
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA structural examination and collision cross section database for over 500 metabolites and xenobiotics using drift tube ion mobility spectrometry†
Xueyun
Zheng
 ,
Noor A.
Aly
,
Yuxuan
Zhou
,
Kevin T.
Dupuis
,
Aivett
Bilbao
,
Vanessa L.
Paurus
,
Daniel J.
Orton
,
Ryan
Wilson
,
Samuel H.
Payne
,
Richard D.
Smith
and
Erin S.
Baker
*
,
Noor A.
Aly
,
Yuxuan
Zhou
,
Kevin T.
Dupuis
,
Aivett
Bilbao
,
Vanessa L.
Paurus
,
Daniel J.
Orton
,
Ryan
Wilson
,
Samuel H.
Payne
,
Richard D.
Smith
and
Erin S.
Baker
*
Biological Sciences Division, Pacific Northwest National Laboratory, 902 Battelle Blvd, P.O. Box 999, MSIN K8-98, Richland, WA 99352, USA. E-mail: erin.baker@pnnl.gov; Tel: +1-509-371-6219
First published on 28th September 2017
Abstract
The confident identification of metabolites and xenobiotics in biological and environmental studies is an analytical challenge due to their immense dynamic range, vast chemical space and structural diversity. Ion mobility spectrometry (IMS) is widely used for small molecule analyses since it can separate isomeric species and be easily coupled with front end separations and mass spectrometry for multidimensional characterizations. However, to date IMS metabolomic and exposomic studies have been limited by an inadequate number of accurate collision cross section (CCS) values for small molecules, causing features to be detected but not confidently identified. In this work, we utilized drift tube IMS (DTIMS) to directly measure CCS values for over 500 small molecules including primary metabolites, secondary metabolites and xenobiotics. Since DTIMS measurements do not need calibrant ions or calibration like some other IMS techniques, they avoid calibration errors which can cause problems in distinguishing structurally similar molecules. All measurements were performed in triplicate in both positive and negative polarities with nitrogen gas and seven different electric fields, so that relative standard deviations (RSD) could be assessed for each molecule and structural differences studied. The primary metabolites analyzed to date have come from key metabolism pathways such as glycolysis, the pentose phosphate pathway and the tricarboxylic acid cycle, while the secondary metabolites consisted of classes such as terpenes and flavonoids, and the xenobiotics represented a range of molecules from antibiotics to polycyclic aromatic hydrocarbons. Different CCS trends were observed for several of the diverse small molecule classes and when urine features were matched to the database, the addition of the IMS dimension greatly reduced the possible number of candidate molecules. This CCS database and structural information are freely available for download at http://panomics.pnnl.gov/metabolites/ with new molecules being added frequently.
Introduction
Over the last decade, interest in metabolomic and exposomic analyses has skyrocketed due to studies demonstrating how small molecules directly influence the phenome.1–5 These analyses have illustrated that changes in small molecules, such as primary metabolites, secondary metabolites and xenobiotics, provide insight into biological and environmental alterations and improve our understanding of disease occurrence and diagnosis. Primary metabolites, secondary metabolites and xenobiotics are all typically low molecular weight molecules (<1500 Da). Primary metabolites are produced directly by the host organism and perform basic life functions such as cell division and growth, respiration, storage, and reproduction.3,6 Therefore, primary metabolites are essential for the life of an organism. On the other hand, secondary metabolites are organic compounds not directly involved in normal growth, development or reproduction but promote better health. In plants, secondary metabolites are a critical component of their survival as they enable adaptation to their environment.7 However, secondary metabolites also play a powerful role in supporting human health as consuming plant-rich diets provides numerous health benefits such as decreasing cholesterol adsorption and oxidative stress.8–10 The combined number of primary and secondary metabolites in different biological systems has been estimated as >2600 metabolites in bacterial Escherichia coli,11 >2000 in yeast Saccharomyces cerevisiae,12 >70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 in human metabolome,13 and >200000 in plants due to their high amounts of secondary metabolites.14 The numerous metabolites in these diverse systems illustrate how important they are for understanding environmental and biological changes. Xenobiotics are also known to have an important role in environmental and biological systems. Xenobiotics are introduced from exposure to exogenous sources such as dietary, medical care, and environmental pollutants, and it is estimated that most people are exposed to more than one thousand xenobiotics per day.15 However, evaluating xenobiotics can be extremely difficult as they are a structurally and physico-chemically diverse group of molecules containing atoms such as chlorine and bromine. Their varying range of concentrations (femtomolar to millimolar) and matrix of analysis (i.e., plasma, urine, saliva, sediment, etc.) also complicates studies. Moreover, xenobiotics are metabolized into numerous side-products as they are processed in the body, some of which affect the activity and efficacy of metabolism.4 Since xenobiotics also influence primary and secondary metabolites during their exposure and metabolism, it is essential to study all of these small molecules simultaneously to understand how their changes affect biological and environmental systems, thereby establishing a genotype–phenotype relationship for diseases.16,17
000 in human metabolome,13 and >200000 in plants due to their high amounts of secondary metabolites.14 The numerous metabolites in these diverse systems illustrate how important they are for understanding environmental and biological changes. Xenobiotics are also known to have an important role in environmental and biological systems. Xenobiotics are introduced from exposure to exogenous sources such as dietary, medical care, and environmental pollutants, and it is estimated that most people are exposed to more than one thousand xenobiotics per day.15 However, evaluating xenobiotics can be extremely difficult as they are a structurally and physico-chemically diverse group of molecules containing atoms such as chlorine and bromine. Their varying range of concentrations (femtomolar to millimolar) and matrix of analysis (i.e., plasma, urine, saliva, sediment, etc.) also complicates studies. Moreover, xenobiotics are metabolized into numerous side-products as they are processed in the body, some of which affect the activity and efficacy of metabolism.4 Since xenobiotics also influence primary and secondary metabolites during their exposure and metabolism, it is essential to study all of these small molecules simultaneously to understand how their changes affect biological and environmental systems, thereby establishing a genotype–phenotype relationship for diseases.16,17
The growing interest in metabolomic and exposomic studies is also inciting a need for new techniques to analyze these diverse molecules. Both targeted and untargeted (global) methods, mainly utilizing mass spectrometry (MS) or nuclear magnetic resonance (NMR) instruments, are normally used in small molecule studies.14 To date MS-based targeted studies, usually with some type of front-end separation (GC or LC), are often preferred for the confident identification of small molecules. However, targeted studies are limited by the list of molecules each sample is matched to, therefore studies are often desired. Unfortunately, there are many challenges identifying the features in global studies since the molecular identity of metabolites or xenobiotics cannot be deduced from genomics information like proteins and transcripts. Further challenges result from the immense chemical and structural diversities of potential small molecule identifications and difficulties understanding the observed fragmentation patterns or even fragmenting the molecules. Therefore, new sensitive techniques are needed to provide additional measurement characteristics, detect a greater range of small molecule concentrations (from femtomolar to millimolar levels), and evaluate the many isomers occurring in each sample which are difficult to distinguish by conventional MS or chromatographic techniques.
Ion mobility spectrometry (IMS) is capable of separating molecules that have the same m/z but different conformational arrangements such as those in metabolomic and exposomic analyses.18–28 IMS is also extremely fast and can provide additional information to present technologies through coupling schemes with front end separations (i.e. LC or GC) and MS, thereby allowing multidimensional sample characterization with increased sensitivity and no additional time needed.29 However, as the desire for IMS measurements of metabolites and xenobiotics continues to grow, so does the need for high quality collision cross section (CCS) values to evaluate the small molecule structures and better develop experimental and theoretical methods.30 Currently, many IMS techniques are available for analyzing small molecules such as drift tube IMS (DTIMS),31–34 traveling wave IMS (TWIMS),35 trapped IMS (TIMS),36 overtone IMS (OIMS),37,38 differential IMS (DIMS),39 field asymmetric IMS (FAIMS),40–42 and transversal modulation IMS (TM-IMS).43 DTIMS, however, has a distinct advantage over many of these techniques since its CCS values are a direct reflection of the 3-dimensional size of the molecules and can be performed without calibration. While calibration works extremely well in many cases, it can introduce error if the wrong calibrant ions are utilized such as has been shown when peptides were used to calibrate lipid CCS values.44,45 This error is extremely detrimental for small molecules where the isomer structures are very similar and can be convoluted if calibration is not done using the correct molecular classes. Complex biological and environmental mixtures, however, present challenges for calibration since their molecular makeup and structures vary so greatly. To date IMS-MS has only been utilized in a few metabolomic and exposomic studies due to the lack of highly accurate small molecule CCS databases for confident molecular identifications.26–28,46 The greatest amount of information available for metabolites is from TWIMS based CCS measurements.44,47–51 However, the TWIMS CCS values were derived through calibration with molecules having known CCS values, such as polyalanine, which can introduce errors if the unknowns deviate from peptide-like structures or have different atom types (i.e. Cl, Br, etc.).44,45 While several small DTIMS based CCS databases have been provided for glycans, lipids, and peptides,52–54 a DTIMS database containing xenobiotics and primary and secondary metabolites analyzed with numerous electric fields and highly accurate gauges is still needed to better understand the small molecules present in complex samples and provide training data to enable better bioinformatics tools to aid in unknown identifications.55–57
In this study we applied DTIMS-MS to characterize the structural trends and CCS values for over 500 small molecules including primary metabolites, secondary metabolites and xenobiotics. The analyzed primary metabolites represented intermediates in key metabolism pathways such as glycolysis, the pentose phosphate pathway and the tricarboxylic acid (TCA) cycle, while the secondary metabolites consisted of classes such as terpenes and flavonoids, and the xenobiotics represented molecules from antibiotics to pesticides. Using these values we detail specific trends for the diverse small molecules classes and illustrate the value added by utilizing the DTIMS measurements in library searches.
Methods
Standards
The chemical standards utilized in this manuscript were mainly purchased from Sigma-Aldrich (St. Louis, MO) at a purity of >95%, however, lipid standards were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) and a few others unavailable from Sigma-Aldrich were purchased from Santa Cruz Biotechnology (Dallas, TX), Cayman Chemical (Ann Arbor, MI), or Toronto Research Chemicals (North York, ON Canada). For the CCS analyses, each chemical was dissolved in nanopure water or analytical grade methanol, chloroform, or acetonitrile depending on their solubility. In most cases methanol was the first solvent tested due to its stable spray performance in both positive and negative mode.Sample preparations
Approximately 1 mg of each standard was weighed out in a Waters glass vial (Milford, MA) and dissolved in 1 mL of solvent. This highly concentrated solution was then diluted with solvent to create a 500 μM stock solution. A final solution concentration of 1 μM was prepared by diluting the 500 μM solution with 80:20 methanol:water and 0.1% formic acid (from Sigma-Aldrich). Each solution was then directly infused into an Agilent 6560 ion mobility-quadrupole time of flight mass spectrometry (IM-QTOF MS) platform.58,59 Primary metabolites, secondary metabolites, nucleotides, oligosaccharides, lipids and peptides were ionized with a nanoESI source where protonated, sodiated and deprotonated forms were observed, while some of the xenobiotics such as polycyclic aromatic hydrocarbons (PAHs) were ionized using atmospheric pressure chemical ionization (APCI) and/or atmospheric pressure photoionization (APPI) where radical, protonated and deprotonated forms were observed.
DTIMS analysis and CCS measurements
An Agilent 6560 IM-QTOF MS (Agilent Technologies, Santa Clara) was utilized for all of the DTIMS measurements in this manuscript. Our instrument was outfitted with the commercial gas kit (Alternate Gas Kit, Agilent) and a precision flow controller (640B, MKS Instruments) to allow for real-time pressure adjustment based on absolute readings of the drift tube pressure using a capacitance manometer (CDG 500, Agilent). This capacitance manometer provided a pressure reading accuracy of 0.2% as opposed to the standard Pirani gauge (∼5%) configured without the gas kit accessory. The thermocouple used to monitor drift gas temperature (Type K, Omega Engineering) was also repositioned to the center of the drift tube to more accurately reflect the mean drift tube temperature. For the DTIMS CCS measurements, ions were passed through the inlet glass capillary, focused by a high pressure ion funnel, and accumulated in an ion funnel trap. Ions were then pulsed into the 78.24 cm long IMS drift tube filled with ∼3.95 Torr of nitrogen gas, where they travelled under the influence of a weak electric field (10–20 V cm−1). Ions exiting the drift tube were refocused by a rear ion funnel prior to QTOF MS detection and their arrival times (tA) were recorded. The reduced mobility (Ko, the mobility scaled for standard temperature and pressure) was then determined from the instrument parameters by plotting tAversus p/V:60where L is the drift length, V is the drift voltage, t0 is the time ion spending outside of the drift cell, T is the drift gas temperature, and p is the drift gas pressure. CCS values (Ω) were then calculated using the reduced mobility and kinetic theory:60

where q is the ion charge, N is the buffer gas number density at STP, μ is the reduced mass of the ion–nitrogen collision, and kB is the Boltzmann constant. All the CCS values were measured using seven stepped electric field voltages to obtain the most accurate slope for calculation of Ko and each sample was analyzed in both positive and negative ion modes. CCS values are listed on http://panomics.pnnl.gov/metabolites/ and in Table S1.† The detailed instrument settings are in Tables S2–S4† and follow those previously published in an interlaboratory study performed at Pacific Northwest National Laboratory (PNNL, Richland, WA), Agilent Technologies (Santa Clara, CA), the University of Natural Resources and Life Sciences (BOKU, Vienna, Austria), and Vanderbilt University (VU, Nashville, TN), where the same instrument set up, sample reparation protocols and experiment conditions were used for all the measurements.61 The relative standard deviation (RSD) across laboratories was observed to be 0.3% for all stepped field measurements and 0.6% for the single field studies. To build the CCS dataset detailed, each sample was studied in triplicate and the RSD values were <1% for all molecules detailed. The Agilent IM-MS Browser software was utilized for all stepped field CCS calculations.

Type 1 diabetes (T1D) metabolomic study
Urine collected from 96 individuals (48 T1D patients and 48 healthy sibling controls) at the Children's National Medical Center (CNMC) was studied to understand the impact of the CCS database. These samples were previously analyzed to see how many features were detected with an automated solid phase extraction (SPE) method coupled to DTIMS-MS.28 For this study, the datasets were reprocessed and aligned to the new CCS database to determine possible molecular candidates for the resulting features. A human subject consent form was designed with sufficient information for the to-be-enrolled sibling pairs and their parents/guardians (for subjects less than 18 years of age) to allow them an assessment of the public benefits of the research and the confidentiality loss or health risks. The consent form and protocol were approved by the Internal Review Boards at Pacific Northwest National Laboratory (PNNL) and CNMC. Initially, the Agilent Mass Profiler software (version B.08.00 B129) was used to automatically extract features from the SPE-DTIMS-MS data files from the 96 patients using the IMFE algorithm, followed by CCS calculation and multi-sample alignment using an m/z tolerance of 15 ppm and 1% for drift time. Results were exported in text format and further analyzed with the R statistical language. Each feature detected in at least 25% of the samples was compared against our CCS database and the Human Metabolome Database (HMDB)13 using just m/z and followed by matching with both m/z and CCS. In all cases, adduct forms ([M + H]+, [M + Na]+ and [M − H]−) were considered independently for each compound since the ion mobility is different for each adduct. Candidate compounds within 15 ppm and either 0.6% or 5% CCS tolerance were considered for further analysis. The annotated features in common between our CCS database and HMDB were then compared.Results and discussion
In this study, we applied DTIMS-MS to characterize the structural trends and CCS values for >500 metabolites and xenobiotics. The workflow for the analysis of each standard is shown in Fig. 1. As detailed in the Methods section, each standard was individually injected into the DTIMS instrument for full evaluation of insource fragmentation and other possible decompositional properties. In only several cases, mixtures of <10 small molecules were used. Each standard was subjected to seven electric fields for the CCS measurements and triplicate analyses were performed in positive and negative polarity to fully understand the reproducibility and polarity differences taking place for each molecule. In our experiments, we found most small molecules had a single conformation and those with two only had a very minor peak, so we only put the single most dominant conformer in the database for each small molecule. It is possible that these small molecules mainly have one stable structure under our soft experimental conditions or if multiple structures exist, they only have subtle differences which were not separable in our DTIMS platform. Thus our methods are thoroughly noted in the ESI† Methods section. For the peptide ions in the database, we did observe multiple dominant conformations in some of the systems and these are all included in the database. All CCS values are available in Table S1† or can be downloaded at http://panomics.pnnl.gov/metabolites/. This website details each molecules CAS number, 2D chemical structure, exact mass, and DTCCSN2 for the potential ionization modes (radical, protonated, sodiated, and deprotonated). Several filters for searching are also available on the website so that the user can select compounds based on a specific pathway or molecular class. All raw DTIMS-MS data files used in making the database are also available on the NIH Metabolomics Workbench website (DataTrack ID: 927), if further evaluation is desired. | ||
| Fig. 1 The workflow for creating the small molecule database. All standards were analyzed separately or in small mixtures (<10 molecules). The standards were subjected to 7 electric fields in the DTIMS cell to calculate CCS values and all analyses were performed in triplicate in both positive and negative mode to evaluate precision and polarity differences. | ||
Primary metabolite, secondary metabolite and xenobiotic structural trends
The DTIMS CCS values in nitrogen gas (DTCCSN2) are shown in Fig. 2 for all molecules in the database with a 1+, 1− or radical charge state. Initially, the CCS values for larger nucleotides, oligosaccharides, lipids and peptides were evaluated to better understand the structural trends occurring in the small molecules. In all cases, the CCS values were measured in the same ways as defined in the Methods section. To make sure our method agreed with what is currently available in literature, popular calibrant molecules such as polyalanine were also analyzed. Fortunately, our values were within <1% error of the other DTIMS studies.45 Further validation of our method was performed in an interlaboratory study where 120 unique ion species were analyzed in four different laboratories and values for all four labs were within 0.3% relative standard deviation for all compounds.61 These results gave us confidence in further characterizing the CCS values from our study. | ||
| Fig. 2 DTCCSN2 values for the small molecules in the database were plotted with larger singly charged nucleotides, oligosaccharides, lipids and peptides to observe structural trends. (A) Illustrates an overview of all ions measured in this work. The different ionization groups are broken out as (B) protonated/radical forms (radicals are shown in black), (C) sodiated forms and (D) deprotonated forms. A single molecule may be included in each of the trendlines if it ionized in all 3 ways. | ||
Upon evaluation of the different classes of larger biological molecules with a single charge (Fig. 2A), we found distinct m/z versus CCS trend lines for each molecular class as has been observed in previous studies.58 These trends illustrate that oligosaccharides and nucleotides have the smallest CCS values for a specific m/z, while lipids are the largest, and peptides fall somewhere in-between. The structural composition of these molecules plays a large role in the DTIMS CCS values as oligosaccharides and nucleotides have cyclic structures making them the smallest while lipids are more linear and exhibit the largest CCS values. It was also interesting to note that the oligosaccharides and nucleotides have almost identical trend lines, which was easily explained since both classes contain cyclic building blocks.
To gain insight into the molecular patterns for the small molecules in the database, they were also plotted with the larger biomolecular classes (Fig. 2A). As shown in Fig. 2A, the small molecule CCS values have a wide band of values that overlapped with the trend lines for the known larger biomolecules, illustrating structural compositions from ring to linear connectivities. CCS values for the molecules between 50 and 800 m/z were extracted and plotted in Fig. 2B–D (depending on their ionization mode) for further evaluation. For the protonated and radical forms in Fig. 2B, the radical species which mainly consisted of PAHs have a trend line that lies on the left side (more compact side) of the z = 1+ protonated species due to the ring structure of these molecules. While there is overlap between the 1+ and radical species, PAHs require a different extraction technique than polar molecules, so this information can also be used in searching the database to eliminate false positives. The sodiated and deprotonated forms of molecules under 800 Da also show a somewhat wide trend line (Fig. 2C and D). The deprotonated trend (Fig. 2D) has a similar width from low to high m/z values, probably due to the always present charge repulsion of deprotonated ions in the gas phase. The sodiated trend (Fig. 2C) however widens at the higher m/z values possibility due to more potential sodium binding locations. The CCS values along with the observed ionization forms of the molecules provide important information for their detection in biological or environmental samples as certain atomic makeups cannot ionize in specific ion modes and some ionize in all three ways.
The small molecules CCS values were then cataloged into primary metabolites, secondary metabolites and xenobiotics for further structural evaluations (Fig. 3). The primary metabolites (e.g., amino acids, fatty acids, organic acids, carbohydrates, etc.) consisted of molecules directly involved in normal growth, development and reproduction. Secondary metabolites (alkaloids, terpenoids, flavonoids, etc.) were molecules not directly involved in these processes but that affect the survivability of an organism.7 Finally, the xenobiotics were foreign chemical substances not naturally produced by or expected within an organism, with the four main types studied in this work being PAHs, antibiotics, pesticides, and herbicides. Most of the primary and secondary metabolites ionized well with ESI in addition to the antibiotics, pesticides and herbicides. The nonpolar nature of some of the xenobiotics (mainly PAHs) required APCI or APPI ionization and detection of the radical and/or protonated forms. However, the metabolites of PAHs, which mainly consist of hydroxyl derivatives (OH-PAHs), could be ionized using the ESI source in negative mode. Overall, the CCS values for the primary metabolites, secondary metabolites and xenobiotics illustrated a wide CCS versus m/z band with molecules at 100 m/z stretching for ∼25 Å2 (Fig. 3A). The primary metabolites, which were the vast majority of small molecules measured in our database, had the broadest distribution of CCS values in both the positive and negative modes. The secondary metabolites had a relatively narrow CCS distribution, consistent with the fact that the secondary metabolites are usually restricted to a narrow set of species within a phylogenetic group. Similarly the xenobiotics also displayed a relatively narrow CCS distribution since many of the pesticides and herbicides have a similar structural composition to that of the PAHs.
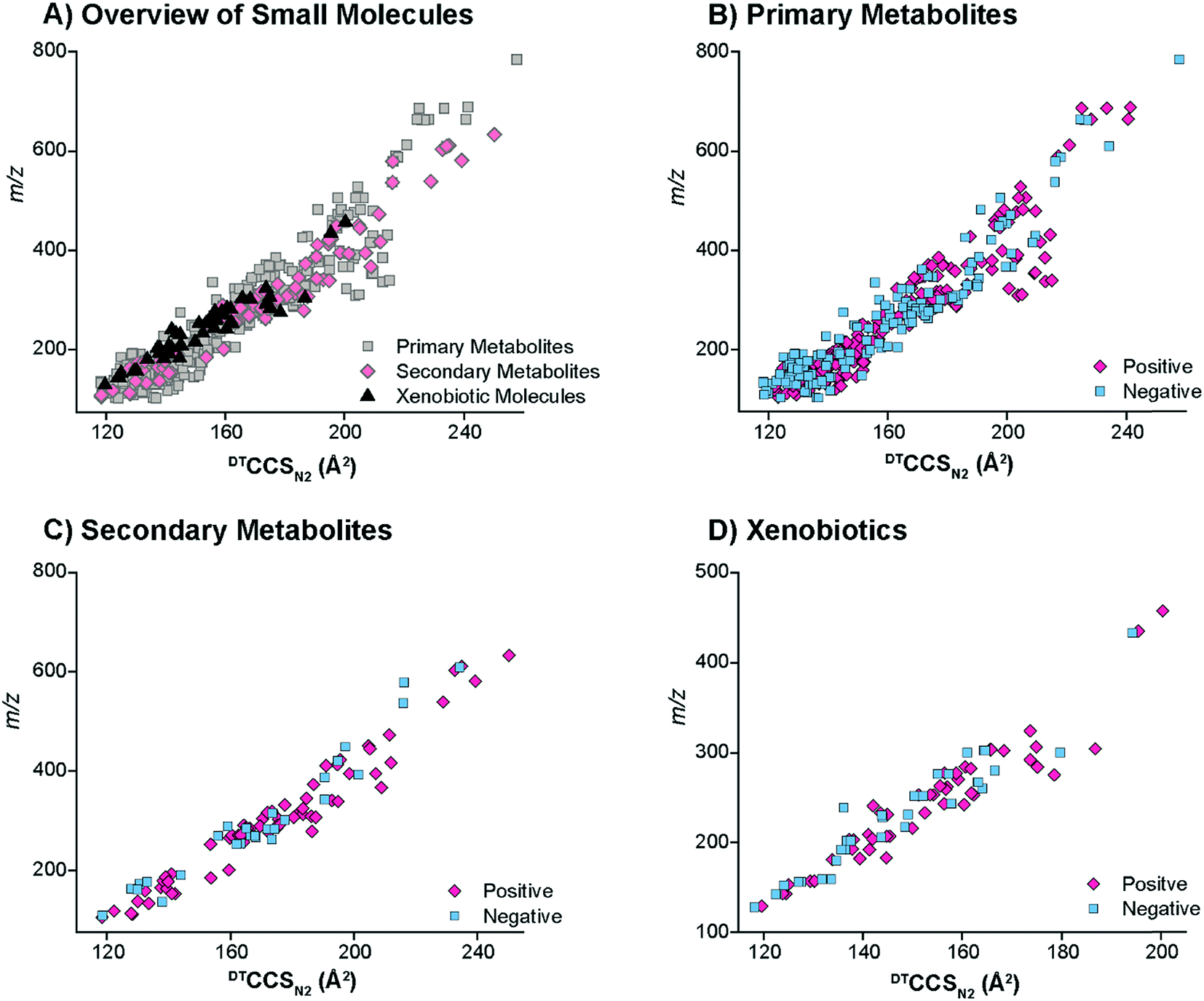 | ||
| Fig. 3 The distinct DTCCSN2 trendlines for the different classes of small molecules examined. (A) The overview of the small molecule CCS values illustrates that the primary metabolites had a wider distribution of CCS values than the secondary metabolites and xenobiotics. The positive and negative CCS values were then examined separately for the (B) primary metabolites, (C) secondary metabolites and (D) xenobiotics. Both polarities seemed to have similar overall trends for the small molecule classes. | ||
Primary metabolite structural evaluations
To understand the structural characteristics of primary metabolites, a more detailed cataloging was performed. Many primary metabolites involved in important metabolic pathways were included in this DTIMS CCS database and more small molecules are being measured and added monthly. The pathways fully characterized in the database include glycolysis, the pentose phosphate pathway, tricarboxylic acid (TCA) cycle and urea cycle (Fig. 4). All metabolites shown in Fig. 4 for the pathways have CCS values in the database except pyruvate (exact mass: 88.02) and urea (exact mass: 60.03), which unfortunately could not be observed by the instrument. By characterizing metabolite intermediates in these central carbon pathways, important application can be probed using IMS-MS, LC-IMS-MS or GC-IMS-MS such as biofuel production and determination of disease states. Interestingly, these four pathways alone have isomers such as glucose-6-phosphate and fructose-6-phosphate in glycolysis and ribose-5-phosphate and ribulose-5-phosphate in the pentose phosphate pathway. Fortunately, distinct CCS values were observed for each of the isomers and are illustrated in the database. We are also planning to characterize additional pathways so more detailed analyses can be performed using the IMS dimension. | ||
| Fig. 4 Four metabolism pathways represented in the DTCCSN2 database. The metabolite intermediates shown for the glycolysis pathway, pentose phosphate pathway, TCA cycle and urea cycle all have values in the CCS database (except urea and pyruvate). Primary metabolites from other pathways are also present in the database but in most cases all intermediates from the pathways are not accounted for. | ||
To investigate primary metabolite m/z versus CCS trends, subgroups including amino acids, fatty acids, steroids, lipid mediators, nucleosides and nucleotides (mononucleotides and dinucleotides), and oligosaccharides (up to trisaccharides) were characterized (Fig. 5). Similar to the larger molecule nucleotides/oligosaccharide relationships (almost identical trend line), the small mono-, di- and trisaccharides have an analogous trend line to the nucleosides and nucleotides. For all subgroups, amino acids represented the smallest primary metabolites as far as mass and CCS size. On the other hand, the fatty acids, lipid mediators and steroids have much larger CCS values than the other primary metabolites with similar m/z values. The distinct ionization species were then separated into the protonated, sodiated and deprotonated forms as shown in Fig. 5B–D to further investigate each trend. From these plots, we noticed immediately that most of the primary metabolites were detected in the sodiated and deprotonated forms, however, only amino acids, nucleotides and steroids were observed as protonated molecules. We also noted that in the protonated form, these three subgroups could be easily separated by their slope, while in the sodiated and deprotonated forms the separation of some groups was possible while others ran together. These trends illustrate the potential of IMS for aiding in the structural determination of unknown molecules since if the m/z and CCS value sit on a specific trendline, the unknown could be identified as similar to that specific subclass of molecules. One interesting structural characteristic observed was the change in CCS values for the steroid group between protonated and sodiated forms (Fig. 5A–C). For the steroids, sodium addition greatly increased their sizes, indicating something very specific to their structural characteristics and sodium binding. This ionization and structural information for the different small molecule subgroups will be an important characteristic to fully identify and validate their presence in biological and environmental studies.
 | ||
| Fig. 5 The DTCCSN2 trends for the distinct primary metabolites examined. (A) Illustrated the overview of primary metabolites present in the database. The three ionization modes were then pulled out to see which primary metabolites showed up as (B) protonated, (C) sodiated or (D) deprotonated ions. | ||
CCS ionization differences, isomer separations, and class distinction
After completing our initial database, we wanted to evaluate structural differences in the characterized molecules (Fig. 6). Initially, we found that molecules from the same class could display significantly different structural characteristics in positive and negative ion modes. For instance, in Fig. 6A, the protonated ion of adenosine 5′-monophosphate (AMP) has the smallest CCS and the sodiated form has the largest size (arrival time: protonated < deprotonated < sodiated). Interestingly, the deprotonated and protonated form switched for cytidine 5′-monophosphate (CMP) and the deprotonated structure became smallest (arrival time: deprotonated < protonated < sodiated). We have observed this order switching for different ion polarities before in a glycan characterization study,62 and these trends were found to be important for compound validation in complex samples. To understand if this trend was based on purine and pyridine rings, we also looked into the IMS profiles for several other nucleotides (guanosine 5′-monophosphate, uridine 5′-monophosphate, and thymidine 5′-monophosphate). Interestingly, only protonated AMP had a smaller structure than its deprotonated form, suggesting that it is structurally different from the other nucleotides and possibility indicating functional differences. | ||
| Fig. 6 Examples of small molecules in the same class or with similar m/z values displaying different IMS characteristics. (A) Shows how two nucleotides have different protonated and deprotonated trends, (B) illustrates differences in the IMS profiles for bile acids and isomeric sugars, and (C) demonstrates structural differences in molecules with the same nominal mass. | ||
Several isomeric metabolites in the CCS database were also evaluated to understand if they could be distinguished. Initially, bile acids were studied. Bile acids are amphipathic molecules with a steroid backbone synthesized from cholesterol exclusively in the liver. Many bile acids are isomers which play important roles in metabolic regulations and therefore are of great interest in metabolomic and microbiome studies.63 To evaluate the ability of DTIMS to improve the identification confidence of unknown bile acids, a list of bile acids and related isomers were included in the CCS database. Our results illustrated that IMS is able to elucidate the structures of certain bile acids. For example, α- and β-muricholic acids, two main bile acids found in mice, were well separated by DTIMS in their deprotonated forms even though their only difference is the orientation of the hydroxyl group at the C6-position (Fig. 6B). This showed that the isomer separations possible with DTIMS provided much needed information in addition to MS studies. Next, we studied ribose-5-phosphate and ribulose-5-phosphate which had much closer DTIMS profiles. These two structural pentose isomers are important in the pentose phosphate pathway where ribulose-5-phosphate can reversibly isomerize to ribose-5-phosphate. The sodiated forms of these isomers were partially distinguished by DTIMS even though they only differentiate by the position of a ketone and alcohol group (Fig. 6B). Interestingly, the large width of the IMS profile for ribose-5-phosphate illustrated that additional conformations are most likely occurring in this standard. These isomers and several others in the database illustrate the chemical complexity and structure diversity of small molecules in complex samples and how our 78 cm drift cell is not always sufficient to completely separate them. However, they all have distinct CCS values and by coupling with front end separations or chemical approaches (i.e. metal complexing with glycans to improve the isomer separations62), there is promise to fully separate these isomers. Further, new high resolution IMS techniques such as TIMS and SLIM IMS are showing great promise for resolving these molecules since they have longer path lengths and separation times, which increase the resolving power of analyses.64,65 Thus, we can utilize our database and DTIMS system to help quickly identify subsets of small molecule candidates in real biological samples and then further validation can be performed with other techniques.
Finally, we were extremely interested in nominal mass isobars from different small molecule classes that can fragment together when they co-elute or are directly injected in shotgun analyses. One of the analytical challenges for small molecule analyses is that many metabolites are similar in mass and hard to distinguish without high resolution MS. For instance, L-tyrosine (exact mass: 181.0739), an amino acid, and glufosinate (exact mass: 181.0503), an herbicide, have the same nominal mass and very close residual masses. They will also be selected in the same MS/MS window (usually the narrowest window is 0.1 Da) so their fragmentation profiles will overlap. However, our DTIMS studies showed that these two molecules have completely different sizes and can be baseline separated in either positive or negative ion modes, with negative mode showing the best distinction (Fig. 6C). These small molecule CCS ionization differences, isomer separations, and class distinction examples illustrate the value IMS structural separations have in providing more information for metabolomic and exposomic studies.
Application of the CCS database to metabolomic studies
Finally, to fully understand the value of our CCS database in small molecule analyses, we tested it on a real case/control study. Previously, due to the lack of a CCS database, we were unable to annotate SPE-DTIMS-MS features found when studying 48 urine samples from T1D patients and 48 from their sibling controls.28 Since confident small molecule identifications require at lest two orthogonal pieces of experimental data, such as exact mass of the molecular ion as well as its retention time, with the new PNNL CCS database we now have the ability to evaluate possible candidate molecular matches for each feature using m/z and CCS values.67 Additionally, it has previously been proposed that the addition of CCS to metabolomics databases will increase identification accuracy compared to traditional analytical methods alone, so this provided the perfect case to see if that was true.66In the case/control datasets, features were grouped across both the m/z and IMS dimension, aligned for multiple patients, and compared to candidate molecules within our PNNL CCS database and the HMDB. Because of differences in mobility for the various adduct forms, [M + H]+, [M + Na]+ and [M − H]− were considered independently for each molecule. From a previous TWIMS interlaboratory study49 and the recent DTIMS interlaboratory study,61 the CCS errors were noted as ∼5% between the laboratories for the TWIMS values and 0.6% for the DTIMS single field values. Thus, using the interlaboratory parameters, we matched features from the urine files to the PNNL CCS database using 15 ppm m/z and either 5% or 0.6% CCS tolerance. The results for these analyses were then compared to m/z only searching of the PNNL database and HMDB (15 ppm tolerance). While 0.3% was noted for the stepped field DTIMS values, the SPE-IMS-MS case/control study was performed using the single field method as the results are acquired to quickly to do a stepped field study so 0.6% matching is needed.
By comparing the HMDB to the PNNL database we observed that for many features the HMDB had more candidates possible since we utilized 35203 of its molecules while our database only contains ∼540. Even though we were undersampled versus the HMDB, we could still see the power of using CCS for feature matching. As shown in Fig. 7 for a subset of 19 features, the number of candidate compounds within an m/z tolerance of 15 ppm was greatly reduced when the 0.6% CCS tolerance was integrated. Interestingly from the matching results, we observed the importance of the lower CCS error (0.6% versus 5%), especially for 2α-mannobiose where at 5% error there were 15 candidate structures and at 0.6% there was only 1. In fact, the 5% CCS matching only decreased the number of candidates from those with just m/z matching for 1 of the 19 features, indicating how structurally similar many of the molecules within the same mass tolerance are and the importance of having accurate CCS values. From this study we also noted that in the case of creatinine, using the CCS value was not even required as only one molecule matched the feature using just m/z matching, even when compared to the HMDB. We would also like to note that four of the identified metabolites (phenylalanine, creatinine, lactose and sucrose) in Fig. 7 were also confirmed with GC-MS at the time of the case/control study (as shown in ref. 28), validating some of our matches. These results exhibit the utility of our CCS database and how the addition of highly accurate CCS values can reduce the number of false positives for metabolite identification and enable higher confidence when the IMS dimension is added to current LC, GC, and MS analyses. Furthermore, this database will provide the scientific community with the structural information needed to perform targeted multidimensional identifications, characterize molecular trends in complex samples, and develop better theoretical prediction software for future unknown evaluations.
 | ||
| Fig. 7 Increasing identification confidence for human urine features by adding CCS information. Features having candidate identifications in both our PNNL database and HMDB were compared. In most cases, the number of candidate molecules within an m/z tolerance of 15 ppm (orange and green bars) was significantly reduced when CCS matching was incorporated (turquoise and purple bars). The importance of highly accurate CCS values (0.6% error) was shown to greatly decrease the number of candidate molecules versus those for 5% error, which were similar to m/z only matching. | ||
Conclusions
In this work, we utilized DTIMS-MS to characterize over 500 small molecules consisting of primary metabolites, secondary metabolites and xenobiotics. All measurements were performed in triplicate with both positive and negative polarities and utilizing nitrogen gas at seven different electric fields, so that DTCCSN2 values could be directly measured and relative standard deviations assessed for each molecule. The analyzed primary metabolites originated from key metabolism pathways such as glycolysis, the pentose phosphate pathway and the TCA cycle, while the secondary metabolites consisted of classes such as terpenes and flavonoids, and the xenobiotics represented a range of molecules from antibiotics to polyaromatic hydrocarbons. Different CCS trends were observed for several of the diverse small molecule classes and allowed insight into their separation. When employed, the CCS database aided in the assignment of features from a case/control T1D study and the highly accurate DTIMS values allowed a tight tolerance to be used for CCS matching (0.6%), which greatly reduced the number of possible candidate molecules for each feature. The CCS database and structural information are freely available for download at http://panomics.pnnl.gov/metabolites/ and more molecules are being added monthly as we perform additional small molecule studies. We believe this CCS structural information will be a valuable resource for the metabolomic and exposomic communities since the values were directly measured for each molecule and do not rely on calibration curves which could induce measurement error. Therefore, these values can aid in developing better calibration mixtures for future small molecules studies with other IMS techniques such as TWIMS and TIMS needing calibrants for accurate CCS values. This CCS database will also be essential for establishing methods for unknown identifications such as determining specific structural groups present or creating better theoretical predictions models.57 Thus, these CCS values will greatly enable future metabolomic and exposomic studies by allowing others to perform more confident multidimensional identifications and create better tools to thoroughly understand small molecule changes in biological and environmental systems.Author contributions
X. Z. and E. S. B. designed the experiments, analyzed the data and wrote the manuscript. X. Z., N. A. A. and Y. Z. prepared the samples. X. Z. and D. J. O. performed the DTIMS instrumental analyses. X. Z., N. A. A., Y. Z., K. T. D., and V. L. P. analyzed the DTIMS raw files, extracted CCS values and uploaded them to the NIH Metabolomics Workbench. R. W., A. B. and S. H. P. created the website for public release of the database. R. D. S. and E. S. B. determined the samples needed for the experiments, supervised different aspects of the project, and edited the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors would like to acknowledge Agilent Technologies and Drs Thomas Metz, Jennifer Kyle, Justin Teeguarden, Albert Rivas Ubach and Ivan Rusyn for aliquots of some of the standards utilized in this database. Portions of this research were supported by grants from the National Institute of Environmental Health Sciences of the NIH (R01 ES022190), National Institute of General Medical Sciences (P41 GM103493), NIH (P42 ES027704), and the Laboratory Directed Research and Development Program at Pacific Northwest National Laboratory. This research utilized capabilities developed by the Pan-omics program (funded by the U.S. Department of Energy Office of Biological and Environmental Research Genome Sciences Program). This work was performed in the W. R. Wiley Environmental Molecular Sciences Laboratory (EMSL), a DOE national scientific user facility at the Pacific Northwest National Laboratory (PNNL). PNNL is operated by Battelle for the DOE under contract DE-AC05-76RL0 1830.References
- K. Suhre, S.-Y. Shin, A.-K. Petersen, R. P. Mohney, D. Meredith, B. Wagele, E. Altmaier, CARDIoGRAM, P. Deloukas, J. Erdmann, E. Grundberg, C. J. Hammond, M. H. de Angelis, G. Kastenmuller, A. Kottgen, F. Kronenberg, M. Mangino, C. Meisinger, T. Meitinger, H.-W. Mewes, M. V. Milburn, C. Prehn, J. Raffler, J. S. Ried, W. Romisch-Margl, N. J. Samani, K. S. Small, H. Erich Wichmann, G. Zhai, T. Illig, T. D. Spector, J. Adamski, N. Soranzo and C. Gieger, Nature, 2011, 477, 54–60 CrossRef CAS PubMed.
- M. Frédérich, B. Pirotte, M. Fillet and P. de Tullio, J. Med. Chem., 2016, 59, 8649–8666 CrossRef PubMed.
- L. Pirhaji, P. Milani, M. Leidl, T. Curran, J. Avila-Pacheco, C. B. Clish, F. M. White, A. Saghatelian and E. Fraenkel, Nat. Methods, 2016, 13, 770–776 CrossRef CAS PubMed.
- C. H. Johnson, A. D. Patterson, J. R. Idle and F. J. Gonzalez, Annu. Rev. Pharmacol. Toxicol., 2012, 52, 37–56 CrossRef CAS PubMed.
- M. T. Smith, R. de la Rosa and S. I. Daniels, Environ. Mol. Mutagen., 2015, 56, 715–723 CrossRef CAS PubMed.
- C. H. Johnson, J. Ivanisevic and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2016, 17, 451–459 CrossRef CAS PubMed.
- M. Wink, Phytochemistry, 2003, 64, 3–19 CrossRef CAS PubMed.
- N. Hounsome, B. Hounsome, D. Tomos and G. Edwards-Jones, J. Food Sci., 2008, 73, R48–R65 CrossRef CAS PubMed.
- K. B. Pandey and S. I. Rizvi, Oxid. Med. Cell. Longevity, 2009, 2, 270–278 CrossRef PubMed.
- D. O. Kennedy and E. L. Wightman, Adv. Nutr., 2011, 2, 32–50 CrossRef CAS PubMed.
- A. C. Guo, T. Jewison, M. Wilson, Y. Liu, C. Knox, Y. Djoumbou, P. Lo, R. Mandal, R. Krishnamurthy and D. S. Wishart, Nucleic Acids Res., 2013, 41, D625–D630 CrossRef CAS PubMed.
- T. Jewison, C. Knox, V. Neveu, Y. Djoumbou, A. C. Guo, J. Lee, P. Liu, R. Mandal, R. Krishnamurthy, I. Sinelnikov, M. Wilson and D. S. Wishart, Nucleic Acids Res., 2012, 40, D815–D820 CrossRef CAS PubMed.
- D. S. Wishart, D. Tzur, C. Knox, R. Eisner, A. C. Guo, N. Young, D. Cheng, K. Jewell, D. Arndt, S. Sawhney, C. Fung, L. Nikolai, M. Lewis, M.-A. Coutouly, I. Forsythe, P. Tang, S. Shrivastava, K. Jeroncic, P. Stothard, G. Amegbey, D. Block, D. D. Hau, J. Wagner, J. Miniaci, M. Clements, M. Gebremedhin, N. Guo, Y. Zhang, G. E. Duggan, G. D. MacInnis, A. M. Weljie, R. Dowlatabadi, F. Bamforth, D. Clive, R. Greiner, L. Li, T. Marrie, B. D. Sykes, H. J. Vogel and L. Querengesser, Nucleic Acids Res., 2007, 35, D521–D526 CrossRef CAS PubMed.
- H. M. Heyman, X. Zhang, K. Tang, E. S. Baker and T. O. Metz, in Encyclopedia of Spectroscopy and Spectrometry, ed. G. E. Tranter and D. W. Koppenaal, Academic Press, Oxford, 3rd edn, 2017, pp. 376–384 Search PubMed.
- C. P. Wild, Cancer Epidemiol., Biomarkers Prev., 2005, 14, 1847–1850 CAS.
- D. Houle, D. R. Govindaraju and S. Omholt, Nat. Rev. Genet., 2010, 11, 855–866 CrossRef CAS PubMed.
- J. K. Jansson and E. S. Baker, Nat. Microbiol., 2016, 1, 16049–16051 CrossRef CAS PubMed.
- T. Wyttenbach and M. Bowers, in Modern Mass Spectrometry, ed. C. Schalley, Springer, Berlin Heidelberg, 2003, ch. 6, vol. 225, pp. 207–232 Search PubMed.
- B. C. Bohrer, S. I. Merenbloom, S. L. Koeniger, A. E. Hilderbrand and D. E. Clemmer, Annu. Rev. Anal. Chem., 2008, 1, 293–327 CrossRef CAS PubMed.
- P. Dwivedi, P. Wu, S. J. Klopsch, G. J. Puzon, L. Xun and H. H. Hill, Metabolomics, 2008, 4, 63–80 CrossRef CAS.
- C. Uetrecht, R. J. Rose, E. van Duijn, K. Lorenzen and A. J. R. Heck, Chem. Soc. Rev., 2010, 39, 1633–1655 RSC.
- F. Lanucara, S. W. Holman, C. J. Gray and C. E. Eyers, Nat. Chem., 2014, 6, 281–294 CrossRef CAS PubMed.
- G. Paglia, M. Kliman, E. Claude, S. Geromanos and G. Astarita, Anal. Bioanal. Chem., 2015, 407, 4995–5007 CrossRef CAS PubMed.
- C. J. Gray, B. Thomas, R. Upton, L. G. Migas, C. E. Eyers, P. E. Barran and S. L. Flitsch, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 1688–1709 CrossRef CAS PubMed.
- C. D. Chouinard, M. S. Wei, C. R. Beekman, R. H. J. Kemperman and R. A. Yost, Clin. Chem., 2016, 62, 124–133 CAS.
- J. C. May, R. L. Gant-Branum and J. A. McLean, Curr. Opin. Biotechnol., 2016, 39, 192–197 CrossRef CAS PubMed.
- T. O. Metz, E. S. Baker, E. L. Schymanski, R. S. Renslow, D. G. Thomas, T. J. Causon, I. K. Webb, S. Hann, R. D. Smith and J. G. Teeguarden, Bioanalysis, 2016, 9, 81–98 CrossRef PubMed.
- X. Zhang, M. Romm, X. Zheng, E. M. Zink, Y.-M. Kim, K. E. Burnum-Johnson, D. J. Orton, A. Apffel, Y. M. Ibrahim, M. E. Monroe, R. J. Moore, J. N. Smith, J. Ma, R. S. Renslow, D. G. Thomas, A. E. Blackwell, G. Swinford, J. Sausen, R. T. Kurulugama, N. Eno, E. Darland, G. Stafford, J. Fjeldsted, T. O. Metz, J. G. Teeguarden, R. D. Smith and E. S. Baker, Clin Mass Spectrom, 2016, 2, 1–10 CrossRef.
- K. E. Burnum-Johnson, S. Nie, C. P. Casey, M. E. Monroe, D. J. Orton, Y. M. Ibrahim, M. A. Gritsenko, T. R. W. Clauss, A. K. Shukla, R. J. Moore, S. O. Purvine, T. Shi, W. Qian, T. Liu, E. S. Baker and R. D. Smith, Mol. Cell. Proteomics, 2016, 15, 3694–3705 CAS.
- J. C. May, C. B. Morris and J. A. McLean, Anal. Chem., 2017, 89, 1032–1044 CrossRef CAS PubMed.
- M. J. Cohen and F. W. Karasek, J. Chromatogr. Sci., 1970, 8, 330–337 CrossRef CAS.
- C. S. Hoaglund, S. J. Valentine, C. R. Sporleder, J. P. Reilly and D. E. Clemmer, Anal. Chem., 1998, 70, 2236–2242 CrossRef CAS PubMed.
- T. Wyttenbach, P. R. Kemper and M. T. Bowers, Int. J. Mass Spectrom., 2001, 212, 13–23 CrossRef CAS.
- K. Tang, A. A. Shvartsburg, H. N. Lee, D. C. Prior, M. A. Buschbach, F. Li, A. V. Tolmachev, G. A. Anderson and R. D. Smith, Anal. Chem., 2005, 77, 3330–3339 CrossRef CAS PubMed.
- S. D. Pringle, K. Giles, J. L. Wildgoose, J. P. Williams, S. E. Slade, K. Thalassinos, R. H. Bateman, M. T. Bowers and J. H. Scrivens, Int. J. Mass Spectrom., 2007, 261, 1–12 CrossRef CAS.
- K. Michelmann, J. A. Silveira, M. E. Ridgeway and M. A. Park, J. Am. Soc. Mass Spectrom., 2015, 26, 14–24 CrossRef CAS PubMed.
- S. M. Zucker, M. A. Ewing and D. E. Clemmer, Anal. Chem., 2013, 85, 10174–10179 CrossRef CAS PubMed.
- M. A. Ewing, C. R. P. Conant, S. M. Zucker, K. J. Griffith and D. E. Clemmer, Anal. Chem., 2015, 87, 5132–5138 CrossRef CAS PubMed.
- J. Rus, D. Moro, J. A. Sillero, J. Royuela, A. Casado, F. Estevez-Molinero and J. Fernández de la Mora, Int. J. Mass Spectrom., 2010, 298, 30–40 CrossRef CAS.
- R. Guevremont, J. Chromatogr. A, 2004, 1058, 3–19 CrossRef CAS PubMed.
- B. M. Kolakowski and Z. Mester, Analyst, 2007, 132, 842–864 RSC.
- L. J. Brown and C. S. Creaser, Curr. Anal. Chem., 2013, 9, 192–198 CAS.
- G. Vidal-de-Miguel, M. Macía and J. Cuevas, Anal. Chem., 2012, 84, 7831–7837 CrossRef CAS PubMed.
- M. F. Bush, Z. Hall, K. Giles, J. Hoyes, C. V. Robinson and B. T. Ruotolo, Anal. Chem., 2010, 82, 9557–9565 CrossRef CAS PubMed.
- M. F. Bush, I. D. G. Campuzano and C. V. Robinson, Anal. Chem., 2012, 84, 7124–7130 CrossRef CAS PubMed.
- J. C. May, C. R. Goodwin, J. A. McLean and A. V. Lyubimov, in Encyclopedia of Drug Metabolism and Interactions, John Wiley & Sons, Inc., 2012, ch. 10 Search PubMed.
- K. Pagel and D. J. Harvey, Anal. Chem., 2013, 85, 5138–5145 CrossRef CAS PubMed.
- J. Hofmann, W. B. Struwe, C. A. Scarff, J. H. Scrivens, D. J. Harvey and K. Pagel, Anal. Chem., 2014, 86, 10789–10795 CrossRef CAS PubMed.
- G. Paglia, J. P. Williams, L. Menikarachchi, J. W. Thompson, R. Tyldesley-Worster, S. Halldórsson, O. Rolfsson, A. Moseley, D. Grant, J. Langridge, B. O. Palsson and G. Astarita, Anal. Chem., 2014, 86, 3985–3993 CrossRef CAS PubMed.
- G. Paglia, P. Angel, J. P. Williams, K. Richardson, H. J. Olivos, J. W. Thompson, L. Menikarachchi, S. Lai, C. Walsh, A. Moseley, R. S. Plumb, D. F. Grant, B. O. Palsson, J. Langridge, S. Geromanos and G. Astarita, Anal. Chem., 2015, 87, 1137–1144 CrossRef CAS PubMed.
- K. M. Hines, D. H. Ross, K. L. Davidson, M. F. Bush and L. Xu, Anal. Chem., 2017, 89, 9023–9030 CrossRef CAS PubMed.
- K. M. Hines, J. C. May, J. A. McLean and L. Xu, Anal. Chem., 2016, 88, 7329–7336 CrossRef CAS PubMed.
- R. S. Glaskin, K. Khatri, Q. Wang, J. Zaia and C. E. Costello, Anal. Chem., 2017, 89, 4452–4460 CrossRef CAS PubMed.
- J. M. Dilger, M. S. Glover and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2017, 28, 1–11 CrossRef PubMed.
- G. B. Gonzales, G. Smagghe, S. Coelus, D. Adriaenssens, K. De Winter, T. Desmet, K. Raes and J. Van Camp, Anal. Chim. Acta, 2016, 924, 68–76 CrossRef CAS PubMed.
- W. B. Struwe, K. Pagel, J. L. P. Benesch, D. J. Harvey and M. P. Campbell, Glycoconjugate J., 2016, 33, 399–404 CrossRef CAS PubMed.
- Z. Zhou, X. Shen, J. Tu and Z.-J. Zhu, Anal. Chem., 2016, 88, 11084–11091 CrossRef CAS PubMed.
- J. C. May, C. R. Goodwin, N. M. Lareau, K. L. Leaptrot, C. B. Morris, R. T. Kurulugama, A. Mordehai, C. Klein, W. Barry, E. Darland, G. Overney, K. Imatani, G. C. Stafford, J. C. Fjeldsted and J. A. McLean, Anal. Chem., 2014, 86, 2107–2116 CrossRef CAS PubMed.
- Y. M. Ibrahim, E. S. Baker, W. F. Danielson Iii, R. V. Norheim, D. C. Prior, G. A. Anderson, M. E. Belov and R. D. Smith, Int. J. Mass Spectrom., 2015, 377, 655–662 CrossRef CAS PubMed.
- E. A. Mason and E. W. McDaniel, in Transport Properties of Ions in Gases, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 137–193 Search PubMed.
- S. M. Stow, T. J. Causon, X. Zheng, R. T. Kurulugama, T. Mairinger, J. C. May, E. Rennie, E. S. Baker, R. D. Smith, J. A. McLean, S. Hann and J. C. Fjeldsted, Anal. Chem., 2017, 89, 9048–9055 CrossRef CAS PubMed.
- X. Zheng, X. Zhang, N. S. Schocker, R. S. Renslow, D. J. Orton, J. Khamsi, R. A. Ashmus, I. C. Almeida, K. Tang, C. E. Costello, R. D. Smith, K. Michael and E. S. Baker, Anal. Bioanal. Chem., 2017, 409, 467–476 CrossRef CAS PubMed.
- T. Li and J. Y. L. Chiang, Pharmacol. Rev., 2014, 66, 948–983 CrossRef CAS PubMed.
- Y. M. Ibrahim, A. M. Hamid, L. Deng, S. V. B. Garimella, I. K. Webb, E. S. Baker and R. D. Smith, Analyst, 2017, 142, 1010–1021 RSC.
- L. Deng, Y. M. Ibrahim, E. S. Baker, N. A. Aly, A. M. Hamid, X. Zhang, X. Zheng, S. V. B. Garimella, I. K. Webb, S. A. Prost, J. A. Sandoval, R. V. Norheim, G. A. Anderson, A. V. Tolmachev and R. D. Smith, ChemistrySelect, 2016, 1, 2396–2399 CrossRef CAS PubMed.
- X. Han, K. Yang and R. W. Gross, Mass Spectrom. Rev., 2012, 31, 134–178 CrossRef CAS PubMed.
- E. L. Schymanski, H. P. Singer, J. Slobodnik, I. M. Ipolyi, P. Oswald, M. Krauss, T. Schulze, P. Haglund, T. Letzel, S. Grosse, N. S. Thomaidis, A. Bletsou, C. Zwiener, M. Ibáñez, T. Portolés, R. de Boer, M. J. Reid, M. Onghena, U. Kunkel, W. Schulz, A. Guillon, N. Noyon, G. Leroy, P. Bados, S. Bogialli, D. Stipaničev, P. Rostkowski and J. Hollender, Anal. Bioanal. Chem., 2015, 407, 6237–6255 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc03464d |
| This journal is © The Royal Society of Chemistry 2017 |