platform.11 Milling facilitated quantitative complexation, but the same encapsulation complexes could be prepared also by refluxing solutions of host and guest. The molecular mechanism for encapsulation complex formation aided by milling has not been reported.
In this work, we investigated the application of ball milling to the synthesis of encapsulation complexes of pyrogallol[4]arene hexamers (Scheme 1). These hexamers have been the subject of numerous studies because of their remarkable self-assembly and high kinetic stability.12–21 A few highlights include mass spectroscopic evidence of intact guest encapsulation complexes of 1b6 in the gas phase and the fluorescence-based measurement of the exchange rate of pyrogallol[4]arene monomers between hexameric capsules in solution.22,23 The Atwood group has shown that a pyrogallol[4]arene can be synthesized using isovaleraldehyde (c.f. undecanal or butanal, used to synthesized 1a and 1b, respectively) in combination with pyrogallol and p-toluenesulfonic acid using liquid-assisted grinding by mortar and pestle.24 They performed solid-state NMR measurements on the resulting crude solid and observed spectra similar to those of pyrogallol[4]arene crystallized from chloroform. More recently, the Cohen group has studied pyrogallol[4]arene powders obtained by evaporation of CH2Cl2 solutions using solid-state NMR.25 They found evidence for solvent encapsulation within hexameric assemblies.
 | ||
| Scheme 1 Solvent-free methods for pyrogallol[4]arene capsule assembly provide access to kinetically trapped guest inclusion complexes that are disfavored at equilibrium in solution. In this work, we show that ball milling can provide solid-state activation to produce the same encapsulation complexes, plus others that are not accessible by any other known method. | ||
Our lab has previously shown that hexamers of 1a are the most kinetically stable known self-assembling molecular capsules governed by hydrogen bonding, with observed ΔG‡guest exchange > 30 kcal mol−1 for the best guests in some nonpolar solvents.26–28 With this high kinetic stability, encapsulation complexes can be formed by cooling molten mixtures of host and guest in the absence of solvent, and subsequently dissolving the assembled capsules.26 The resulting encapsulation complexes are thermodynamically unstable but kinetically trapped.27 Heating is required to reach equilibrium, at which solvent occupancy of the capsules is preferred. Because of the high kinetic stability and well characterized nature of these capsular assemblies, we hypothesized that they would be an ideal proving ground to test the efficacy and scope of mechanochemical capsule synthesis in the solid state. Unlike past examples of supramolecular mechanochemistry, our results are the first to show that solid-state mechanical activation can be used to obtain an equilibrium population of encapsulation complexes that differs substantially from the equilibrium positions observed in solution. This method allowed the first supramolecular synthesis of encapsulation complexes of [2.2]paracyclophane in 1a6, for which we could find no other method of preparation. Mechanistic studies and 13C CP-MAS NMR experiments support a mechanism for milling-induced reorganization in the solid state that leads to an equilibrium population rich in capsular assemblies. These assemblies have sufficient kinetic stability that they can be dissolved intact for subsequent studies in solution.
Results and discussion
Method optimization and scope of encapsulation
Past work in our lab and by others has shown that pyrogallol[4]arene hexamers are competent to encapsulate alkanes, alkenes, arenes, amines, and carboxylic acids that have suitable size and shape to fill the capsules' interiors, sometimes aided by the co-encapsulation of solvent molecules.12,17,26–31 The encapsulation of amines and carboxylic acids is observed at equilibrium in solution, whereas solvent occupancy of the capsules is typical for alkanes, alkenes, and arenes at the mM concentrations used for most solution phase studies.26–28,30 These guests can be loaded efficiently into the capsules using the melting and cooling cycle described above, followed by dissolution in aprotic solvents for solution studies under nonequilibrium conditions.26,27First, we sought to determine if and to what extent ball milling could induce the encapsulation of these guests by solid-state activation, and under what conditions. We chose not to use Atwood's method of liquid-assisted grinding to synthesize the pyrogallol[4]arene and to attempt to obtain guest-filled capsules at the same time.24 Our rationale was that we wanted to focus only on solid-state supramolecular transformations and Atwood's method requires catalytic p-toluenesulfonic acid, which could interfere with guest encapsulation. For our experiments, we synthesized pyrogallol[4]arenes with long (1a) and short (1b) alkyl groups using conventional methods (structures shown in Scheme 1).26,32
A series of guests was chosen based on the known encapsulation capabilities of the pyrogallol[4]arene hexamer so as to test the scope of milling-induced encapsulation complex assembly (Table 1).12,26,27,30 We also sought to determine whether this method could provide novel encapsulation complexes inaccessible by other means. Samples were prepared by loosely mixing pyrogallol[4]arene 1a with a guest (both were powders) either in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio by mass or in a stoichiometric ratio for encapsulation complex formation. The samples were placed in a 5 mL stainless steel milling chamber (screw closure with Teflon washer). A stainless steel milling ball (7 mm diameter) was then inserted into the metal chambers, which were sealed and milled at 30 Hz using a Retsch MM400 ball mill. Milling was paused and samples were removed periodically for dissolution in CDCl3 and immediately analyzed by 1H NMR. Because of slow guest exchange in solution, simple signal integration in the 1H NMR spectra was used to quantify the percent loading of the guest into the capsule (for details and spectra, see the ESI†).
:1 ratio by mass or in a stoichiometric ratio for encapsulation complex formation. The samples were placed in a 5 mL stainless steel milling chamber (screw closure with Teflon washer). A stainless steel milling ball (7 mm diameter) was then inserted into the metal chambers, which were sealed and milled at 30 Hz using a Retsch MM400 ball mill. Milling was paused and samples were removed periodically for dissolution in CDCl3 and immediately analyzed by 1H NMR. Because of slow guest exchange in solution, simple signal integration in the 1H NMR spectra was used to quantify the percent loading of the guest into the capsule (for details and spectra, see the ESI†).
| Entry | Guest (max. no. encap.)a | Conditionsb | % Encap. obs.c |
|---|---|---|---|
| a The maximum number of each guest molecule that can be encapsulated in 1a6. b All samples were milled at 30 Hz at room temperature for 50 min, except entry 9 was milled for 60 min. c % Encapsulation observed is calculated by integration of 1H NMR spectra after dissolving samples in CDCl3. d 0.3 g of pyrogallol[4]arene and 0.3 g of guest were mixed before milling. e 0.5 g pyrogallol[4]arene and stoichiometric amount of guest were combined before milling. f ND = not determined; multiple encapsulation species observed (see Fig. S10 in the ESI). | |||
| 1 | 1-Adamantanecarboxylic acid (3) | 0.3 g 1a + 0.3 g guestd | 31 ± 10% |
| 2 | 1-Adamantanecarboxylic acid (3) | Stochiometric 6:3 ratio of 1a to gueste |
22 ± 5% |
| 3 | Anthracene (3) | Stochiometric 6:3 ratio of 1a to gueste |
65 ± 15% |
| 4 | Carbazole (4) | 0.3 g 1a + 0.3 g guestd | 0% |
| 5 | Coumarin (5) | 0.3 g 1a + 0.3 g guestd | 0% |
| 6 | 1,5-Diaminonaphthalene (4) | 0.3 g 1a + 0.3 g guestd | 20 ± 15% |
| 7 | 1,5-Diaminonaphthalene (4) | Stoichiometric 6:4 ratio of 1a to gueste |
30 ± 5% |
| 8 | Fluoranthene (3) | 0.3 g 1a + 0.3 g guestd | >95% |
| 9 | Fluoranthene (3) | Stochiometric 6: 3 ratio of 1a to gueste | 85 ± 8% |
| 10 | Fluorene (4) | 0.3 g 1a + 0.3 g guestd | 50 ± 15% |
| 11 | Fluorene (4) | 0.3 g 1b + 0.3 g guestd | 13 ± 3% |
| 12 | Fluorene (4) | Stochiometric 6:4 ratio of 1a to gueste |
81 ± 6% |
| 13 | [2.2]Paracyclophane (3) | 0.3 g 1a + 0.3 g guestd | 12 ± 3% |
| 14 | [2.2]Paracyclophane (3) | Stochiometric 6:3 ratio of 1a to gueste |
9 ± 3% |
| 15 | Pyrene (3) | 0.3 g 1a + 0.3 g guestd | NDf |
| 16 | Pyrene (3) | Stochiometric 6:3 ratio of 1a to gueste |
NDf |
Optimization studies revealed that the majority of the experiments reached an encapsulation maximum in less than 50 minutes of ball milling. Plots of % guest encapsulation vs. time for fluorene, 1-adamantanecarboxylic acid, and [2.2]paracyclophane showed a plateau with increased milling time, indicating that milling-induced assembly of these encapsulation complexes is an equilibrium process in the solid state (Fig. S21, S22, S28, and S33 in the ESI†). In the case of fluorene, quadruplicate measurements appear to show a subtle trend of maximum guest encapsulation at approximately 40 minutes of milling, followed by a gradual, slight decline in guest encapsulation to reach a plateau at 90–120 minutes (Fig. S22†). It is well established that the thermodynamic stability of polymorphs can change as crystals are ground from the micro to the nanoscale.33,34 At smaller sizes, polymorph stability is increasingly dominated by the surface domains of the crystals. For this reason, milling-induced changes in micro- and nanocrystallinity may well be expected to change the thermodynamic preferences for supramolecular complex assembly. All guests except fluoranthene reached maximum encapsulation in less than 50 minutes when milled with decyl pyrogallol[4]arene 1a (Table 1; individual data plots are shown in the ESI†). Fluoranthene showed greater encapsulation when milled for 60 minutes as compared with 50 minutes. We examined the effects of milling a series of potential guest molecules, decyl and propyl pyrogallol[4]arenes 1a and 1b, changes in the molar ratio of guest to host, and the addition of NaCl as a milling additive.
Solution 1H NMR studies after milling and dissolution show efficient encapsulation of fluorene, fluoranthene, anthracene, pyrene, 1,5-diaminonaphthalene, and 1-adamantanecarboxylic acid. The first four of these guests—the arenes—can be efficiently loaded into the capsules instead by melting and cooling as reported previously,26,27 but none of these encapsulation complexes is thermodynamically stable in CDCl3 (Scheme 1). Milling followed by dissolution was also effective at overcoming the kinetic barrier to encapsulation of 1,5-diaminonaphthalene and 1-adamantanecarboxylic acid. These guests are retained in the capsule at equilibrium in CDCl3,30 but the initial rate of their spontaneous loading into the capsule in solution is slower than that obtained by milling. Coumarin and carbazole are found encapsulated to a small extent using the melting–cooling cycle,26 but we were unable to observe their encapsulation as a result of ball milling.
To confirm that the dissolved capsules from milling are indeed hexamers and that guests are included in the capsules' interiors, we performed DOSY NMR measurements in CDCl3. Diffusion coefficients of 2.36 × 10−10 M2 s−1 were obtained both for hexamers from ball milling and from the melting–cooling cycle and their guests (Fig. S7–S9†). This value matches past reports of similar pyrogallol[4]arene hexamers.29,35
When testing [2.2]paracyclophane as guest, we observed efficient loading into the pyrogallol[4]arene hexamer by ball milling (Scheme 2). Encapsulation of [2.2]paracyclophane using a melting–cooling cycle fails; decomposition occurs below the melting point (285–288 °C) and no encapsulation is observed. To test whether [2.2]paracyclophane is encapsulated at equilibrium in CDCl3, we tracked changes over time in the 1H NMR spectrum of its encapsulation complex produced by ball milling. A plot of % encapsulation against time showed clear first-order kinetics of guest exchange for solvent with a half-life t1/2 = 26 ± 4 h at 295 K (Fig. S35†). At equilibrium, no encapsulated [2.2]paracyclophane could be detected. Similarly, samples prepared in CCl4, which is a less favorable guest than CDCl3,27 showed no encapsulation of [2.2]paracyclophane at equilibrium. This result is significant because it is, to the best of our knowledge, the first example of a molecular encapsulation complex for which supramolecular mechanochemistry is the only known method of preparation.
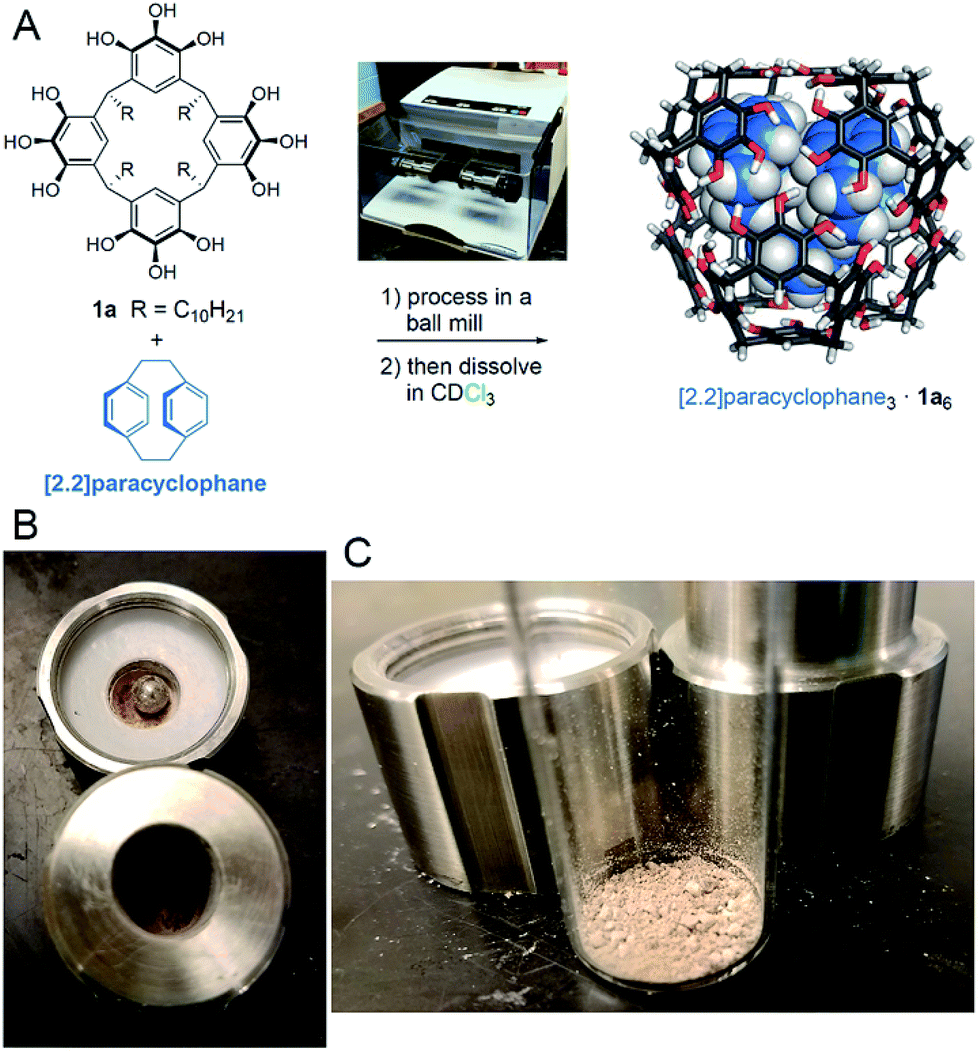 | ||
| Scheme 2 (A) Milling of 1a with [2.2]paracyclophane followed by dissolution results in the formation of its encapsulation complex in the pyrogallol[4]arene hexamer. (B, C) The appearance of mixture of 1a and [2.2]paracyclophane after milling. | ||
Next, we tested the ability of ball milling to induce encapsulation complex formation from a stoichiometric mixture of host and guest (Table 1). The maximum number of each guest molecule that can be encapsulated in a pyrogallol[4]arene hexamer was determined by experiment and molecular modeling (see Table S1 in the ESI†).26,27 Stoichiometric amounts of host and guest were combined and milled for 50 minutes at 30 Hz, followed by dissolution in CDCl3 and characterization by 1H NMR. These stoichiometric conditions showed a good efficiency of capsule loading, up to 85% in the case of fluoranthene, comparable to when excess guest was used. The utility of ball milling is evident. The use of a melting–cooling cycle to load the pyrogallol[4]arene hexamers fails if the guest is not present in excess. Ball mill-induced assembly in the solid state provides the only known stoichiometric method for synthesis of these encapsulation complexes.
Salts or other abrasives are sometimes added during milling to aid mechanical grinding. Part of the rationale for testing an additive in our context is that there was some tendency of the milled compounds to be compacted at the ends of the milling chamber and to have an appearance that was not completely homogeneous (Scheme 2). We tested the use of sodium chloride as an additive during the milling process. When NaCl was present during milling, the mechanical induction of encapsulation was completely prevented with every guest tested: anthracene, fluoranthene, and pyrene. Separately, we examined multiple samples from one milled mixture of pyrene and 1a without NaCl and observed no significant differences in the 1H NMR spectra after dissolution, indicating that the visual appearance of inhomogeneity of the milled powders is predominantly macroscopic.
All of the milling experiments discussed above involved periodically stopping the milling process and removing samples from the chamber before resuming. To test whether disturbing the samples in this way affects the outcome, we prepared independent experiments for each time point, milling fresh samples continuously and measuring % encapsulation only once at the end of the experiment (Fig. S23†). To our surprise, these data were much more scattered than those obtained from periodically sampling and resuming the same experiment (Fig. S21 and S22†). Samples tended to be compacted at the ends of the milling chamber with extended milling; periodically opening and disturbing the milled mixture was beneficial to maintaining homogeneity and obtaining efficient grinding.
Last, we investigated the potential for synergy or competition in the mechanically induced encapsulation/co-encapsulation of 1-adamantanecarboxylic acid and 1,5-diaminonaphthalene. 0.1 g of 1-adamantanecarboxylic acid, 0.1 g of 1,5-diaminonaphthalene and 0.2 g of pyrogallol[4]arene 1a were milled at 30 Hz for 50 minutes. Samples were dissolved in CDCl3 and analyzed by 1H NMR. Both molecules were observed encapsulated, although there was no evidence of co-encapsulation and the efficiency of capsule loading was less than for either guest alone.
Kinetics of milling-induced encapsulation complex formation
Having established the scope and capabilities of this mechanochemical approach to pyrogallol[4]arene encapsulation complex synthesis, we next sought to study the relationships between host structure, guest structure, and the kinetics of mechanically induced assembly. For this study, 0.5 g of pyrogallol[4]arene 1a was combined with a stoichiometric amount of guest, the mixed powders were placed in the milling chamber, and the sample was oscillated at 30 Hz (conditions A; Table 2). Oscillation was stopped periodically and samples were removed for dissolution and analysis by 1H NMR. Percent encapsulation was determined by comparing integrated NMR signals for encapsulated guests with free guests and the host methine signal (4.4 ppm) at each time interval (see example data for fluorene and fluoranthene, Fig. 1). Initial rates were calculated using the slope from plotting % encapsulation vs. time with early data points. The formation of the encapsulation complexes was always most rapid at the earliest time points; no induction periods were observed in any experiments.| Entry | Guest (max. no. encap.) | Conditions | Initial rated/% encap. min−1 | Final % encap. obs. |
|---|---|---|---|---|
| a 0.5 g of pyrogallol[4]arene 1a combined with a stoichiometric amount of guest at milled at 30 Hz. b Solid-state passive diffusion after 2 min of ball milling. c Solid-state passive diffusion without ball milling or any other form of agitation. d Initial rate constants were calculated for the first two time points for each guest for three trials then averaged (see ESI). e Initial rate measured after ball milling had ceased. f Not applicable; the experiment was stopped after 7 days with 33 ± 10% observed guest encapsulation. g Not applicable; the experiment was stopped after 90 days with 28% observed guest encapsulation. All experiments, except entry 7, were run at least in duplicate. | ||||
| 1 | 1-Adamantanecarboxylic acid (3) | Aa | 21 ± 6 | 22 ± 5% |
| 2 | Anthracene (3) | Aa | 3.1 ± 0.7 | 65 ± 15% |
| 3 | 1,5-Diaminonaphthalene (4) | Aa | 0.6 ± 0.1 | 30 ± 5% |
| 4 | Fluoranthene (3) | Aa | 2.6 ± 0.5 | 83 ± 6% |
| 5 | Fluorene (4) | Aa | 35 ± 10 | 81 ± 6% |
| 6 | Fluorene (4) | Bb | (7 ± 1) × 10−4e | n/af |
| 7 | Fluorene (4) | Cc | 2 × 10−4 | n/ag |
| 8 | [2.2]Paracyclophane (3) | Aa | 2.5 ± 1 | 9 ± 3% |
 | ||
| Fig. 1 (A) Solution 1H NMR stacked plot (CDCl3 added after ball milling) showing progressive encapsulation of fluorene (a′–d′ signals) in pyrogallol[4]arene hexamers 1a6 as a function of total ball milling duration. (B) Percent encapsulation of fluorene and fluoranthene in 1a6 observed by dissolution and 1H NMR, as a function of ball milling duration prior to dissolution. | ||
An examination of the relationship between guest structure and the efficacy of ball milling at giving rise to encapsulation reveals little in the way of obvious trends (Chart 1). Guests with hydrogen bonding functional groups, and therefore the potential to facilitate changes in hydrogen bonding among pyrogallol[4]arene units, are both among the slowest and fastest to be encapsulated. Likewise for the nonpolar arenes, there is no clear relationship between the rate of milling-induced encapsulation and the size, shape, or melting point of the guest.
 | ||
| Chart 1 The initial rate of ball mill-induced encapsulation of six guest molecules. | ||
In contrast to the structure of the guest, host structure plays an important role in determining the efficacy of ball mill-induced encapsulation. When we used propyl pyrogallol[4]arene 1b in place of decyl pyrogallol[4]arene 1a, the efficiency with which ball milling induced encapsulation of fluorene was greatly diminished. Fluorene encapsulation in 1a exceeded 50% after milling for only a few minutes (Table 2 entry 5 and Fig. 1B), but a similar experiment with 1b showed that the rate of fluorene encapsulation complex formation was approximately 15-fold slower (Fig. S24†). The flexible alkane groups of 1a are important for facilitating the solid-state reorganization that leads to guest encapsulation. These alkanes may provide disordered, semi-mobile domains through which guest molecules—fluorene, in this case—can diffuse with mechanical assistance. Propyl pyrogallol[4]arene 1b, which is relatively more rigid, is less amenable to reorganization in the solid state.
In the course of investigating the changes that take place during milling (discussed below in Mechanistic studies), we made the observation that samples of pyrogallol[4]arene and guest continue to change in the solid state even after milling has ceased. Aging of solid samples is known to allow solid-state reorganization in some systems.36 Samples of milled mixtures of host and guest were dissolved in CDCl3 and characterized by 1H NMR as described above, but with an additional aging of the samples between milling and dissolution (method B; Table 2). Although milling was much more effective at giving rise to guest encapsulation upon dissolution, aging of the samples after a short duration of milling also led to increased guest encapsulation that correlated with the duration of aging (Table 2 entry 6 and Fig. S39†). A short duration of milling partially homogenizes the solid mixture and presumably reduces the size of micro- and nanocrystalline domains, creating conditions for enhanced encapsulation from solid-state diffusion. To further test this hypothesis, we combined stoichiometric amounts of 1a and fluorene in a glass vial and mixed the powders only by shaking the vial briefly by hand. After storing the mixture at 23 °C in the dark for 3 months, a sample was removed, dissolved in CDCl3, and the 1H spectrum was acquired immediately. Integration of the spectrum showed 28% encapsulation of fluorene. A control experiment showed no encapsulation when the combined mixture of solids was not stored for a prolonged period. These results clearly show that diffusive mixing takes place in the solid state and that this mixing leads to guest encapsulation, even with no milling-induced changes in micro- or nanocrystallinity. Clearly, this solid-state transformation cannot be explained only by a change in the equilibrium position as micro- and nanocrystalline domains are ground to smaller sizes.
The results of these studies show that ball milling has a powerful ability to assist in the formation of molecular encapsulation complexes of pyrogallol[4]arene, some of which are inaccessible by other means. However, a critical question is unanswered. What is the mechanism of this mechanically activated molecular capsule assembly (Scheme 3)? Is the guest-filled capsule fully formed in the solid state by milling (mechanism A)? Or does milling give rise to an activated state that is not the encapsulation complex, but that favors the kinetically controlled formation of this encapsulation complex (as opposed to the more stable encapsulation complex of solvent) during dissolution (mechanism B)? Perhaps both of these mechanisms operate together, in competition?
 | ||
| Scheme 3 Two alternative mechanisms for ball mill induced molecular capsule assembly in the solid state. Mechanism A involves direct formation of the complete, or nearly complete, encapsulation complex in the solid state, followed by dissolution of this complex. Mechanism B is characterized by a mechanically induced reordering of the solid into an “activated state” that leads to kinetically controlled encapsulation complex formation during dissolution, when the solvent can also function as a competing guest. | ||
Mechanistic studies
To investigate the solid-state transformation responsible for this ball mill-induced pyrogallol[4]arene encapsulation complex formation, we turned first to solid-state NMR measurements. For these experiments, we elected to study the encapsulation of fluorene, which was particularly amenable to stoichiometric capsule synthesis by milling and whose encapsulation complex has a relatively simple NMR spectrum.To prepare for 13C CP-MAS NMR measurements, we first used solution HSQC to assign 13C shifts for free and bound guests. These measurements were carried out in CCl4 using C6D6 as an external solvent lock in a coaxial NMR tube insert. The use of CCl4 instead of CDCl3 slows the kinetics of guest release (i.e. exchange for encapsulated solvent) at ambient temperature sufficiently that no significant changes are seen in the 1H and 13C NMR spectra over the course of 24 hours, more than enough time for the HSQC measurements. Based on HSQC spectral analysis, we determined that carbon atoms a and d of fluorene show the best resolved upfield shifts upon encapsulation (Fig. 2A) and hoped to use them for studying guest encapsulation by solid-state NMR.
 | ||
| Fig. 2 (A) HSQC spectrum correlating 13C and 1H shifts for free and encapsulated fluorene in 1a6. (B) Stacked plot of 13C CP-MAS NMR data. The bottom three spectra show the effects of milling 1a alone for fixed durations. The middle three spectra in red show the effects of milling a stoichiometric amount of 1a and fluorene. The top spectrum is pre-assembled fluorene4·1a6. This sample is a powder resulting from the evaporation of a CHCl3 solution of 70% fluorene-filled 1a6 capsules. | ||
13C CP-MAS NMR measurements were collected using freshly prepared samples to avoid the effects of sample aging (Fig. 2B). Pyrogallol[4]arene was milled alone and a separate experiment was performed by milling a stoichiometric amount of fluorene and 1a. Samples were removed at 1, 15, and 50 minutes and solid state NMR spectra were acquired. As a control, we prepared the fluorene4·1a6 encapsulation complex in solution (“pre-assembled fluorene4·1a6) by milling a stoichiometric mixture for 50 minutes, adding a few mL of CHCl3 to obtain dissolution, and immediately evaporating to give a powder. Removal of a small sample of this powder and characterization by solution NMR in CDCl3 showed 70% of the fluorene was encapsulated. (The remaining 1a6 capsules were solvent-filled and 30% of the fluorene was not encapsulated). We have previously shown that, when pyrogallol[4]arene encapsulation complexes are present in solution, evaporation and re-dissolution in another nonpolar solvent does not lead to guest leakage.28 The 13C CP-MAS NMR spectrum of this pre-assembled control was compared with the milled samples described above that had never been dissolved, yielding vital mechanistic information on hexameric capsule formation. Unfortunately, precise peak assignments could not be made due to overlapping resonances from host and guest and also due to the variability of chemical shifts in the solid state. Clear signals for encapsulated guest carbon atoms a′ and d′ are not visible, but this is also the case in the spectrum of the pre-assembled fluorene4·1a6. Nevertheless, we believe the structural features in the 13C CP-MAS data are sufficient for analysis. When 1a is milled alone without guest (Fig. 2 blue), the aromatic resonances are broad, heterogeneous and largely featureless. This is expected for an amorphous powder where many structural conformations of the 1a could be adopted in the solid state. When pyrogallol[4]arene 1a and fluorene are milled together, the 13C CP-MAS spectra clearly show prominent NMR signals matching the pre-assembled fluorene4·1a6 even after 1 minute, and these signals are absent when pyrogallol[4]arene is milled alone. Most notably, features at 138 ppm, 132 ppm, and 114 ppm are consistent with pre-formed capsules. However, the NMR spectra of freshly milled host–guest samples show remaining signs of disorder and heterogeneity; other signals present in the milled mixture of pyrogallol[4]arene 1a and guest likely result from additional states involving partial organization of host and guest. Based on this evidence, and cognizant that the CDCl3 used for dissolution is the thermodynamically preferred capsule occupant, we propose that milling leads directly to a substantial population of nearly or fully assembled fluorene4·1a6 encapsulation complexes in the solid state (mechanism A in Scheme 3).
To further shed light on the mechanism of milling-induced encapsulation, we studied the effect of the choice of solvent of dissolution on the distribution of encapsulation complexes as observed by solution 1H NMR. Here, we hypothesized that if ball milling leads directly to a given yield of the complete (and nearly complete) pyrogallol[4]arene hexamer encapsulation complexes in the solid state (mechanism A of Scheme 3), then the solvent of dissolution should have little impact on the observed % guest encapsulation. Alternatively, if the milled solid is an activated state that is not the complete capsule (mechanism B), then a change of solvents should substantially alter the outcome of a “race” between solvent and guest to fill the capsule during dissolution and the completion of capsule assembly.
We prepared stoichiometric mixtures of 1a and fluorene and milled them at 30 Hz for 50 minutes. Samples from the same milling chamber were removed and dissolved separately in either CDCl3 or CCl4. Our past work has shown that CCl4 is a less favorable occupant of this capsule than CDCl3 and that it greatly slows the kinetics of guest exchange.27 In the first trial, we observed 38% fluorene encapsulation in CDCl3 and 45% in CCl4. A second trial reversed this order, giving 54% in CDCl3 and 44% in CCl4. The variability between trials was larger than any difference in % encapsulation. In our opinion, this evidence is again most consistent with mechanism A. If mechanism B operates, then the activated state must be sufficiently well organized for the identity of the solvent to have little impact on the competition between assembling the capsule around guest vs. solvent molecules.
Conclusions
Mechanochemistry has grown from its origins as a degradative technique to a synthetic method with a rapidly expanding pool of applications in covalent bond formation and molecular self-assembly. In this work, we have shown that supramolecular mechanochemistry in the form of ball milling is capable of unprecedentedly complex solid-state synthesis of molecular capsules, as exemplified by the preparation of up to 10-component (including host and guest) encapsulation complexes of 1a6. It is especially significant that ball milling, in this context, is not merely accelerating the formation of molecular encapsulation complexes that are also accessible by solution synthesis. Rather, ball milling of 1a and guest gives rise to soluble encapsulation complexes that will, over time, release their guests in exchange for encapsulated solvent. The application of this solid-state methodology has given rise to a kinetically stable encapsulation complex of [2.2]paracyclophane that we were unable to prepare using any other known method. This example shows that solid-state synthesis has the capacity to extend supramolecular chemistry to the formation of new structures that may be otherwise inaccessible by self-assembly in solution.Mechanistic studies and 13C CP-MAS NMR measurements show that host and guest contribute to a solid-state reorganization during ball milling that results in a significant proportion of complete or nearly complete encapsulation complexes. While the guest molecules tested did not provide a clear relationship between structure and the efficacy of this ball milling method, it was noted that the hydrocarbon domains of the host 1a greatly facilitated assembly as compared with 1b. Presumably, the presence of disordered domains in the solid state attributable to the linear alkanes facilitates the reorganization, molecular recognition, and self-assembly in response to mechanical forces or solid-state diffusion, or both.
We expect that solid-state synthesis will continue to expand the scope of structures accessible by molecular self-assembly and that additional studies on other systems will shed light on the relationships between structure and the efficiency of solid-state supramolecular synthesis.
Experimental section
General information
Reagents, solvents, guest molecules, and synthetic precursors were purchased from commercial suppliers at ACS Reagent Grade or equivalent purity and used without further purification. Pyrogallol, butrylaldehyde, anthracene, fluoranthene, pyrene, and fluorene were obtained from Acros Organics. Hydrochloric acid, ethanol, and methanol were obtained from Fisher Scientific. Undecanal and 1,5-diaminonaphthalene were obtained from TCI. Carbazole and coumarin were obtained from Sigma-Aldrich. 1-Adamantanecarboxylic acid and [2.2]paracyclophane were obtained from Alfa Aesar and Combi-blocks respectively. Deuterated solvents for NMR spectroscopy were purchased from Cambridge Isotopes. CDCl3 was filtered through basic alumina prior to use. Pyrogallol[4]arenes 1a and 1b were synthesized using published procedures.26,321H and 13C solution NMR spectra were acquired using a Bruker Avance IIIHD 600 MHz, a Varian Unity INOVA 500 MHz, or a Varian Mercury 400 MHz spectrometer. Residual solvent peaks were used as internal standards: CHCl3 (δH = 7.26 ppm; δC = 77.16 ppm), benzene (δH = 7.16 ppm; δC = 128.06 ppm). NMR measurements performed in CCl4 used a coaxial NMR tube insert and C6D6 for an external solvent lock and chemical shift referencing.13C solid-state NMR experiments
13C solid-state NMR experiments were conducted on a 600 MHz Bruker Avance IIIHD spectrometer equipped with a 4 mm HX double resonance MAS probe. Samples were spun at the magic angle at 10 kHz. 13C chemical shifts were referenced externally to TMS by setting the downfield adamantane resonance to 38.56 ppm. 1H → 13C Cross Polarization Magic Angle Spinning (CP-MAS) conditions were optimized using crystalline glycine. For CP-MAS experiments, typical experimental conditions used were an initial 2.5 μs proton π/2 pulse, a 5 ms ramped (50%) spin-lock pulse at a maximum of 80 kHz on the 1H channel, and a square contact pulse set to the 1 spinning side band of the Hartmann–Hahn profile on the 13C channel. A 300 ppm spectral width was used with 20 ms acquisition time, a 7 second relaxation delay, and 8192 scan averages. All CP data were collected using 100 kHz Spinal-64 proton decoupling during acquisition. All spectra were processed using 40 Hz exponential line broadening before Fourier transform.Solid-state encapsulation complex assembly by ball milling
Samples of pyrogallol[4]arene 1a or 1b plus a guest molecule, either in a 1:1 ratio by mass or in a stoichiometric ratio for encapsulation complex formation, were combined as a loose mixture of powders and placed in a 5 mL stainless steel milling chamber. A stainless steel milling ball (7 mm diameter) was added and the chambers was closed and tightened firmly. The chamber was placed in a Retsch MM400 ball mill and oscillated at 30 Hz. Milling was stopped periodically and samples were removed for analysis. Sample analysis was performed by dissolving a sample in a solvent suitable for NMR spectroscopy, typically CDCl3, and recording spectra immediately. Alternatively, samples were studied using 13C CP-MAS NMR as described above. Detailed information on individual experiments, relevant spectra, and kinetic data are reported in the ESI.†
Molecular modeling
Molecular models of guest encapsulation complexes in pyrogallol[4]arene hexamers were prepared based on the crystallographic coordinates of the 1b6 hexamer as solved by Gerkensmeier et. al., CCDC 101190.32 Residual solvent molecules were removed from the cavity, guest molecules were placed on the interior, and the structure was energy minimized in Spartan'08 using AM1 and with pyrogallol[4]arene atoms frozen.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully thank San Diego State University and its University Grants Program for financial support of this research and NIH IMSD for fellowship support to K. L. T. and C. A. G. (5R25GM058906). Acknowledgment is made to the donors of the American Chemical Society Petroleum Research Fund for support of the early stages of this research.References
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- S. L. Craig, Nature, 2012, 487, 176–177 CrossRef CAS PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2011, 41, 413 RSC.
- J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- C. R. Hickenboth, J. S. Moore, S. R. White, N. R. Sottos, J. Baudry and S. R. Wilson, Nature, 2007, 446, 423–427 CrossRef CAS PubMed.
- B. Ravnsbæk and T. M. Swager, ACS Macro Lett., 2014, 3, 305–309 CrossRef.
- V. Declerck, E. Colacino, X. Bantreil, J. Martinez and F. Lamaty, Chem. Commun., 2012, 48, 11778 RSC.
- E. Tullberg, D. Peters and T. Frejd, J. Organomet. Chem., 2004, 689, 3778–3781 CrossRef CAS.
- T. Friščić, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC.
- M. Szymański, M. Wierzbicki, M. Gilski, H. Jędrzejewska, M. Sztylko, P. Cmoch, A. Shkurenko, M. Jaskólski and A. Szumna, Chem.–Eur. J., 2016, 22, 3148–3155 CrossRef PubMed.
- L. Avram and Y. Cohen, J. Am. Chem. Soc., 2003, 125, 16180–16181 CrossRef CAS PubMed.
- L. Avram and Y. Cohen, Org. Lett., 2003, 5, 1099–1102 CrossRef CAS PubMed.
- H. Kumari, S. R. Kline, W. G. Wycoff, R. L. Paul, A. V. Mossine, C. A. Deakyne and J. L. Atwood, Angew. Chem., Int. Ed., 2012, 51, 5086–5091 CrossRef CAS PubMed.
- P. Jin, S. J. Dalgarno, C. Barnes, S. J. Teat and J. L. Atwood, J. Am. Chem. Soc., 2008, 130, 17262–17263 CrossRef CAS PubMed.
- L. Avram and Y. Cohen, J. Am. Chem. Soc., 2004, 126, 11556–11563 CrossRef CAS PubMed.
- L. C. Palmer and J. Rebek, Org. Lett., 2005, 7, 787–789 CrossRef CAS PubMed.
- H. D. F. Winkler, E. V Dzyuba, J. A. W. Sklorz, N. K. Beyeh, K. Rissanen and C. A. Schalley, Chem. Sci., 2011, 2, 615 RSC.
- S. Negin, M. M. Daschbach, O. V Kulikov, N. Rath and G. W. Gokel, J. Am. Chem. Soc., 2011, 133, 3234–3237 CrossRef CAS PubMed.
- E. S. Barrett, T. J. Dale and J. Rebek, Chem. Commun., 2007, 4224–4226 RSC.
- S. J. Dalgarno, S. A. Tucker, D. B. Bassil and J. L. Atwood, Science, 2005, 309, 2037–2039 CrossRef CAS PubMed.
- N. K. Beyeh, M. Kogej, A. Åhman, K. Rissanen and C. A. Schalley, Angew. Chem., Int. Ed., 2006, 45, 5214–5218 CrossRef CAS PubMed.
- E. S. Barrett, T. J. Dale and J. Rebek, J. Am. Chem. Soc., 2008, 130, 2344–2350 CrossRef CAS PubMed.
- J. Antesberger, G. W. V. Cave, M. C. Ferrarelli, M. W. Heaven, C. L. Raston and J. L. Atwood, Chem. Commun., 2005, 892 RSC.
- L. Avram, A. Goldbourt and Y. Cohen, Angew. Chem., Int. Ed., 2016, 55, 904–907 CrossRef CAS PubMed.
- M. Kvasnica, J. C. Chapin and B. W. Purse, Angew. Chem., Int. Ed., 2011, 50, 2244–2248 CrossRef CAS PubMed.
- J. C. Chapin, M. Kvasnica and B. W. Purse, J. Am. Chem. Soc., 2012, 134, 15000–15009 CrossRef CAS PubMed.
- J. C. Chapin and B. W. Purse, Supramol. Chem., 2014, 26, 517–520 CrossRef CAS.
- Y. Cohen, T. Evan-Salem and L. Avram, Supramol. Chem., 2008, 20, 71–79 CrossRef CAS.
- V. Guralnik, L. Avram and Y. Cohen, Org. Lett., 2014, 16, 5592–5595 CrossRef CAS PubMed.
- S. Yi, E. Mileo and A. E. Kaifer, Org. Lett., 2009, 11, 5690–5693 CrossRef CAS PubMed.
- T. Gerkensmeier, W. Iwanek, C. Agena, R. Fröhlich, S. Kotila, C. Näther and J. Mattay, Eur. J. Org. Chem., 1999, 2257–2262 CrossRef CAS.
- A. Navrotsky, L. Mazeina and J. Majzlan, Science, 2008, 319, 1635–1638 CrossRef CAS PubMed.
- A. M. Belenguer, G. I. Lampronti, A. J. Cruz-Cabeza, C. A. Hunter and J. K. M. Sanders, Chem. Sci., 2016, 7, 6617–6627 RSC.
- L. Avram and Y. Cohen, Org. Lett., 2006, 8, 219–222 CrossRef CAS PubMed.
- M. J. Cliffe, C. Mottillo, R. S. Stein, D.-K. Bučar and T. Friščić, Chem. Sci., 2012, 3, 2495 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra, and tabulated data. See DOI: 10.1039/c7sc03821f |
| This journal is © The Royal Society of Chemistry 2017 |