
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Programmed serial stereochemical relay and its application in the synthesis of morphinans†
Kun
Ho (Kenny) Park
,
Rui
Chen
and
David Y.-K.
Chen
 *
*
Department of Chemistry, Seoul National University, Gwanak-1 Gwanak-ro, Gwanak-gu, Seoul 151-742, South Korea. E-mail: davidchen@snu.ac.kr
First published on 30th August 2017
Abstract
Herein we report a rationally designed, serial point-to-axial and axial-to-point stereoinduction and its integration into multi-step and target-oriented organic synthesis. In this proof-of-concept study, the configurational stability of several carefully designed atropisomeric intermediates and the fidelity of their unconventional stereoinductions were systematically investigated. The highly functionalized prepared synthetic intermediate was further applied in a novel chemical method to access the morphinans and it is potentially applicable to other structurally related alkaloids.
Introduction
Thoughtful orchestration of substrate and reagent controlled stereochemical induction is essential in any stereoselective synthesis.1 To date, while the fundamental principles behind stereochemical induction are well-grounded, some forms of stereoinduction are much less conventional than others. For example, if we consider “point” and “axial” as two of the most common forms of stereochemical elements, one can appreciate that while the intermolecular point-to-point (for example, oxazaborolidine-mediated reduction)2 and axial-to-point (for example, Noyori BINAP hydrogenation)3 stereoinductions are routinely practiced, intramolecular point-to-axial, axial-to-point, and axial-to-axial stereoinductions are considerably more rare.4 In the asymmetric synthesis of longithorone A5a and rhazinilam,5b unconventional yet highly effective stereoinductions were elegantly illustrated by the Shair and Zakarian groups, respectively (Scheme 1a). Furthermore, we pondered if a series of these unconventional forms of stereoinduction could be logically sequenced as part of a multi-step synthetic scheme, and in doing so demonstrate their utility in the context of target-oriented synthesis (Scheme 1b).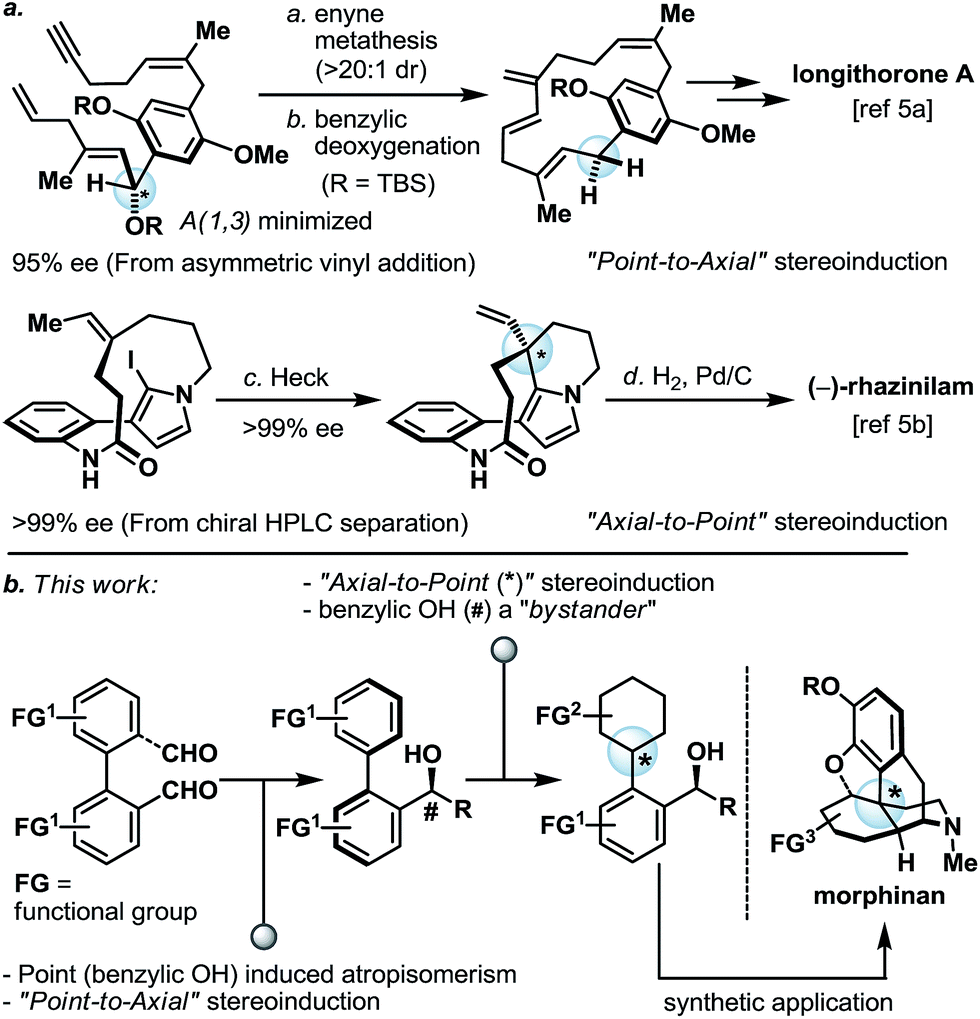 | ||
| Scheme 1 (a) Selected examples of unconventional stereochemical induction in target-oriented organic synthesis; (b) proposed point-to-axial-to-point serial stereochemical relay and its application in the synthesis of morphinans. TBS = tert-butyldimethylsilyl. | ||
In preparation for the proposed studies, it is important to recognize that while a “point” stereochemical element can be readily generated and/or identified, an “axial” stereochemical element is only observable if the system can attain a sufficiently high rotational energy barrier. In line with this requirement, we began our proof-of-concept studies by leveraging the well-established biaryl system as the source of the “axial” stereochemical element (Scheme 1b). Furthermore, the proposed biaryl system harbors additional functional group (FG) handles which allow for the subsequent programmed-stereoinduction events.
Results and discussion
We first investigated if a “point” stereochemistry that resides in close proximity to the biaryl axis can render, and hence induce, an axial stereochemical property i.e. a point-to-axial stereoinduction. In this context, the synthesis of biaryl aldehyde 4 was originally envisioned based on the desymmetrization of the readily accessible (and potentially optically active) dialdehyde 1 (Scheme 2).6 However, a more direct approach was later realized through one of the earliest illustrations of CH-activation chemistry pioneered by Dyker,7 and the as-obtained biaryl system 3 underwent smooth benzylic oxidation (DDQ) to afford the targeted aldehyde 4 together with its equilibrating hemiacetal 4′ in 65% yield over the two steps. The phenolic aldehyde 4 (and 4′) was next subjected to a selection of organometallic reagents, and gratifyingly, each organometallic addition reaction afforded a separable mixture of two stereoisomers in high yield (5a/5a′–5d/5d′, 85–97%). Recognizing that the biaryl aldehyde 4 does not have atropisomeric properties at ambient temperature,8 this finding indicated that the newly formed biaryls 5a/5a′–5d/5d′ exhibit restricted rotation and constitute an illustration of point-induced atropisomerism.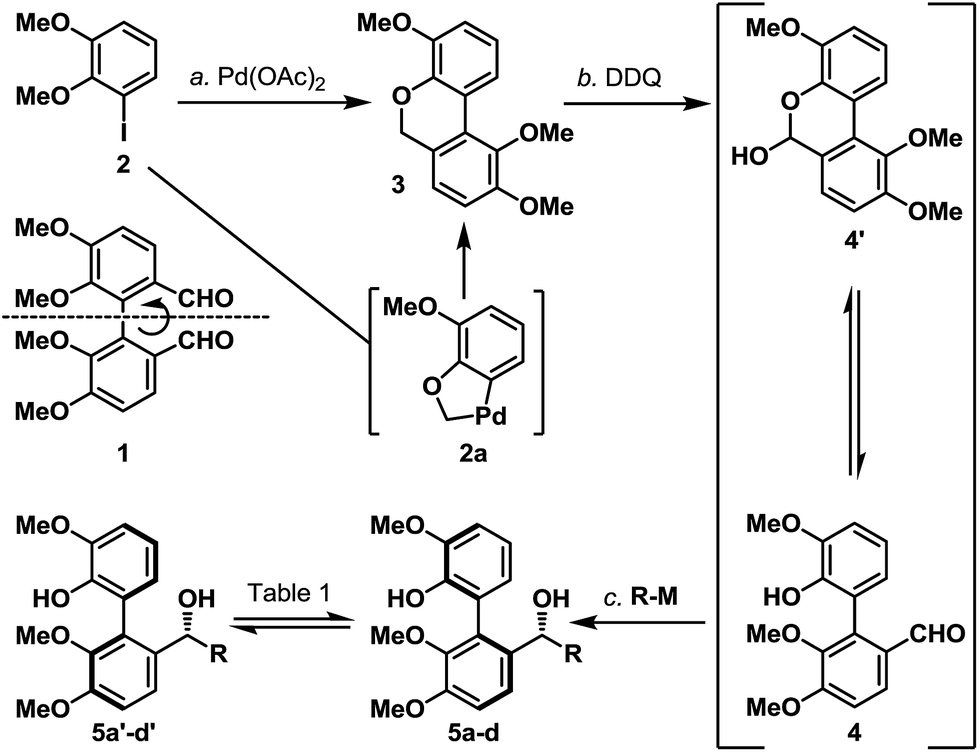 | ||
Scheme 2 Synthesis of biaryl benzylic alcohols 5a/5a′ to 5d/5d′ exhibiting atropisomeric properties. Reagents and conditions: (a) Pd(OAc)2 (0.04 equiv.), K2CO3 (4.0 equiv.), TBAB (1.0 equiv.), DMF, 120 °C, 16 h, 86%; (b) DDQ (1.0 equiv.), CH2Cl2/pH 9.2 buffer (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 0 °C, 1.5 h, 76%; (c) for 5a/5a′: methylmagnesium bromide (3.0 M in Et2O, 3.3 equiv.), THF, −78 to 23 °C, 16 h, 92% (5a:5a′ ∼ 1:1); for 5b/5b′: phenylmagnesium bromide (1.0 M in THF, 4.7 equiv.), THF, −78 to 23 °C, 16 h, 85% (5b:5b′ ∼ 6:1); for 5c/5c′: t-butyllithium (1.7 M in pentane, 3.5 equiv.), THF, −78 to 23 °C, 16 h, 92% (5c:5c′ ∼ 4:1); for 5d/5d′: allylmagnesium bromide (1.0 M in THF, 2.6 equiv.), THF, −78 to 23 °C, 16 h, 97% (5d:5d′ ∼ 3:1). DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone; DMF = N,N′-dimethylformamide; OAc = acetate; TBAB = tetra-n-butylammonium bromide. :1), 0 °C, 1.5 h, 76%; (c) for 5a/5a′: methylmagnesium bromide (3.0 M in Et2O, 3.3 equiv.), THF, −78 to 23 °C, 16 h, 92% (5a:5a′ ∼ 1:1); for 5b/5b′: phenylmagnesium bromide (1.0 M in THF, 4.7 equiv.), THF, −78 to 23 °C, 16 h, 85% (5b:5b′ ∼ 6:1); for 5c/5c′: t-butyllithium (1.7 M in pentane, 3.5 equiv.), THF, −78 to 23 °C, 16 h, 92% (5c:5c′ ∼ 4:1); for 5d/5d′: allylmagnesium bromide (1.0 M in THF, 2.6 equiv.), THF, −78 to 23 °C, 16 h, 97% (5d:5d′ ∼ 3:1). DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone; DMF = N,N′-dimethylformamide; OAc = acetate; TBAB = tetra-n-butylammonium bromide. | ||
Before pressing onto our next objective, namely the “axial-to-point” stereochemical induction, two crucial criteria had to be considered. First, the substrate must exhibit sufficient atropisomeric configurational stability during the axial-to-point stereoinduction event. Second, any preparatory transformations leading to the axial-to-point stereoinduction event must preserve the atropisomeric property within the substrate. In order to assess the configurational stability of the organometallic addition products (5a/5a′–5d/5d′), chromatographically separated and atropisomerically pure biaryl benzylic alcohols were subjected to thermal conditions and their atropisomerization was monitored by 1H NMR analysis (Table 1). Our qualitative analysis revealed that all of the substrates exhibited good configurational stability from low to moderately elevated temperatures (up to 70 °C), with the t-butyl substrates 5c and 5c′ demonstrating extended stability up to 100 °C (for details, see the ESI†).
| Compd. | Temp. °C | |||||
|---|---|---|---|---|---|---|
| 40 | 70 | 90 | 100 | 110 | 120 | |
| (1H NMR analysis after 1 h at each temperature up to 110 °C) | ||||||
| a Ratio determined by 1H NMR integration. For details, see the ESI.† | ||||||
| 5a | 1:0 |
1:0.10 |
1:0.43 |
1:0.88 |
0.79:1 |
0.69:1 |
| 5a′ | 0:1 |
0.07:1 |
0.16:1 |
0.39:1 |
0.60:1 |
0.70:1 |
| 5b | 1:0 |
1:0.13 |
1:0.43 |
1:0.98 |
0.68:1 |
0.63:1 |
| 5b′ | 0:1 |
0.06:1 |
0.23:1 |
0.44:1 |
0.58:1 |
0.65:1 |
| 5c | 1:0 |
1:0 |
1:0 |
1:0.09 |
1:0.35 |
0.53:1 |
| 5c′ | 0:1 |
0:1 |
0:1 |
0.06:1 |
0.18:1 |
0.52:1 |
| 5d | 1:0 |
1:0 |
1:0.36 |
0.93:1 |
0.71:1 |
0.71:1 |
| 5d′ | 0:1 |
0:1 |
0.53:1 |
0.70:1 |
0.70:1 |
0.70:1 |
Next, we envisaged that the proposed axial-to-point stereoinduction would take place via a dearomatization process, in which the dearomatized product or its chemically transformed derivative would possess new “point” stereochemical element(s).9 After contemplating a variety of well-documented dearomatization protocols, hypervalent iodine mediated oxidative dearomatization appeared the most appealing owing to its operational ease, substrate scope and product versatility.10 Accordingly, benzylic alcohols 5a/5a′–5d/5d′ (except 5c/5c′, which remained as benzylic alcohols) were protected as their corresponding TBS ethers to prevent any unwanted side reactions and to enhance their atropisomeric stabilities, followed by treatment with PIFA in the presence of methanol (5d/5d′ in Scheme 3a, for 5a/5a′–5d/5d′ see the ESI†). Much to our surprise, each of the isomerically pure phenols 6d and 6d′ underwent oxidative dearomatization to afford an identical mixture of two dienone products 7d and 7d′ (7d:7d′ ∼ 1:1). The fact that 7d and 7d′ were observed as two distinct diastereoisomers based on our 1H NMR analysis suggested that the epimerization of the biaryl axis must take place during the course of the reaction. However, a postulated transition state (6-PIFA-TS, Scheme 4a), formed through an associative mechanism by invoking a coordinated phenol–aryliodide complex, should increase the rotational energy barrier and thus render higher configurational stability. While seeking a more concrete explanation, we also became aware of a recent report from the Pappo laboratory describing that several optically pure BINOL substrates underwent metal-catalyzed racemization under single-electron-transfer (SET) conditions (Scheme 4b), although the precise mechanistic origin behind this racemization process remained unclear.11 Very recently, NMR and DFT studies reported by the Koltunov group also suggested an acid-mediated atropisomerization of optically pure BINOL via several enone intermediates (e.g.BINOL-H+) that structurally closely resemble those generated during the hypervalent iodine and metal–salt mediated phenol oxidations (Scheme 4c).12
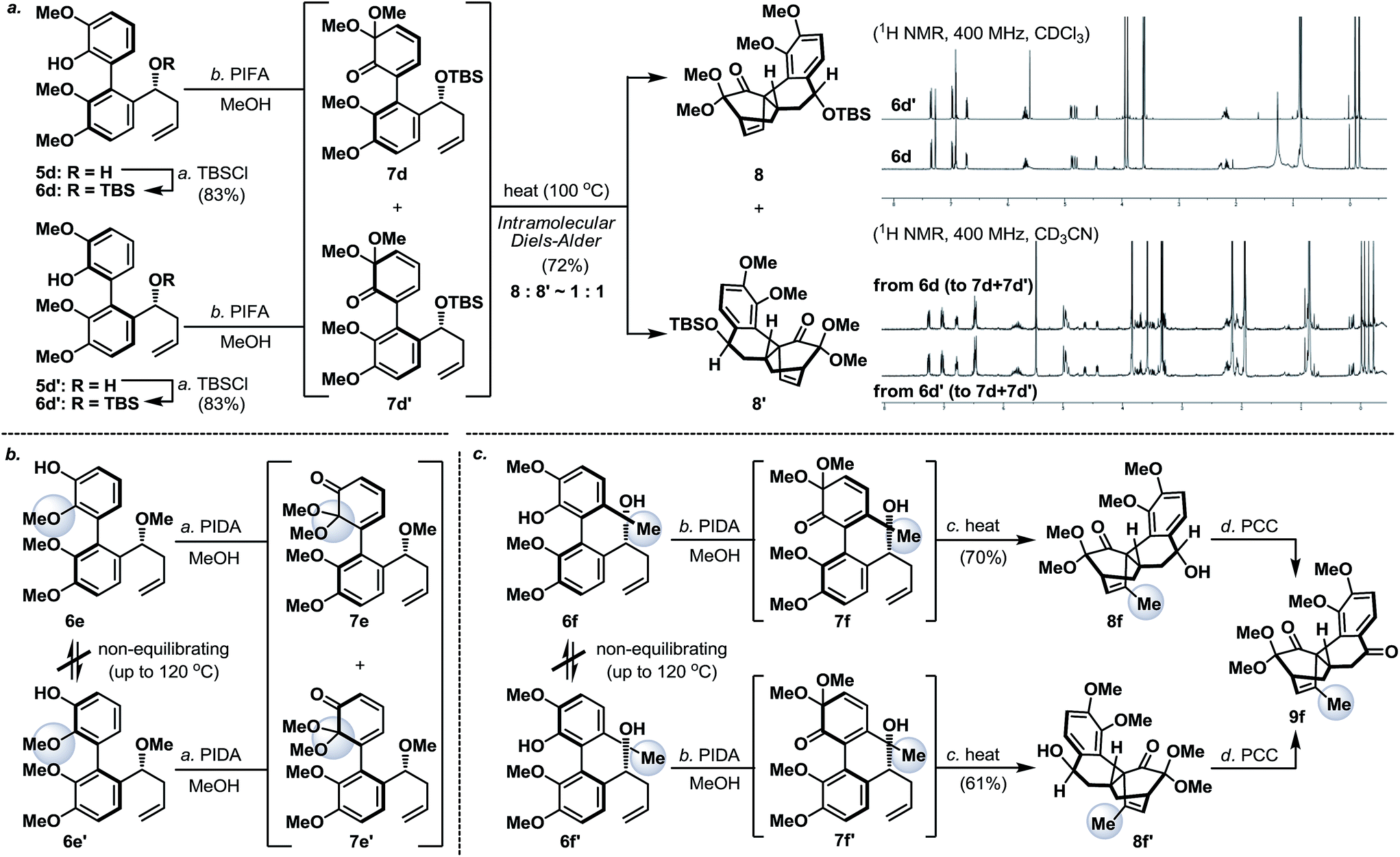 | ||
| Scheme 3 (a) Oxidative dearomatization of atropisomerically pure biaryl phenols 6d and 6d′; (b) oxidative dearomatization and configurational stability studies of biaryl phenols 6e and 6e′; and (c) oxidative dearomatization and configurational stability studies of biaryl phenols 6f and 6f′. PCC = pyridinium chlorochromate; PIDA = [bis(acetoxy)iodo]benzene; PIFA = [bis(trifluoroacetoxy)iodo]benzene; and TBSCl = tert-butyldimethylsilyl chloride. | ||
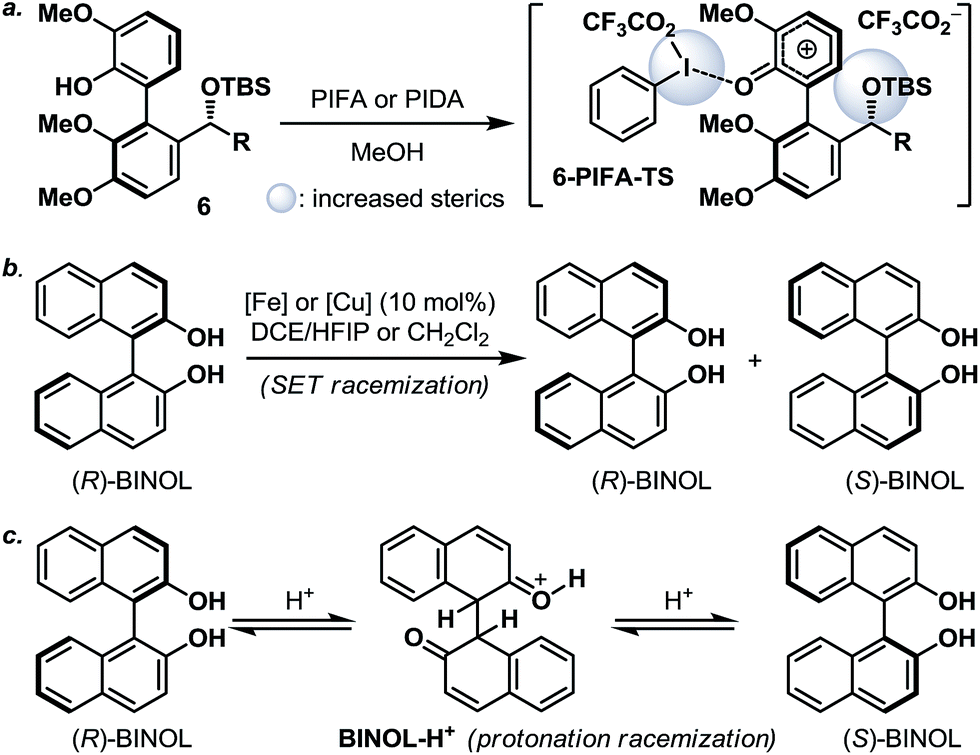 | ||
| Scheme 4 (a) Oxidative dearomatization of 6via an associative mechanism; (b) racemization of (R)-BINOL under SET conditions; and (c) acid promoted racemization of (R)-BINOL. BINOL = 1,1′-bi-2-naphthol; DCE = 1,2-dichloroethane; and HFIP = hexafluoroisopropanol. | ||
Continuing our studies, several related phenolic biaryl substrates were conceived in order to understand and to preserve the atropisomeric information (Scheme 3b and c). In this context, the phenolic substrates 6e and 6e′ with the phenol and methoxy positions switched compared to those in 6d and 6d′ demonstrated much improved thermal stability, however, when separately treated with PIDA in the presence of methanol, also afforded an identical mixture of dienones 7e and 7e′ (∼1:1). Gratifyingly, phenols 6f and 6f′ with an additional methyl substituent were found to retain their atropisomeric purity upon PIDA-mediated oxidative dearomatization, and the atropisomerically pure dienones 7f and 7f′ faithfully underwent intramolecular Diels–Alder reactions to afford tetracycles 8f and 8f′ in 70% and 61% yield, respectively, with their stereochemical relationship confirmed upon oxidation with PCC. An analogous intramolecular Diels–Alder reaction could also be realized with a 1:1 mixture of dienones 7d and 7d′ to afford a near 1:1 mixture of tetracycles 8 and 8′ (Scheme 3a). Considering that dienones 7d and 7d′ are likely to be configurationally labile at elevated temperatures (see Table 1), this latter result implied that the benzylic OTBS stereocenter offered essentially no stereoinduction during the intramolecular Diels–Alder process. Furthermore, the selective formation of the Diels–Alder product 8f from 7f (and 8f′ from 7f′) strongly suggested that the stereoinduction arose exclusively from the atropisomeric property of 7f (and 7f′) (Scheme 5). In retrospect, the benzylic “point” stereochemical directing element was positioned in proximity to the biaryl-axis for the first “point-to-axial” stereoinduction, while it was distant from the second “axial-to-point” stereoinduction event to suppress its stereo-directing ability. From a design perspective, a single stereo-directing element at each stereochemistry inducing step is synthetically more attractive to avoid any complicated synergistic stereo-directing phenomena.
 | ||
| Scheme 5 Atropisomerism dictated stereoinduction leading to the stereocontrolled formation of Diels–Alder products 8f and 8f′. | ||
To conclude our studies in remote stereoinductions, we found that substrates 6g and 6h (racemic or optically active) with their OH/OTBS stereocenter relocated in proximity to the intramolecular Diels–Alder transition-state could render a significantly higher level of point-to-point stereoinduction than 6d/6d′ (Scheme 6), an observation that was consistent with our previous synthetic studies towards platencin.13
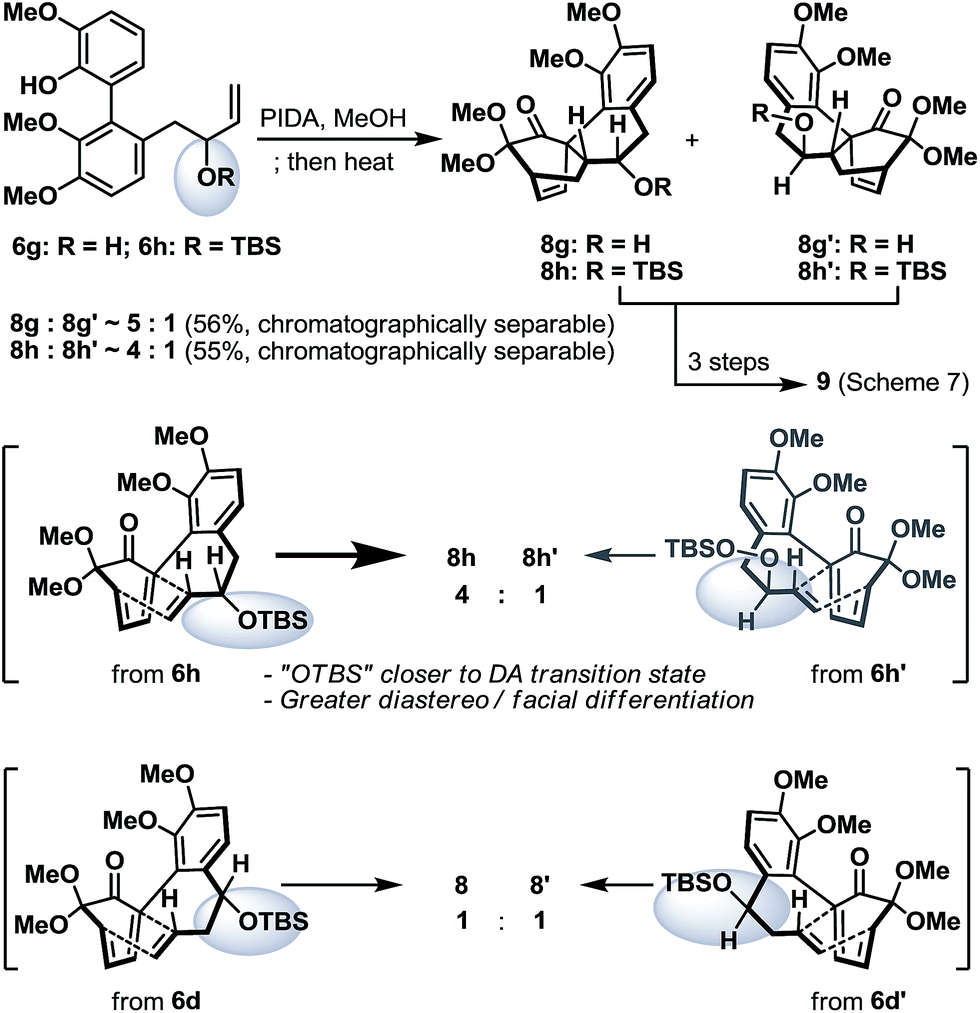 | ||
| Scheme 6 Oxidative dearomatization and intramolecular Diels–Alder reaction of biaryl phenols 6g and 6h. | ||
Synthetic applications
Finally, we turned our attention to the synthetic utility of the synthesized advanced intermediates. To this end, we recognized that the highly functionalized phenanthrene-type tetracycle 8/8′ bearing a quaternary stereocenter shared some similarities with the core structure of the morphinan family of natural products.14 Further structural analysis implied that the [2.2.2]-bicyclic domain within 8 (or 8′) had to be ruptured while retaining its quaternary stereocenter. As shown in Scheme 7, Diels–Alder product 8 (and 8′) was readily converted to diketone 9via a regioselective hydroboration and an oxidative work-up with PCC,15 followed by an acid-induced elimination. It is worth noting that, while tetracycles 8 and 8′ could be used in combination in this racemic illustration, enantio- and diastereo-isomerically pure 8 (or 8′) will be required to access optically pure intermediates. The more sterically accessible ketone in 9 was converted to its corresponding oxime and thermally equilibrated to a single geometric isomer 10, which was subsequently tosylated (TsCl, 87% yield from 9) to set the stage for the Beckmann rearrangement.16 Under optimized conditions, tosyloxime 11 underwent regioselective ring expansion under the influence of ZnCl2 to afford lactam 12 in 73% yield. Further cleavage of the bridgehead C–N bond in 12 was realized through a Hoffmann elimination17 of its quaternary ammonium salt derivative 13 to furnish dimethylamine 14. Unmasking the dimethoxy acetal in 14 under acidic conditions, very surprisingly, afforded a 1,2-migratory product 15 (after treatment with ethyl chloroformate).18 This undesired bond migration was easily rectified upon partial reduction of diene 14,19 followed by acidic deketalization to yield hydroxy ketone 16 in 66% overall yield. α-Deoxygenation and N-demethylation were sequentially realized through the action of SmI2 and 1-chloroethyl chloroformate and the as-obtained ketone 18 represents a valuable common intermediate that is also amenable for a synthetic method to access the hasubananes (bridgehead cyclization “a”).20 Advancing tricyclic ketone 18 next called for the formation of a five-membered oxacycle that resides in several flagship morphinans. To this end, phenolic ketone 18 was converted to its dioxolane derivative followed by a dioxolane-directed phenolic demethylation.22 Dioxolane removal followed by treatment of the resulting phenolic ketone (19) with pyridinium tribromide23 and subsequent heating smoothly delivered an inconsequential mixture of oxa-tetracycle 20 and arylbromide 20a. Next, relocation of the bridgehead olefin24 in 20/20a first converged both compounds through hydrogenation and dioxolane formation, and on further exposure to NBS under radical conditions afforded styrene 21. Reductive detosylation of 21 under Birch-type conditions took place with concomitant piperidine formation21s and furnished synthetic dihydrocodeinone upon dioxolane removal (HCl, MeOH).25 Dihydrocodeinone serves as a valuable intermediate to readily access a diverse array of naturally occurring and designed morphinans, including but not limited to dihydrocodeine, codeine, morphine, thebaine and oxycodone.26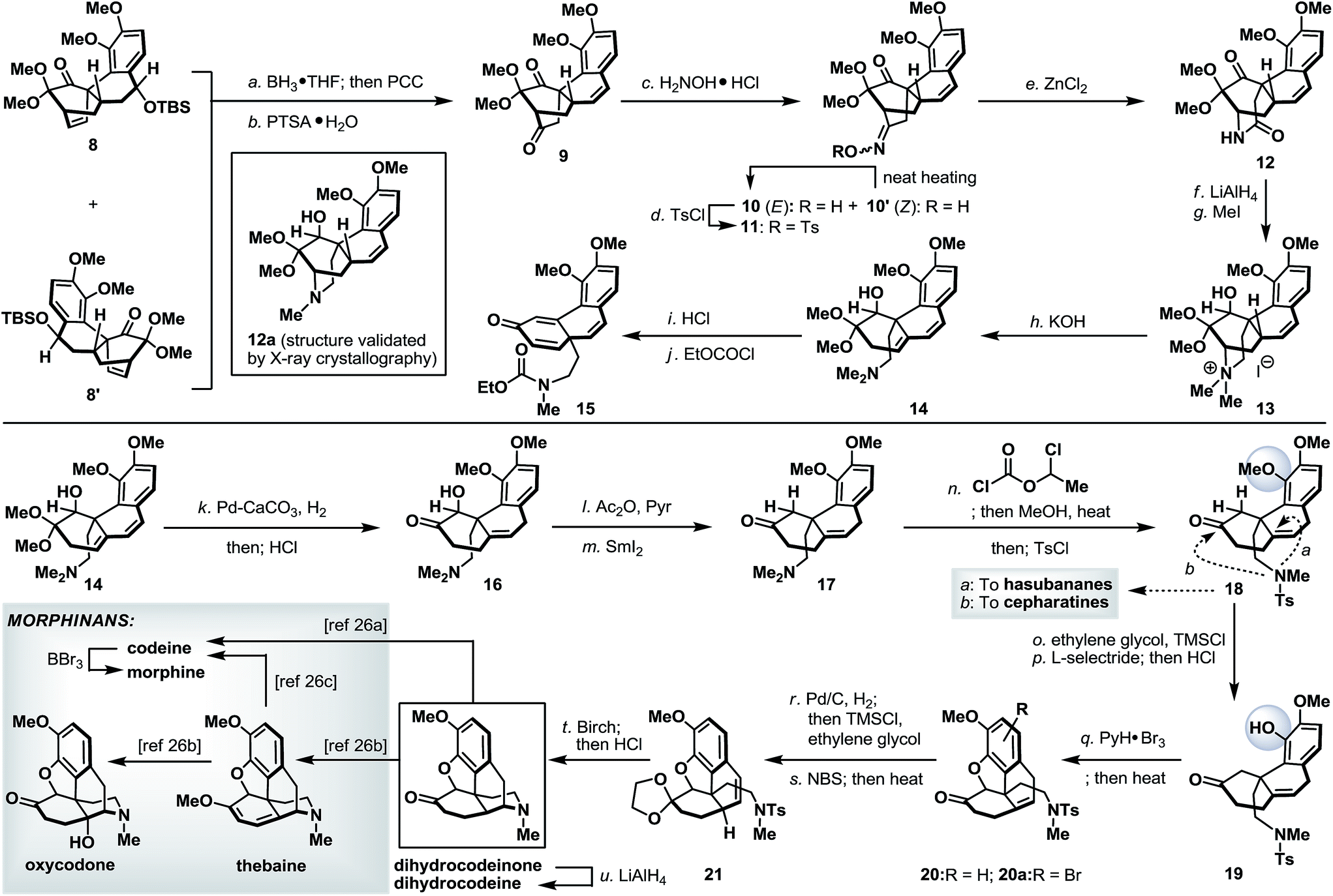 | ||
| Scheme 7 Synthesis of morphinans. Reagents and conditions: (a) BH3·THF (5.0 equiv.), THF, −78 to 70 °C, 4 h; then PCC (5.0 equiv.), 23 °C, 5 h, 41%; (b) p-TsOH·H2O (1.0 equiv.), toluene, 110 °C, 20 min, 95%; (c) H2NOH·HCl (3.0 equiv.), NaOAc (3.0 equiv.), MeOH, 23 to 70 °C, 16 h (heat to neat), 90%; (d) TsCl (1.5 equiv.), NaOH (2.5 equiv.), acetone/H2O (1:1), 23 °C, 2 h, 97%; (e) ZnCl2 (2.0 equiv.), MeCN, 100 °C, 4 h; then ZnCl2 (2.0 equiv.), 100 °C, 4 h, 73%; (f) LiAlH4 (10 equiv.), THF, 0 to 70 °C, 8 h; (g) MeI (14 equiv.), CH2Cl2, 23 °C, 10 h, 42% for two steps; (h) KOH (20% in MeOH), 70 °C, 10 h, 80%; (i) HCl (10% aq.)/MeOH (4:1), 23 °C, 1 h, 67%; (j) EtOCOCl (5.0 equiv.), NaHCO3 (20 equiv.), 1,2-dichloroethane, 100 °C, 2 h, 93%; (k) Pd/CaCO3 (1.0 equiv.), MeOH, H2 (1 atm), 23 °C, 16 h, (1,4:1,2-hydrogenated compound ∼ 5:1); then HCl (10% aq.)/MeOH (2:1), 23 °C, 1 h, 66% for two steps; (l) Ac2O (10 equiv.), Et3N (10 equiv.), DMAP (0.1 equiv.), CH2Cl2, 23 °C, 4 h; (m) SmI2 (0.1 M in THF), THF/MeOH (1:1), −78 °C, 0.5 h, 78% for two steps; (n) 1-chloroethyl chloroformate (20 equiv.), NaHCO3 (20 equiv.), 1,2-dichloroethane, 100 °C, 2 h; then MeOH, 60 °C, 1.5 h; then TsCl (2.0 equiv.), DMAP (0.4 equiv.), Et3N (2.3 equiv.), CH2Cl2, 23 °C, 1.5 h, 64% for two steps; (o) TMSCl (2.0 equiv.), ethylene glycol/CH2Cl2 (1:1), 23 to 50 °C, 5 h, 92%; (p) L-selectride (1.0 M in THF, 5.0 equiv.), THF, 80 °C, 24 h; then HCl (4.0 N aq.)/MeOH (1:15), 23 to 45 °C, 3 h, 77% for two steps; (q) PyH·Br3 (2.0 equiv.), CH2Cl2/AcOH (2:5), 23 °C, 30 min; then LiBr (5.0 equiv.), Et3N (10.0 equiv.), MeCN, 60 °C, 20 min, 53%; (r) Pd/C (10% wt/wt, 2.0 equiv.), EtOAc/MeOH (1:3), H2, 23 °C, 30 min, 90%; then TMSCl (excess), ethylene glycol/CH2Cl2 (1:1), 23 to 50 °C, 5 h, 80%; (s) NBS (1.05 equiv.), benzoyl peroxide (0.5 equiv.), CCl4, 80 °C, 1 h; then Et3N (7.8 equiv.), 80 °C, 15 min, 60%; (t) Li (excess), NH3, −78 °C, 10 min, 69%; then HCl (4.0 N aq.)/MeOH (1:15), 70 °C, 6 h, 75%; and (u) LiAlH4 (excess), THF, 0 °C, 45 min, 74%. DMAP = N,N′-dimethylaminopyridine; EtOAc = ethyl acetate; L-selectride = lithium tri-sec-butylborohydride; NBS = N-bromosuccinimide; PyH·Br3 = pyridinium tribromide; TMSCl = trimethylsilyl chloride; p-TsOH·H2O = para-toluenesulfonic acid monohydrate; TsCl = para-toluenesulfonyl chloride; and Ts = para-toluenesulfonyl. | ||
Conclusions
In summary, we have demonstrated proof-of-concept, rationally designed serial point-to-axial-to-point stereoinductions and examined the stereochemical fidelity of these processes. During this investigation, an unexpected atropisomeric epimerization upon hypervalent iodine-mediated oxidative dearomatization of isomerically pure biaryl phenols was discovered and systematically investigated. This finding bears important ramifications in related oxidative generation11 and transformations of designed and naturally occurring phenolic biaryl systems and their atropisomeric integrity. While the undesired atropisomeric epimerization could be suppressed by increasing the steric pressure about the biaryl axis of the substrate (e.g.6f/6f′), more in depth mechanistic studies are in progress to render a substrate-independent solution.27 Further application of the developed synthetic strategy also enabled a novel synthetic method to access the morphinan family of natural products and potential access to other related alkaloid structures. Conceptually related programmed serial-stereoinductions, particularly those involving unconventional intramolecular axial-to-point and point-to-axial processes, are currently being designed and implemented in the context of target-oriented synthesis in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper is dedicated to Professor William S. Knowles on the occasion of his centennial. This work was supported by the Seoul National University Foreign Faculty Fund, the New Faculty Resettlement Fund, the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (No. 2012R1A2A2A01002895, 2013R1A1A2057837, and 2014-011165, Center for New Directions in Organic Synthesis), and Novartis. Rui Chen and Kenny Park were supported by the BK21Plus Program, Ministry of Education. We thank Youngwook Park and Sungmin Song for the preliminary studies. We thank Professor Doron Pappo, Ben-Gurion University of the Negev, Israel, for helpful discussions regarding the racemization/epimerization of phenolic biaryl/binaphthyl systems under oxidative conditions.Notes and references
- E. M. Carreira and L. Kvaerno, Classics in Stereoselective Synthesis, Wiley-VCH, Weinheim, 2009 Search PubMed.
- For a review on the use of chiral oxazaborolidines in the asymmetric reduction of carbonyl compounds, see: E. J. Corey and C. J. Helal, Angew. Chem., Int. Ed., 1998, 37, 1986 CrossRef CAS.
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008 CrossRef CAS PubMed.
- For a recent review on axial-to-central chirality transfer in cyclization processes, see: (a) D. Campolo, S. Gastaldi, C. Roussel, M. P. Bertrand and M. Nechab, Chem. Soc. Rev., 2013, 42, 8434 RSC. For a recent review on the application of chiral allenes in axial-to-central chirality transfer, see: (b) R. K. Neff and D. E. Frantz, Tetrahedron, 2015, 71, 7 CrossRef CAS.
- (a) M. E. Layton, C. A. Morales and M. D. Shair, J. Am. Chem. Soc., 2002, 124, 773 CrossRef CAS PubMed; (b) Z. Gu and A. Zakarian, Org. Lett., 2010, 12, 4224 CrossRef CAS PubMed.
- For the preparation of related optically enriched dialdehydes and a desymmetrization process, see: (a) W.-W. Chen, Q. Zhao, M.-H. Xu and G.-Q. Lin, Org. Lett., 2010, 12, 1072 CrossRef CAS PubMed; (b) C. Zhu, Y. Shi, M.-H. Xu and G.-Q. Lin, Org. Lett., 2008, 10, 1243 CrossRef CAS PubMed.
- G. Dyker, Angew. Chem., Int. Ed., 1992, 31, 1023 CrossRef.
- A biaryl aldehyde structurally closely related to 4 was found to undergo racemization at room temperature and hence does not possess atropisomeric properties. See: A. I. Meyers and R. J. Himmelsbach, J. Am. Chem. Soc., 1985, 107, 682 CrossRef CAS.
- For an example of the oxidative dearomatization of a biaryl system while maintaining atropchirality, see: Y. Yasui, K. Suzuki and T. Matsumoto, Synlett, 2004, 619 CAS.
- (a) For the oxidation of phenolic compounds with organohypervalent iodine reagents, see: R. M. Moriaty and O. Prakash, Org. React. 2001, 57, 327 Search PubMed; (b) For a review on the chemistry of masked o-benzoquinones, including Diels–Alder reactions, see: C.-C. Liao and R. K. Peddiniti, Acc. Chem. Res., 2002, 35, 856 CrossRef CAS PubMed.
- S. Narute, E. Parnes, F. D. Toste and D. Pappo, J. Am. Chem. Soc., 2016, 138, 16553 CrossRef CAS PubMed.
- A. M. Genaev, G. E. Salnikov, A. V. Shernyukov, Z. Zhu and K. Y. Koltunov, Org. Lett., 2017, 19, 532 CrossRef CAS PubMed.
- G. Y. C. Leung, H. Li, Q.-Y. Toh, A. M.-Y. Ng, R. J. Sum, J. E. Bandow and D. Y.-K. Chen, Eur. J. Org. Chem., 2011, 183 CrossRef CAS.
- For recent books and review chapters, see: (a) A. Brossi, in The Chemistry and Biology of Isoquinoline Alkaloids, ed. J. D. Phillipson, M. R. Roberts and M. H. Zenk, Springer-Verlag, Berlin, 1985, pp. 171–189 Search PubMed; (b) H. Nagase, in Chemistry of Opioids; Top. Curr. Chem., Springer-Verlag, Berlin Heidelberg, 2011, vol. 299, pp. 1–312 Search PubMed.
- For pioneering discovery and examples of a one-pot hydroboration-PCC oxidation, see: (a) H. C. Brown, S. U. Kulkarni and C. G. Rao, Synthesis, 1980, 151 CrossRef CAS; (b) E. J. Parish, S. Parish and H. Honda, Synth. Commun., 1990, 20, 3265 CrossRef CAS.
- T. Tatsumi, Beckmann rearrangement, ed. R. A. Sheldon and H. Bekkum, Wiley-VCH, Weinheim, New York, 2001, p. 185–204 Search PubMed.
- A. C. Cope and E. R. Trumbull, Org. React., 1953, 7, 99–197 Search PubMed.
- For 1,2-migratory processes in related phenanthrene systems, see: (a) T. Matsumoto, Y. Tanaka, H. Terao, Y. Takeda and M. Wada, Chem. Pharm. Bull., 1993, 41, 1960 CrossRef CAS; (b) R.-J. Zhang, Q. Lei, Y.-M. Pan, H.-S. Wang and Y. Zhang, Chin. J. Struct. Chem., 2010, 29, 1327 CrossRef CAS.
- For related examples of migratory 1,4-hydrogenation, see: (a) C. J. Flann and L. E. Overman, J. Am. Chem. Soc., 1987, 109, 6115 CrossRef CAS; (b) K. S. Feldman, M.-J. Wu and D. P. Rotella, J. Am. Chem. Soc., 1989, 111, 6457 CrossRef CAS; (c) D. M. Filippo, I. Izzo, S. Raimondi, F. D. Riccardis and G. Sodano, Tetrahedron Lett., 2001, 42, 1575 CrossRef; (d) F. Li, N. C. Warshakoon and M. J. Miller, J. Org. Chem., 2004, 69, 8836 CrossRef CAS PubMed.
- M. Matsui, in The Alkaloids, ed. A. Brossi, Academic Press, New York, 1988, vol. 33, pp. 307–347 Search PubMed.
- For the synthesis of morphine, codeine, thebaine, dihydrocodeinone and neopine, see: (a) M. Gates and G. Tschudi, J. Am. Chem. Soc., 1952, 74, 1109 CrossRef CAS; (b) M. Gates and G. Tschudi, J. Am. Chem. Soc., 1956, 78, 1380 CrossRef CAS; (c) D. Elad and D. Ginsburg, J. Am. Chem. Soc., 1954, 76, 312 CrossRef CAS; (d) R. Grewe and H. Fischer, Chem. Ber., 1963, 96, 1520 CrossRef CAS; (e) R. Grewe and W. Friedrichsen, Chem. Ber., 1967, 100, 1550 CrossRef CAS PubMed; (f) D. H. R. Barton, D. S. Bhakuni, R. James and G. W. Kirby, J. Chem. Soc. C, 1967, 128 RSC; (g) G. C. Morrison, R. O. Waite and J. J. Shavel, Tetrahedron Lett., 1967, 8, 4055 CrossRef; (h) T. Kametani, M. Ihara, K. Fukumoto and H. Yagi, J. Chem. Soc. C, 1969, 2030 RSC; (i) M. A. Schwartz and I. S. Mami, J. Am. Chem. Soc., 1975, 97, 1239 CrossRef CAS PubMed; (j) H. C. Beyerman, T. S. Lie, M. Maat, H. H. Bosman, E. Buurman and E. J. M. Bijsterveld, Recl. Trav. Chim. Pays-Bas, 2010, 95, 24 CrossRef; (k) T. S. Lie, L. Maat and H. C. Beyerman, Recl. Trav. Chim. Pays-Bas, 2010, 98, 419 CrossRef; (l) K. C. Rice, J. Org. Chem., 1980, 45, 3135 CrossRef CAS; (m) D. A. Evans and C. H. Mitch, Tetrahedron Lett., 1982, 23, 285 CrossRef CAS; (n) W. H. Moos, R. D. Gless and H. Rapoport, J. Org. Chem., 1983, 48, 227 CrossRef CAS; (o) J. D. White, G. Caravatti, T. B. Kline, E. Edstrom, K. C. Rice and A. Brossi, Tetrahedron, 1983, 39, 2393 CrossRef CAS; (p) J. E. Toth and P. L. Fuchs, J. Org. Chem., 1987, 52, 473 CrossRef CAS; (q) J. E. Toth, P. R. Hamann and P. L. Fuchs, J. Org. Chem., 1988, 53, 4694 CrossRef CAS; (r) M. A. Tius and M. A. Kerr, J. Am. Chem. Soc., 1992, 114, 5959 CrossRef CAS; (s) K. A. Parker and D. Fokas, J. Am. Chem. Soc., 1992, 114, 9688 CrossRef CAS; (t) K. A. Parker and D. Fokas, J. Org. Chem., 1994, 59, 3927 CrossRef CAS; (u) K. A. Parker and D. Fokas, J. Org. Chem., 2006, 71, 449 CrossRef CAS PubMed; (v) C. Y. Hong, N. Kado and L. E. Overman, J. Am. Chem. Soc., 1993, 115, 11028 CrossRef CAS; (w) C. Y. Hong and L. E. Overman, Tetrahedron Lett., 1994, 35, 3453 CrossRef CAS; (x) J. Mulzer, G. Duerner and D. Trauner, Angew. Chem., Int. Ed., 1996, 35, 2830 CrossRef CAS; (y) J. Mulzer, J. W. Bats, B. List, T. Opatz and D. Trauner, Synlett, 1997, 441 CrossRef CAS; (z) D. Trauner, J. W. Bats, A. Werner and J. Mulzer, J. Org. Chem., 1998, 63, 5908 CrossRef CAS PubMed; (a a) J. Mulzer and D. Trauner, Chirality, 1999, 11, 475 CrossRef CAS; (a b) J. D. White, P. Hrnciar and F. Stappenbeck, J. Org. Chem., 1997, 62, 5250 CrossRef CAS; (a c) J. D. White, P. Hrnciar and F. Stappenbeck, J. Org. Chem., 1999, 64, 7871 CrossRef CAS; (a d) H. Nagata, N. Miyazawa and K. Ogasawara, Chem. Commun., 2001, 1094 RSC; (a e) D. F. Taber, T. D. Neubert and A. L. Rheingold, J. Am. Chem. Soc., 2002, 124, 12416 CrossRef CAS PubMed; (a f) B. M. Trost and W. Tang, J. Am. Chem. Soc., 2002, 124, 14542 CrossRef CAS PubMed; (a g) B. M. Trost, W. Tang and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 14785 CrossRef CAS PubMed; (a h) K. Uchida, S. Yokoshima, T. Kan and T. Fukuyama, Org. Lett., 2006, 8, 5311 CrossRef CAS PubMed; (a i) K. Uchida, S. Yokoshima, T. Kan and T. Fukuyama, Heterocycles, 2009, 77, 1219 CrossRef CAS; (a j) A. T. Omori, K. J. Finn, H. Leisch, R. J. Carroll and T. Hudlicky, Synlett, 2007, 2859 CAS; (a k) M. Verin, E. Barre, B. Iorga and C. Guillou, Chem.–Eur. J., 2008, 14, 6606 CrossRef PubMed; (a l) H. Tanimoto, R. Saito and H. Chida, Tetrahedron Lett., 2008, 49, 358 CrossRef CAS; (a m) H. Leisch, A. T. Omori, K. J. Finn, J. Gilmet, T. Bissett, D. Ilceski and T. Hudlicky, Tetrahedron, 2009, 65, 9862 CrossRef CAS; (a n) P. Magnus, N. Sane, B. P. Fauber and V. Lynch, J. Am. Chem. Soc., 2009, 131, 16045 CrossRef CAS PubMed; (a o) G. Stork, A. Yamashita, J. Adams, G. R. Schulte, R. Chesworth, Y. Miyazaki and J. J. Farmer, J. Am. Chem. Soc., 2009, 131, 11402 CrossRef CAS PubMed; (a p) H. Koizumi, S. Yokoshima and T. Fukuyama, Chem.–Asian J., 2010, 5, 2192 CrossRef CAS PubMed; (a q) T. Erhard, G. Ehrlich and P. Metz, Angew. Chem., Int. Ed., 2011, 50, 3892 CrossRef CAS PubMed; (a r) J. Duchek, G. Piercy, J. Gilmet and T. Hudlicky, Can. J. Chem., 2011, 89, 709 CrossRef CAS; (a s) J. Li, G.-L. Liu, X.-H. Zhao, J.-Y. Du, H. Qu, W.-D. Chu, M. Ding, C.-Y. Jin, M.-X. Wei and C.-A. Fan, Chem.–Asian J., 2013, 8, 1105 CrossRef CAS PubMed; (a t) V. Varghese and T. Hudlicky, Synlett, 2013, 24, 369 CrossRef CAS; (a u) M. Ichiki, H. Tanimoto, S. Miwa, R. Saito, T. Sato and N. Chida, Chem.–Eur. J., 2013, 19, 264 CrossRef CAS PubMed; (a v) V. Varghese and T. Hudlicky, Angew. Chem., Int. Ed., 2014, 53, 4355 CrossRef CAS PubMed; (a w) M. Geffe and T. Opatz, Org. Lett., 2014, 16, 5282 CrossRef CAS PubMed; (a x) M. Tissot, R. J. Phipps, C. Lucas, R. M. Leon, R. D. M. Pace, T. Ngouansavanh and M. J. Gaunt, Angew. Chem., Int. Ed., 2014, 53, 13498 CrossRef CAS PubMed; (a y) Q. Li and H. Zhang, Chem.–Eur. J., 2015, 21, 16379 CrossRef CAS PubMed; (b `) S. Chu, N. Munster, T. Balan and M. D. Smith, Angew. Chem., Int. Ed., 2016, 55, 14306 CrossRef CAS PubMed; (b a) L. Rycek, J. J. Hayward, M. A. Latif, J. Tanko, R. Simionescu and T. Hudlicky, J. Org. Chem., 2016, 81, 10930 CrossRef CAS PubMed; (b b) H. Umihara, S. Yokoshima, M. Inoue and T. Fukuyama, Chem.–Eur. J., 2017, 23, 6993 CrossRef CAS PubMed.
- H. Wu, L. N. Thatcher, D. Bernard, D. A. Parrish, J. R. Deschamps, K. C. Rice, A. D. Mackerell Jr and A. Coop, Org. Lett., 2005, 7, 2531 CrossRef CAS PubMed.
- A. Kimishima, H. Umihara, A. Mizoguchi, S. Yokoshima and T. Fukuyama, Org. Lett., 2014, 16, 6244 CrossRef CAS PubMed.
- We found that the bridgehead trisubstituted olefin in 20/20a was resistant to isomerization under a variety of acidic and basic conditions. A similar finding was observed by Trost and co-worker: B. M. Trost and W. Tang, J. Am. Chem. Soc., 2003, 125, 8744 CrossRef CAS PubMed.
- For a comparison with previously reported morphinan syntheses, see the ESI† (page 70).
- For the preparation of codeine and morphine from dihydrocodeinone, see: (a) D. D. Weller and H. Rapoport, J. Med. Chem., 1976, 19, 1171 CrossRef CAS PubMed; (b) I. Iijima, K. C. Rice and J. V. Silverton, Heterocycles, 1977, 6, 1157 CrossRef CAS. For the preparation of thebaine and oxycodone from dihydrocodeinone, see: (c) B. R. Selfridge, X. Wang, Y. Zhang, H. Yin, P. M. Grace, L. R. Watkins, A. E. Jacobson and K. C. Rice, J. Med. Chem., 2015, 58, 5038 CrossRef CAS PubMed. For the conversion of thebaine to codeinone, see: (d) R. B. Barber and H. Rapoport, J. Med. Chem., 1976, 19, 1175 CrossRef CAS PubMed.
- We believe that by unravelling the precise origin of the epimerization process, judicious design and choice of reagent(s) and reaction conditions could provide a substrate-independent solution. Theoretical investigation of the racemization/epimerization of phenolic biaryl/binaphthyl systems under oxidative conditions is currently in progress.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1526432. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03189k |
| This journal is © The Royal Society of Chemistry 2017 |