
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of electron transfer on the stability of hydrogen bonds†
Tyler M.
Porter
 ,
Gavin P.
Heim
and
Clifford P.
Kubiak
*
,
Gavin P.
Heim
and
Clifford P.
Kubiak
*
Department of Chemistry and Biochemistry, University of California San Diego, 9500 Gilman Drive, La Jolla, California 92093-0358, USA. E-mail: ckubiak@ucsd.edu
First published on 30th August 2017
Abstract
The measurement of the dimerization constants of hydrogen-bonded ruthenium complexes (12, 22, 32) linked by a self-complementary pair of 4-pyridylcarboxylic acid ligands in different redox states is reported. Using a combination of FTIR and UV/vis/NIR spectroscopies, the dimerization constants (KD) of the isovalent, neutral states, 12, 22, 32, were found to range from 75 to 130 M−1 (ΔG0 = −2.56 to −2.88 kcal mol−1), while the dimerization constants (K2−) of the isovalent, doubly-reduced states, (12)2−, (22)2−, (32)2−, were found to range from 2000 to 2500 M−1 (ΔG0 = −4.5 to −4.63 kcal mol−1). From the aforementioned values and the comproportionation constant for the mixed-valent dimers, the dimerization constants (KMV) of the mixed-valent, hydrogen-bonded dimers, (12)−, (22)−, (32)−, were found to range from 0.5 × 106 to 1.2 × 106 M−1 (ΔG0 = −7.78 to −8.31 kcal mol−1). On average, the hydrogen-bonded, mixed-valent states are stabilized by −5.27 (0.04) kcal mol−1 relative to the isovalent, neutral, hydrogen-bonded dimers and −3.47 (0.06) kcal mol−1 relative to the isovalent, dianionic hydrogen bonded dimers. Electron exchange in the mixed valence states imparts significant stability to hydrogen bonding. This is the first quantitative measurement of the strength of hydrogen bonds in the presence and absence of electronic exchange.
Main text
Electron transfer reactions are among the simplest yet most important reactions in chemistry and biology. The transfer of electrons lies at the heart of any chemical reaction and all biological energy transformations fundamentally depend on electron transfer through proteins and protein assemblies. In the last several decades, extensive experimental and theoretical investigations have been performed to elucidate the nature of electron transfer (ET) in biological energy transfer processes.1–14 Electron flow through proteins typically occurs in a site-to-site manner between redox centers separated by distances of 10 to 20 Å.12,13 Larger distances require coupling several of these site-to-site reactions such that distances upwards of 25 Å can be traversed.2,6,12–14 ET multistep mechanisms are often mediated by intervening amino acid side chains where donor–acceptor ET is favored over tunneling across bridges.2,13 ET across such groups typically proceeds across weak, non-covalent interactions as demonstrated by Gray et al. in work on mutant azurins.7,8,12,13The study of ET processes across weak, non-covalent interactions thus has important implications in understanding the nature of long range ET in biological systems, but the importance of non-covalent interactions also extends throughout the chemical sciences and affects the stability of artificial supramolecular structures,15,16 and selectivity of catalysts.17–24 In this report, we examine the fundamental relationship between non-covalent molecular interactions and ET to gain new understanding of electron transfer processes ubiquitous in biological and artificial systems.2,6–9,12,24–33
While several examples of hydrogen-bonded mixed valency have emerged over the last decade,27,34–37 our laboratory has focused on oxo-centered triruthenium clusters featuring isonicotinic acid as a bridging ligand (Fig. 1). Near-IR (NIR) spectroscopic analysis showed the appearance of intervalence charge transfer (IVCT) bands in the singly reduced, hydrogen-bonded dimers, (12)−, (22)−, (32)−, indicative of moderately coupled mixed-valent anions.38,39 In an effort to better understand the nature of ET across weak, non-covalent interactions, we compared the strength of hydrogen bonds in dimers of 1–3 in the presence and absence of electron exchange.
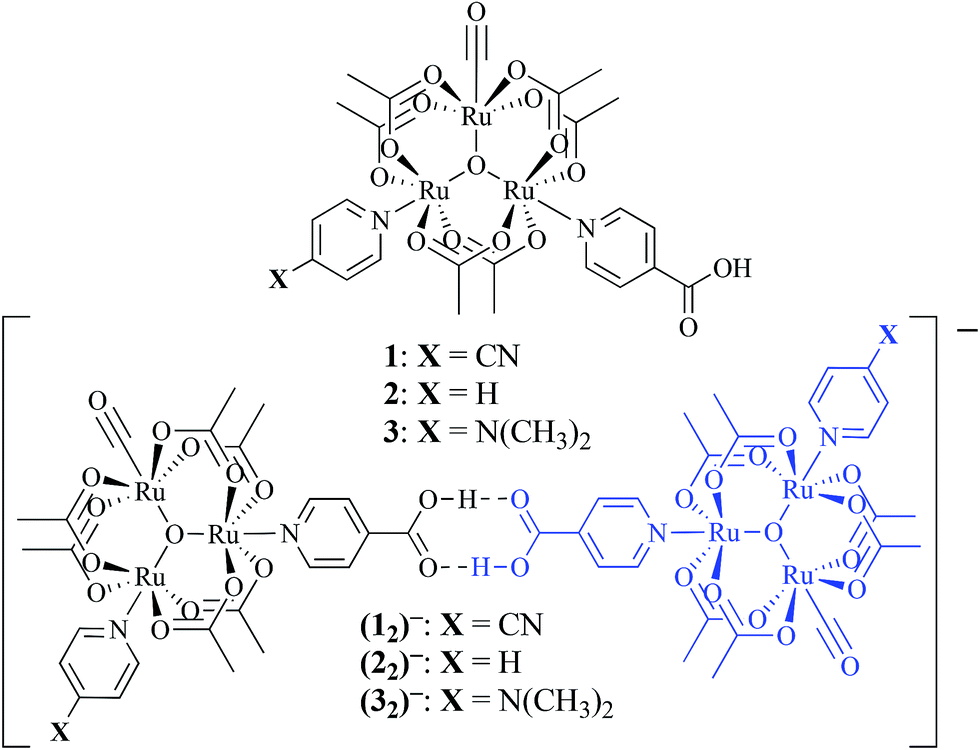 | ||
| Fig. 1 (top) Oxo-centered triruthenium cluster of the type [Ru3(μ3-O)(OAc)6(CO)(L1)(ina)] where L1 = 4-cyanopyridine (cpy, 1), pyridine (py, 2), or 4-dimethylaminopyridine (dmap, 3) and ina = isonicotinic acid. (bottom) Dimerization interaction upon a one electron reduction to generate the hydrogen-bonded, mixed-valent ions, (12)−, (22)−, (32)−. | ||
Non-covalent, mixed-valent complexes such as (12)−, (22)−, (32)−, can be described in general by four dimerization equilibria (Fig. 2). Here KD and K2− are the two isovalent equilibrium constants, which describe the self-dimerization of the neutral and one-electron reduced clusters respectively, KC is the comproportionation constant, and KMV is the equilibrium dimerization constant of the mixed-valent state. These terms offer thermodynamic information on the formation and stability of hydrogen-bonded species in the three possible redox states. The direct comparison of KMV to KD or K2− allows determination of the relative degree of stability gained from charge transfer across a hydrogen bond.
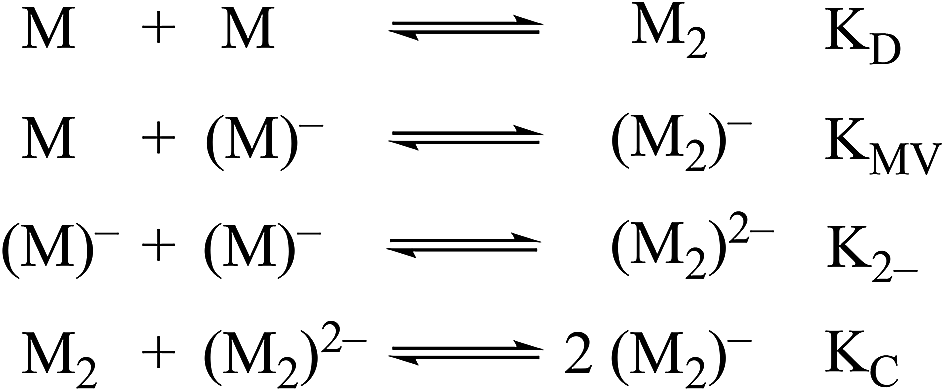 | ||
| Fig. 2 Dimerization equilibria of non-covalent, mixed-valent complexes. | ||
While several spectroscopic methods for the determination of association constants have been established,24,40–44 it is clear that determination of any three of the constants, KMV, KC, K2− and KD, provides the fourth by eqn (1).
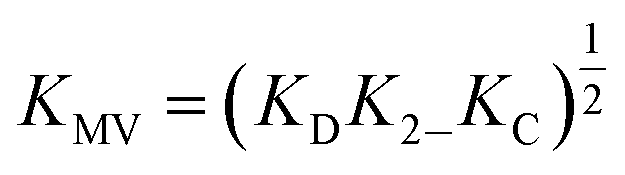 | (1) |
K
D, K2−, and KC can be readily obtained from established spectroscopic and electrochemical methods. The neutral dimerization constant (KD) was measured by FTIR spectroscopy as the acidic proton of complexes 1–3 was not resolvable in the 1H NMR but the ν(COOH) bands for the monomer (1748 cm−1) and dimer species (1711 cm−1) were well resolved in the FTIR spectrum in methylene chloride (DCM) at 25 °C (Fig. S1–S3†).24,40–44 Using a variable path length, CaF2 windowed cell set to 2.0 mm, the FTIR spectra of complexes 1–3, and their hydrogen-bonded dimers were recorded in DCM across a range of concentrations from 2.3 mM to 0.25 mM. After solvent subtraction, the ν(COOH) bands of the monomeric (1748 cm−1) and dimeric (1711 cm−1) complexes were fit as two, well resolved Gaussian functions (Fig. S4–S6†) to obtain the integrated spectral area of each band (Table S1†). KD was then determined from the eqn (2) where a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 self-dimerization model was used (Fig. S7†).40
:1 self-dimerization model was used (Fig. S7†).40
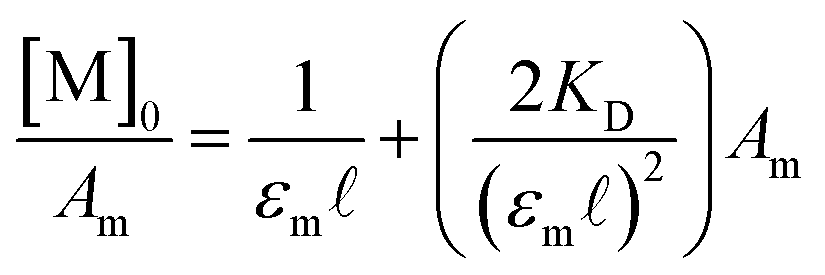 | (2) |
Here, [M]0 is the stoichiometric concentration of the solute, Am is the integrated spectral area of the monomer band, εm is the extinction coefficient and ![[small script l]](https://www.rsc.org/images/entities/char_e146.gif) the cell path length.40 Previous studies have shown that the electronic couplings in complexes (12)−, (22)−, (32)−, and Ru3O clusters in general have a large dependence on the electron-donating nature of the ancillary pyridine ligand.45–48 While similar trends in the equilibrium dimerization constant would be expected, no general trend in KD is observed and the dimerization constants remain largely independent of the ancillary ligand (Table 1, KD (M−1): 1: 119 (6), 2: 75 (5), 3: 130 (8)). In addition to treatment of the monomer band, KD can also be determined by consideration of the dimer band through eqn (3).
the cell path length.40 Previous studies have shown that the electronic couplings in complexes (12)−, (22)−, (32)−, and Ru3O clusters in general have a large dependence on the electron-donating nature of the ancillary pyridine ligand.45–48 While similar trends in the equilibrium dimerization constant would be expected, no general trend in KD is observed and the dimerization constants remain largely independent of the ancillary ligand (Table 1, KD (M−1): 1: 119 (6), 2: 75 (5), 3: 130 (8)). In addition to treatment of the monomer band, KD can also be determined by consideration of the dimer band through eqn (3).
 | (3) |
| Complex | K D (M−1) | K 2− (103 M−1) | K C (106) | K MV (106 M−1) |
|
|
|---|---|---|---|---|---|---|
a Value for K2− was only determined in THF solutions with Co(cp*)2 used as a chemical reductant.
b
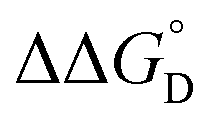 = = 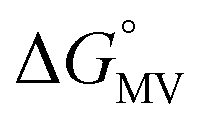 − − 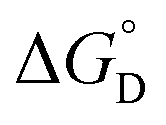 and and 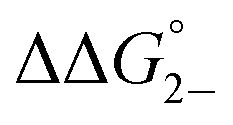 = = 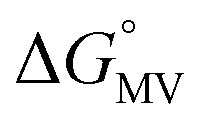 − −  . .
|
||||||
| 1 | 119 (6) | 2.0 (0.4) | 1.09 (0.04) | 0.5 (0.1) | −4.95 (0.07) | −3.3 (0.1) |
| 2 | 75 (5) | 2.2 (0.3) | 3.2 (0.1) | 0.7 (0.1) | −5.4 (0.1) | −3.4 (0.1) |
| 3 | 130 (8) | 2.5 (0.3) | 4.8 (0.2) | 1.2 (0.1) | −5.43 (0.06) | −3.68 (0.08) |


While calculation of KD should remain independent of band choice, when the dimer ν(COOH) band is used (Fig. S7†), a larger degree of uncertainty is found between the values (Table S3,†KD (M−1): 1: 450 (70), 2: 240 (90), 3: 600 (200)). This discrepancy is attributed to uncertainty found in the integrated spectral areas arising from errors in integration (Fig. S10–S12†) compounded by solvent subtraction (Fig. S8†). Regardless, further support of these results can be found by extrapolation to infinite dilution through eqn (4) as detailed by Luck (Fig. S9†).49
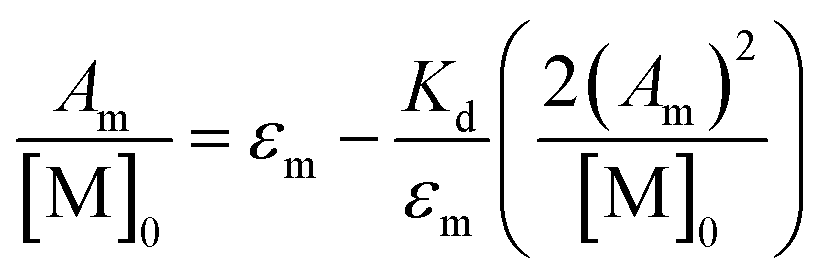 | (4) |
Here all values have their usual meanings, and excellent agreement is found upon comparison to those values determined by eqn (3) (Table S4,†KD (M−1): 1: 120 (7), 2: 73 (5), 3: 126 (9)). All three results support the notion that 1–3 form weak hydrogen bonds in solution at 25 °C ( (kcal mol−1), 1: −2.83 (0.02), 2: −2.56 (0.04), 3: −2.88 (0.04)).
(kcal mol−1), 1: −2.83 (0.02), 2: −2.56 (0.04), 3: −2.88 (0.04)).
Previous 1H DOSY NMR experiments have shown that fully reduced solutions of 1–3 consist of hydrogen bonded dimers, supporting a K2− ≥ 103.38 These findings are confirmed through the determination of K2− by UV/vis/NIR spectroscopy. Applying the same methodology for the determination of KD, the absorption spectra of (12)2−, (22)2−, (32)2−, (Fig. S10–S12†) displays a broadened, intra-cluster-charge-transfer (ICCT) band in the visible region for both the anionic monomer, (1)−, (2)−, (3)−, and the dianionic hydrogen-bonded dimer, (12)2−, (22)2−, (32)2−, species (Fig. S10–S12†). Upon comparison of the electronic spectra of similar, homoleptic clusters [Ru3(μ3-O)(OAc)6(CO)(L1)2]− where L1 = cpy, py, or dmap (Fig. S13†) which are incapable of dimerizing, it is clear to see that the broadened ICCT band consists of both monomeric and dimeric contributions.38,39,47,48,50In lieu of determining spectral areas, the peak heights of the monomeric band ((1)−: 612 nm, (2)−: 487 nm, (3)−: 550 nm) were used with eqn (2) and K2− was found to range from 2000 to 2500 M−1 (Fig. S14;†Table 1, K2− (M−1): (1)−: 2000 (400), (2)−: 2200 (300), (3)−: 2500 (300)). Unlike KD, K2− was found to increase linearly with increasing electron-donating nature of the ancillary ligand (Fig. S15†). These values are further confirmed through eqn (4), where values are nearly identical within experimental error (Fig. S14; Table S6,† K2− (M−1): (1)−: 2000 (400), (2)−: 2200 (300), (3)−: 2700 (300)) and indicate the formation of moderately strong hydrogen bonds in solution (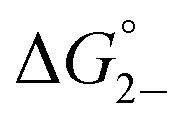 (kcal mol−1), (1)−: −4.5 (0.1), (2)−: −4.56 (0.08), (3)−: −4.63 (0.07)).
(kcal mol−1), (1)−: −4.5 (0.1), (2)−: −4.56 (0.08), (3)−: −4.63 (0.07)).
The comproportionation constant (KC), is largely a measure of the thermodynamic stability of the mixed-valent (1−) state with respect to the isovalent states (0 and 2−) and can be determined from the electrochemical splitting (ΔE) of the 0/− and −/2− redox couples measured in a cyclic voltammogram (CV) through eqn (2).51
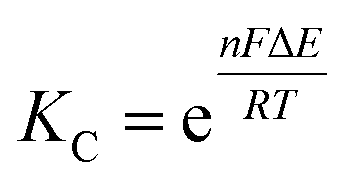 | (5) |
For complexes 1–3, the values for KC were determined from the electrochemical splittings of the return oxidation features observed in the cyclic voltammograms (CV, Fig. S16–S18†). At 23 °C in dichloromethane; for all clusters, KC is found to be on the order of 106 (1: KC = 1.09 (0.04) × 106, 2: KC = 3.2 (0.1) × 106, 3: KC = 4.8 (0.2) × 106) and increases with increasing pKa of the ancillary pyridyl ligand.
Utilizing eqn (1), KMV was found to be on average four orders of magnitude larger than KD and three orders of magnitude larger than K2−. KMV was found to range from 0.5–1.2 × 106 M−1 (Table 1) in DCM and increases linearly with increasing electron-donating nature of the ancillary pyridyl ligand (Fig. S15,† pKa: 1, cpy = 1.9; 2, py = 5.1; 3, dmap = 9.2). This effect can be explained through a ligand-field description; as stronger donor ligands are used, the Ru3O d-manifold is raised into closer energetic alignment with the isonicotinic acid π* levels, giving rise to more resonant delocalization across the hydrogen bonded dimers.52 This description is consistent with the direct mixing of metal center and bridging ligand wave-functions providing an indirect method for donor–acceptor overlap.39 The difference in free energies obtained between KMV,KD, and K2− (ΔΔG°, Table 1) reveal the relative stabilities of the mixed-valent states relative to the two isovalent hydrogen-bonded states. On average, a stabilization of −5.27 (0.04) kcal mol−1 (1850 (10) cm−1) and −3.47 (0.06) kcal mol−1 (1210 (20) cm−1) is gained upon the formation of mixed-valent, hydrogen-bonded dimers, (12)−, (22)−, (32)−, relative to the neutral, 12, 22, 32 and dianionic, (12)2−, (22)2−, (32)2−, states respectively (Fig. 3).
 | ||
| Fig. 3 Relative free energy diagram of hydrogen-bond formation, showing the additional stabilization of hydrogen bonds participating in electron delocalization. | ||
While the bonds joining the dimers of Ru3 clusters in the mixed valence states (12)−, (22)−, (32)− fulfill the definition of hydrogen bonds, their significantly larger than normal stabilities are derived from electron exchange. It is apparent that significant mixing of the metal and bridging ligand molecular orbitals in the mixed-valent states provide larger-than-expected electronic couplings for metal centers typically considered too far apart or too weakly interacting to show significant electronic interactions.39 To our knowledge, this is the first determination of the significant increase in the strength of hydrogen bonds when they participate in delocalization of an electron.
Methods
Preparation and purification
Complexes 1–3 were synthesized following previously reported procedures.38,39 The isonicotinic acid was used as received from MP Biomedical Inc. while the decamethyl ferrocene and decamethyl cobaltocene were used as received from Sigma-Aldrich. The cyclohexane stabilized dichloromethane (DCM), and tetrahydrofuran (THF) were purchased from VWR International LLC, deoxygenated and dried over alumina columns on a custom built solvent system under an argon atmosphere and stored over activated 4 Å molecular sieves in a nitrogen filled glove box.Chemical reductions
Stock solutions of 0.60 mM of 1–3 and 3.60 mM of Co(cp*)2 were prepared in dry THF under an inert atmosphere. From the stock solution of 1–3, five aliquots were prepared for each sample directly into an air tight 10 mm path length quartz cuvette ranging in concentrations from 0.13 mM to 0.03 mM. The absorption spectrum of each aliquot was recorded prior to reduction to determine the exact molarity of each aliquot. A stoichiometric amount of Co(cp*)2 was then injected into each aliquot, using a Hamilton gas-tight microsyringe, to fully reduce the samples by one electron. After injection the cell was sealed and the absorption spectrum was promptly collected.Infrared data collection and analysis
Infrared spectra were collected on a Bruker Equinox 55 FTIR spectrometer using a SPECAC variable path length IR cell with CaF2 windows set to a path length of 2.0 mm. Solutions were prepared in a glove box under a nitrogen atmosphere using pre-dried DCM and subsequently analyzed. After solvent subtraction, ν(COOH) bands were fit to two, well resolved Gaussian functions using the Igor Pro software to obtain integrated spectral areas used in the equilibrium analysis. It is important to note that calculation of the dimerization constant should remain independent of band choice; however, discrepancy between the two values in this experiment (Table S3†) are attributed to errors in solvent subtraction resulting from a slight DCM absorbance between 1730 and 1700 cm−1 coinciding with the dimeric ν(COOH) stretch at 1711 cm−1 (Fig. S8†).UV/visible data collection and analysis
UV-visible spectra were collected on a Shimadzu UV-3600 UV/vis/NIR spectrometer. Samples for determination of K2− were diluted directly into air tight 10 mm path length quartz cuvettes from stock solutions of 1–3. Samples from the determination of KD were taken directly from FTIR solutions and enclosed in a 1.0 mm path length, Hellma Analytics QS® high precision cell.Electrochemical measurements
Electrochemistry was performed on a BASi Epsilon potentiostat, in dried degassed DCM with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6, recrystallized from MeOH vacuum dried at 80 °C) used as a supporting electrolyte. Cyclic voltammograms (CVs) and differential pulse voltammograms (DPVs) were recorded at 298 K with ∼2.7 mM analyte concentrations using a three electrode setup consisting of a glassy carbon working electrode (3 mm diameter), a Pt auxiliary electrode, and an Ag/AgCl wire reference electrode. All samples were referenced to the ferrocene +/0 redox couple using an internal standard of decamethyl ferrocene (E1/2 = −0.59 vs. Fc+/0).Author contributions
T. M. P. designed and performed the experiments. G. P. H. aided in data collection and synthesis. C. P. K oversaw the project. All authors analyzed the data and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Prof. Charles W. Machan, Dr Steven A. Chabolla, Dr Jane S. Henderson, and Dr Mark M. Reineke for insightful conversation about these systems. The authors thank Dr Anthony Mrse in the UCSD NMR facility for assistance. We gratefully acknowledge support from NSF CHE-1461632.References
- D. Xia, et al., Crystal Structure of the Cytochrome bc1 Complex from Bovine Heart Mitochondria, Science, 1997, 277, 60 CrossRef CAS PubMed.
- C. Shih, et al., Tryptophan-Accelerated Electron Flow Through Proteins, Science, 2008, 320, 1760 CrossRef CAS PubMed.
- L. A. Sazanov and P. Hinchliffe, Structure of the Hydrophilic Domain of Respiratory Complex I from Thermus Thermophilus, Science, 2006, 311, 1430 CrossRef CAS PubMed.
- M. Saraste, Oxidative Phosphorylation at the fin de siecle, Science, 1999, 283, 1488 CrossRef CAS PubMed.
- S. Iwata, et al., Complete Structure of the 11-Subunit Bovine Mitochondrial Cytochrome bc1 Complex, Science, 1998, 281, 64 CrossRef CAS PubMed.
- P. Hinchliffe and L. A. Sazanov, Organization of Iron-Sulfur Clusters in Respiratory Complex I, Science, 2005, 309, 771 CrossRef CAS PubMed.
- H. B. Gray and J. R. Winkler, Electron tunneling through proteins, Q. Rev. Biophys., 2003, 36, 341–372 CrossRef CAS PubMed.
- H. B. Gray and J. R. Winkler, Long-range electron transfer, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3534–3539 CrossRef CAS PubMed.
- Z. Zhang, et al., Electron transfer by domain movement in cytochrome bc1, Nature, 1998, 392, 677–684 CrossRef CAS PubMed.
- S. Iwata, C. Ostermeier, B. Ludwig and H. Michel, Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans, Nature, 1995, 376, 660–669 CrossRef CAS PubMed.
- J. P. Abrahams, A. G. W. Leslie, R. Lutter and J. E. Walker, Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria, Nature, 1994, 370, 621–628 CrossRef CAS PubMed.
- J. R. Winkler and H. B. Gray, Electron Flow through Metalloproteins, Chem. Rev., 2014, 114, 3369–3380 CrossRef CAS PubMed.
- H. B. Gray and J. R. Winkler, Electron flow through proteins, Chem. Phys. Lett., 2009, 483, 1–9 CrossRef CAS PubMed.
- F. Sun, et al., Crystal Structure of Mitochondrial Respiratory Membrane Protein Complex II., Cell, 2005, 121, 1043–1057 CrossRef CAS PubMed.
- R. Noyori, M. Tokunaga and M. Kitamura, Stereoselective Organic Synthesis via Dynamic Kinetic Resolution, Bull. Chem. Soc. Jpn., 1995, 68, 36–55 CrossRef CAS.
- B. M. Trost, M. R. Machacek and A. Aponick, Predicting the Stereochemistry of Diphenylphosphino Benzoic Acid (DPPBA)-Based Palladium-Catalyzed Asymmetric Allylic Alkylation Reactions: A Working Model, Accounts Chem. Res., 2006, 39, 747–760 CrossRef CAS PubMed.
- R. Noyori and S. Hashiguchi, Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes, Accounts Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, Asymmetric Transfer Hydrogenation of Aromatic Ketones Catalyzed by Chiral Ruthenium(II) Complexes, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS.
- A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Exploiting Non-covalent pi Interactions for Catalyst Design, Nature, 2017, 543, 637–646 CrossRef CAS PubMed.
- X. Zhang, G. O. Jones, J. L. Hedrick and R. M. Waymouth, Fast and selective ring-opening polymerizations by alkoxides and thioureas, Nat. Chem., 2016, 8, 1047–1053 CrossRef CAS PubMed.
- M. Rakowski DuBois and D. L. DuBois, The roles of the first and second coordination spheres in the design of molecular catalysts for H2 production and oxidation, Chem. Soc. Rev., 2009, 38, 62–72 RSC.
- C. P. Casey and H. Guan, Cyclopentadienone Iron Alcohol Complexes: Synthesis, Reactivity, and Implications for the Mechanism of Iron-Catalyzed Hydrogenation of Aldehydes, J. Am. Chem. Soc., 2009, 131, 2499–2507 CrossRef CAS PubMed.
- R. R. Knowles and E. N. Jacobsen, Attractive noncovalent interactions in asymmetric catalysis: links between enzymes and small molecule catalysts, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS PubMed.
- C. W. Machan, et al., Supramolecular Assembly Promotes the Electrocatalytic Reduction of Carbon Dioxide by Re(I) Bipyridine Catalysts at a Lower Overpotential, J. Am. Chem. Soc., 2014, 136, 14598–14607 CrossRef CAS PubMed.
- T. J. Meyer, Chemical Approaches to Artificial Photosynthesis, Accounts Chem. Res., 1989, 22, 163–170 CrossRef CAS.
- J. Bonin, C. Costentin, M. Robert, J.-M. Savéant and C. Tard, Hydrogen-Bond Relays in Concerted Proton–Electron Transfers, Accounts Chem. Res., 2012, 45, 372–381 CrossRef CAS PubMed.
- M. Tadokoro, et al., Mixed-Valence States Stabilized by Proton Transfer in a Hydrogen-Bonded Biimidazolate Rhenium Dimer, Angew. Chem., Int. Ed., 2007, 46, 5938–5942 CrossRef CAS PubMed.
- M. D. Ward, Photo-Induced Electron and Energy Transfer in Non-Covalently Bonded Supramolecular Assemblies, Chem. Soc. Rev., 1997, 26, 365–375 RSC.
- T. J. Meyer, M. H. V. Huynh and H. H. Thorp, The Possible Role of Proton-Coupled Electron Transfer (PCET) in Water Oxidation by Photosystem II, Angew. Chem., Int. Ed., 2007, 46, 5284–5304 CrossRef CAS PubMed.
- M. H. V. Huynh and T. J. Meyer, Proton-Coupled Electron Transfer, Chem. Rev., 2007, 107, 5004–5064 CrossRef CAS PubMed.
- D. R. Weinberg, et al., Proton-Coupled Electron Transfer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- S. Hammes-Schiffer, Proton-Coupled Electron Transfer: Moving Together and Charging Forward, J. Am. Chem. Soc., 2015, 137, 8860–8871 CrossRef CAS PubMed.
- E. C. Gentry and R. R. Knowles, Synthetic Applications of Proton-Coupled Electron Transfer, Accounts Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed.
- H. Sun, J. Steeb and A. E. Kaifer, Efficient Electronic Communication between Two Identical Ferrocene Centers in a Hydrogen-Bonded Dimer, J. Am. Chem. Soc., 2006, 128, 2820–2821 CrossRef CAS PubMed.
- M. Pichlmaier, R. F. Winter, M. Zabel and S. Záliš, Electron Transfer Across Multiple Hydrogen Bonds: The Case of Ureapyrimidinedione-Substituted Vinyl Ruthenium and Osmium Complexes, J. Am. Chem. Soc., 2009, 131, 4892–4903 CrossRef CAS PubMed.
- L. A. Wilkinson, L. McNeill, A. J. H. M. Meijer and N. J. Patmore, Mixed Valency in Hydrogen Bonded ‘Dimers of Dimers’, J. Am. Chem. Soc., 2013, 135, 1723–1726 CrossRef CAS PubMed.
- L. Jin, Y. Matsuda, K. Uemura and M. Ebihara, Mixed Valency in Quadruple Hydrogen-Bonded Dimers of Bis(biimidazolate)dirhodium Complexes, Inorg. Chem., 2015, 54, 2331–2338 CrossRef PubMed.
- J. C. Goeltz and C. P. Kubiak, Mixed Valency across Hydrogen Bonds, J. Am. Chem. Soc., 2010, 132, 17390–17392 CrossRef CAS PubMed.
- G. Canzi, et al., On the Observation of Intervalence Charge Transfer Bands in Hydrogen-Bonded Mixed-Valence Complexes, J. Am. Chem. Soc., 2014, 136, 1710–1713 CrossRef CAS PubMed.
- D.-Y. Kao, W.-T. Shu and J.-S. Chen, A Consistent Determination of the Dimerization Constants of the Self-Association of 2,2-Dimethyl-3-ethyl-3-pentanol in Carbon Tetrachloride from its Infrared Spectral Data, J. Chin. Chem. Soc., 2005, 52, 1171–1178 CrossRef CAS.
- D. P. N. Satchell and J. L. Wardell, Dimerization of Carboxylic Acids in o-Dichlorobenzene, Trans. Faraday Soc., 1965, 61, 1199–1201 RSC.
- V. G. H. Lafitte, et al., Quadruply Hydrogen Bonded Cytosine Modules for Supramolecular Applications, J. Am. Chem. Soc., 2006, 128, 6544–6545 CrossRef CAS PubMed.
- J. T. Harris and M. E. A. Hobbs, Study of the Association of Some Organic Acids by Infrared Absorption Measurements, J. Am. Chem. Soc., 1954, 76, 1419–1422 CrossRef CAS.
- M. Andujar-Sanchez, A. Cámara-Artigas and V. Jara-Perez, Thermodynamic Study of the Dimerization of 8-Anilino-1-Naphthalenesulfonic Acid by Isothermal Titration Calorimetry, J. Chem. Thermodyn., 2010, 42, 337–341 CrossRef CAS.
- J. C. Salsman, S. Ronco, C. H. Londergan and C. P. Kubiak, Tuning the Electronic Communication and Rates of Intramolecular Electron Transfer of Dimers of Trinuclear Ruthenium Clusters: Bridging and Ancillary Ligand Effects, Inorg. Chem., 2006, 45, 547–554 CrossRef CAS PubMed.
- J. C. Goeltz, C. J. Hanson and C. P. Kubiak, Rates of Electron Self-Exchange Reactions between Oxo-Centered Ruthenium Clusters Are Determined by Orbital Overlap, Inorg. Chem., 2009, 48, 4763–4767 CrossRef CAS PubMed.
- J. C. Goeltz, E. E. Benson and C. P. Kubiak, Electronic Structural Effects in Self-Exchange Reactions, J. Phys. Chem. B, 2010, 114, 14729–14734 CrossRef CAS PubMed.
- T. M. Porter, G. C. Canzi, S. A. Chabolla and C. P. Kubiak, Tuning Electron Delocalization and Transfer Rates in Mixed-Valent Ru3O Complexes through “Push–Pull” Effects, J. Phys. Chem. A, 2016, 120, 6309–6316 CrossRef CAS PubMed.
- P. L. Huyskens, W. A. P. Luck and T. Zeegers-Huyskens, Intermolecular Forces An Introduction to Modern Methods and Results, Springer-Verlag Berlin, 1991, pp. 161–170 Search PubMed.
- J. C. Goeltz, C. J. Hanson and C. P. Kubiak, Rates of Electron Self-Exchange Reactions between Oxo-Centered Ruthenium Clusters Are Determined by Orbital Overlap, Inorg. Chem., 2009, 48, 4763–4767 CrossRef CAS PubMed.
- D. E. Richardson and H. Taube, Mixed-Valence Molecules: Electronic Delocalization and Stabilization, Coord. Chem. Rev., 1984, 60, 107–129 CrossRef CAS.
- C. P. Kubiak, Inorganic Electron Transfer: Sharpening a Fuzzy Border in Mixed Valency and Extending Mixed Valency across Supramolecular Systems, Inorg. Chem., 2013, 52, 5663–5676 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc03361c |
| This journal is © The Royal Society of Chemistry 2017 |