
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Harnessing fungal nonribosomal cyclodepsipeptide synthetases for mechanistic insights and tailored engineering†
Charlotte
Steiniger
a,
Sylvester
Hoffmann
a,
Andi
Mainz
a,
Marcel
Kaiser
bc,
Kerstin
Voigt
d,
Vera
Meyer
e and
Roderich D.
Süssmuth
 *a
*a
aFachgebiet Biologische Chemie, Institut für Chemie, Technische Universität Berlin, Strasse des 17. Juni 124, 10623 Berlin, Germany. E-mail: roderich.suessmuth@tu-berlin.de
bParasite Chemotherapy, Medical Parasitology & Infection Biology, Swiss Tropical and Public Health Institute, Socinstrasse 57, 4051 Basel, Switzerland
cUniversity of Basel, Petersplatz 1, 4003 Basel, Switzerland
dJena Microbial Resource Collection (JMRC), Leibniz-Institut für Naturstoff-Forschung und Infektionsbiologie, Hans-Knöll-Institut, Adolf-Reichwein-Straße 23, 07745 Jena, Germany
eFachgebiet Angewandte und Molekulare Mikrobiologie, Institut für Biotechnologie, Technische Universität Berlin, Gustav-Meyer-Allee 25, 13355 Berlin, Germany
First published on 25th September 2017
Abstract
Nonribosomal peptide synthetases represent potential platforms for the design and engineering of structurally complex peptides. While previous focus has been centred mainly on bacterial systems, fungal synthetases assembling drugs like the antifungal echinocandins, the antibacterial cephalosporins or the anthelmintic cyclodepsipeptide (CDP) PF1022 await in-depth exploitation. As various mechanistic features of fungal CDP biosynthesis are only partly understood, effective engineering of NRPSs has been severely hampered. By combining protein truncation, in trans expression and combinatorial swapping, we assigned important functional segments of fungal CDP synthetases and assessed their in vivo biosynthetic capabilities. Hence, artificial assembly line components comprising of up to three different synthetases were generated. Using Aspergillus niger as a heterologous expression host, we obtained new-to-nature octa-enniatin (4 mg L−1) and octa-beauvericin (10.8 mg L−1), as well as high titers of the hybrid CDP hexa-bassianolide (1.3 g L−1) with an engineered ring size. The hybrid compounds showed up to 12-fold enhanced antiparasitic activity against Leishmania donovani and Trypanosoma cruzi compared to the reference drugs miltefosine and benznidazole, respectively. Our findings thus contribute to a rational engineering of iterative nonribosomal assembly lines.
Introduction
Nonribosomal peptide synthetases (NRPSs) assemble a great variety of linear and cyclic peptides, which display a diverse spectrum of valuable pharmacologically useful bioactivities. Amongst those biosynthetic machineries, iterative-type NRPSs from filamentous fungi are a remarkable subgroup, which produce N-methylated cyclodepsipeptides (CDPs) of different ring sizes. Hexa-CDPs such as enniatin B (Fusarium ssp.) and beauvericin (Beauveria bassiana) as well as octa-CDPs like PF1022A (Rosellinia sp.) and bassianolide (B. bassiana) (Fig. 1b) show various antibacterial, anticancer, anthelmintic and insecticidal activities.1 This turns them into promising study candidates for further drug discovery and development. Iterative CDP synthetases consist of two modules, each containing domains for adenylation (A), transfer (T) and condensation (C) of the substrate, as well as a terminal T2b–C3 bidomain (Fig. 1a). The first module (M1) activates D-α-hydroxycarboxylic acids (D-HA) and has been shown to be promiscuous concerning aliphatic and aromatic D-HAs to varying extents.2–4 The D-HA is subsequently coupled to an L-α-amino acid (L-AA), which is activated and N-methylated by the second module (M2) containing an auxiliary N-methyltransferase (Mt) domain. Module 2 has also been reported to process different N-Me-L-amino acid (N-Me-L-AA) building blocks to a certain degree.5,6 Concerning substrate elongation, it has been proposed that the growing depsipeptide chain is elongated either in dipeptidol units (parallel mode)7,8 or in a stepwise fashion using single HA or AA building blocks (linear mode; Fig. 1a).9 Recently, the linear mode has been strongly favored, stating that the growing chain is shuttling between the T1 and T2a/b domains which alternately switch their donor and acceptor roles.10 For efficient macrocyclization of the full-length linear depsipeptide precursor, the N-terminal C1 and C-terminal C3 domain have previously been proposed to interact by forming a cavern.9 In contrast, the C3 domain alone was very recently found to mediate macrocyclization,10 which is in agreement with the established function of so-called termination C (Cterm) domains in fungal linear NRPSs.11 Still, various biosynthetic features of CDP synthetases are only partially understood, including mechanistic insights into how hexa- and octa-CDPs are assembled. | ||
| Fig. 1 Alternative CDP biosynthesis models and structures. (a) Models of CDP biosynthesis. Fungal CDP synthetases produce six- and eight-membered CDPs, with module 1 incorporating D-HAs and module 2 activating L-AAs. The building blocks are coupled until the chain length is long enough to be cyclized by the C3 domain. In the looping model (top), the depsipeptide chain grows by attachment of a single building block (HA or AA), shuttling between T1 and T2a/b. The role of T2b is still unclear. In the parallel model (bottom), the depsipeptide chain grows by addition of dipeptidols. a: acceptor site, d: donor site. (b) Cognate, non-methylated and hybrid CDP products. Iterative units are colored. | ||
Due to their modular architecture and high homology, CDP synthetases appear well suited to study domain-specific contributions by combinatorial exchanges, while generating novel peptide structures. Consequently, fungal NRPSs offer an excellent opportunity for engineering approaches. Inspiration comes from various engineering attempts in bacterial systems, including module fusion,12 substitution,13 extension14 and deletion15 of NRPS segments. Initial attempts in fungal NRPS-engineering have shown encouraging results in vivo, leading to novel nonribosomal peptides (NRPs) including derivatives of enniatin and beauvericin with altered D-HA side chains.16–18 Since the chemical synthesis of CDPs has some drawbacks and limitations, biotechnological production in heterologous microbial cell factories is a promising alternative for sustainable CDP production.17,19 However, as long as our mechanistic understanding of fungal NRPS assembly remains limited, an effective engineering of fungal iterative NRPSs is unfeasible. We thus focused on the functional analysis of various catalytic steps of CDP synthetases, whilst concomitantly establishing an in vivo expression system for new-to-nature CDP derivatives with altered bioactivities. While we were preparing this manuscript, Yu et al. proposed a linear CDP biosynthesis model, which complements our findings.10
Experimental
Selection and design of swapping and truncation sites
Structural information about flexible T–C linker regions was obtained from the T5C6 bidomain of tyrocidine synthetase from Brevibacillus brevis (TycC5–6; PDB 2JGP).20 Using the TycC5–6 T–C bidomain as a template, structural models of EnSYN-, BeSYN- and BaSYN-T1–C2 and T2b–C3 bidomains were generated (SWISS-MODEL21). The homology models were aligned to TycC5–6 (PyMOL, Schrödinger, LLC) to identify promising swapping sites in the linker regions. The structurally identified swapping regions were annotated in a multiple sequence alignment with the synthetases PF-, En-, Be- and BaSYN in order to define the distinct T1–C2 and T2b–C3 swapping sites (ESI Fig. 1†). The latter site was also used for the construction of EnSYNΔC3 (ESI Fig. 2†). For T2a–T2b swapping, the determined T2b–C3 swapping site was annotated for the T2a–T2b linker region in a multiple sequence alignment. Structural information about the C3 domain was obtained from the T–Cterm bidomain of TqaA from Penicillium aethiopicum (PDB 5EJD, ESI Fig. 3†).22 Based on TqaA–Cterm, the borders of the two subdomains CNTD and CCTD were defined (ESI Fig. 4†) and annotated in a multiple sequence alignment with PF-, En-, Be- and BaSYN (ESI Fig. 5†) in order to define the distinct C3sub-swapping sites (ESI Fig. 1†).For construction of the SYNΔC1 constructs, structural information about the C–A linker region was obtained from the termination module (C–A–T–Te) of surfactin A-synthetase from Bacillus subtilis (SrfA-C; PDB 2VSQ).23 The identified truncation region was annotated in a multiple sequence alignment with PF-, En-, Be- and BaSYN to define the distinct C1 truncation site (ESI Fig. 2†). For truncation of the Mt domain insertion, the boundaries were identified in a multiple sequence alignment of the PF-, En-, Be- and BaSYN-A1 and -A2 domains as well as the gramicidin A synthetase A domain (GrsA).24 Structural information about A domains was obtained from the crystal structure of GrsA (PDB 1AMU)24 to identify secondary structure elements flanking the loop region in which Mt domains are commonly embedded. The loop region was annotated in a multiple sequence alignment of the PF-, En-, Be- and BaSYN-A2 domains to identify the distinct truncation sites (ESI Fig. 2†). Based on GrsA, structural models of the EnSYN- and BaSYN-A1 domain and truncated A2 domains were generated (SWISS-MODEL) and aligned (PyMOL) to confirm the construction of a loop in A2 similar to A1 (ESI Fig. 6†). Further experimental details are provided in the ESI.†
Results and discussion
Providing the basis for an efficient fungal NRPS engineering
Due to low synthesis yields, many engineered in vivo NRPS systems are of limited value for deriving extensive conclusions on mechanisms or for a sustainable production.13,14,17 In order to lay a robust foundation for engineering approaches, we investigated the individual contributions of the C1, C3, T2a/b and Mt domains to CDP biosynthesis (Fig. 1a). A comparison of the seven conserved core motifs of canonical C domains25 indicated that they all differ significantly between the C1, C2 and C3 domains, suggesting different roles in CDP biosynthesis (ESI Tables 1–3†). This inspired us to construct truncated versions of the wild-type enniatin synthetase (EnSYN), beauvericin synthetase (BeSYN) and bassianolide synthetase (BaSYN) lacking the C1 domain. All expression assays were performed in vivo in Escherichia coli (DE3), harboring the fungal phosphopantetheinyl transferase (PPTase) npgA derived from A. nidulans DM3365 on a second plasmid. The co-expressed NpgA displayed a 14-fold enhanced production of the fungal CDP products in comparison to the endogenous PPTase and 4.8-fold increase when compared to Sfp (ESI Fig. 7†). Crude ethyl acetate extracts of the cell pellets were analyzed by MALDI-TOF-MS and LC-ESI-MS. Unlike previous approaches in Saccharomyces cerevisiae,10 the GST-tagged C1 deletion constructs (SYNΔC1; ∼324 kDa) were successfully produced in E. coli co-expressing NpgA (ESI Fig. 8†). Most importantly, all SYNΔC1 constructs maintained production of their cognate CDPs (Fig. 2a), while production yields dropped to 1–55% compared to the wild-type enzymes (Fig. 2c). This clearly indicated that the C1 domain plays an important role in CDP assembly, but that, in contrast to the cavern model,9 the C1 domain is not involved in macrocyclization.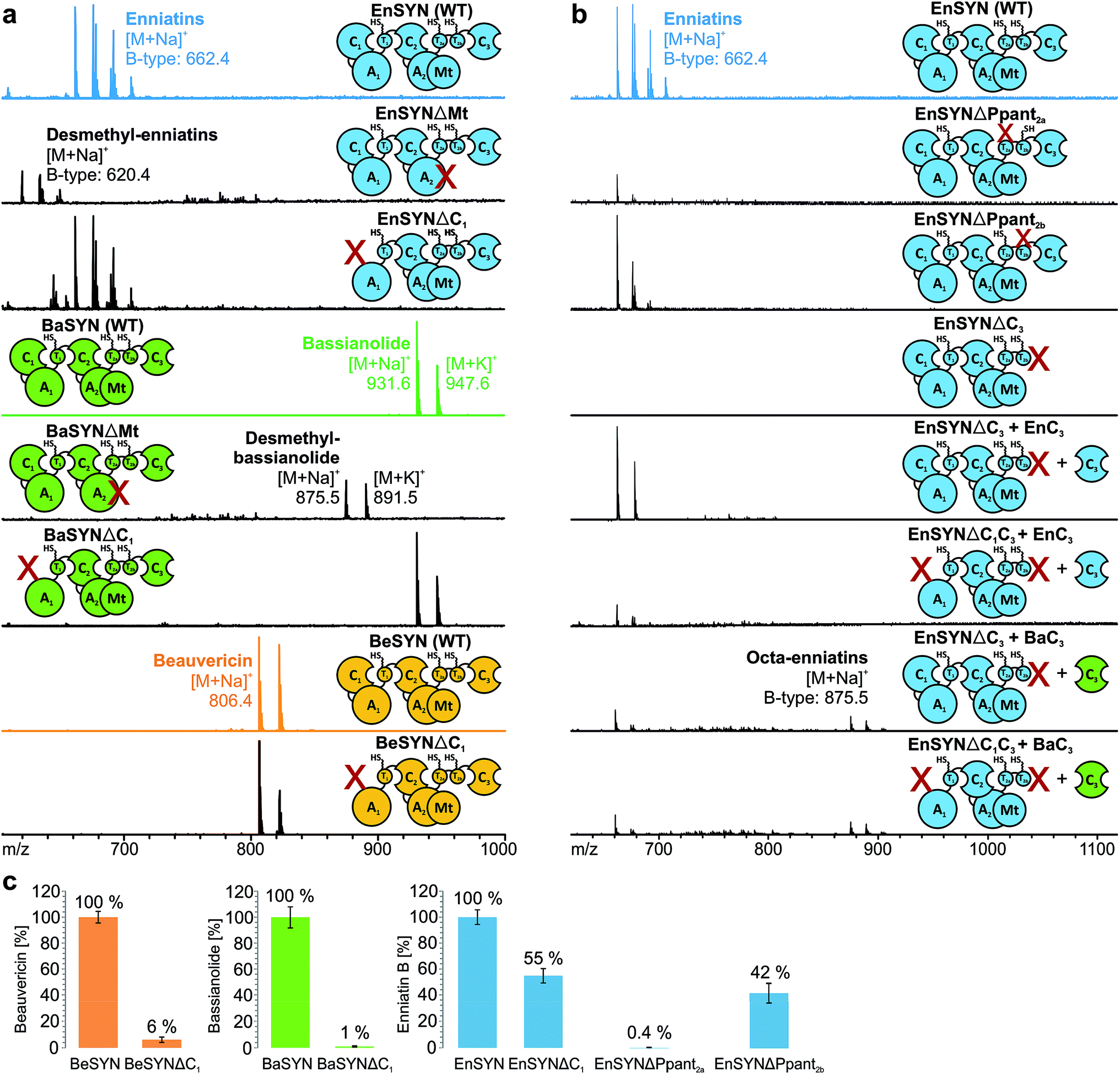 | ||
| Fig. 2 Mechanistic studies on fungal CDP synthetases. In vivo CDP production of (a) wild-type, mutated (EnSYNΔPpant2a/b) and truncated synthetases (SYNΔC1, SYNΔMt) as well as (b) in trans combinations synthesized in E. coli and monitored by MALDI-TOF-MS. (c) CDP yields in E. coli strains containing wild-type, ΔC1- and ΔPpant2a/b-synthetases determined by LC-ESI-MS (n = 3 cultures, standard deviation). | ||
In accordance with a recent in trans expression of the BeSYN-C3 domain,10 we generated truncated versions of EnSYN lacking the C3 domain (EnSYNΔC3) and monitored in vivo production of enniatins with and without co-expression of the excised EnSYN-C3 domain. EnSYNΔC3 was unable to produce enniatins, however, CDP production could be restored in trans by co-expression with the free-standing EnSYN-C3 domain (Fig. 2b). The EnSYN-C3 domain could also restore enniatin production of a synthetase construct devoid of both the C1 and C3 domains (EnSYNΔC1C3; Fig. 2b). Surprisingly, a combination of EnSYNΔ(C1)C3 with a heterologous C3 domain derived from the octa-CDP-producing BaSYN even enabled the production of new-to-nature octa-enniatin B (comprising one D-Hiv-Val unit more; Fig. 2b). To the best of our knowledge, octa-enniatin has not been reported to date. Hence, the function of the C3 domain can clearly be assigned to macrocyclization and ring size determination, which supports recent findings.10 Additionally, we aimed at challenging the flexibility of fungal CDP synthetases concerning non-methylated AAs. Therefore, we constructed variants of EnSYN and BaSYN devoid of the Mt domain (EnSYNΔMt, BaSYNΔMt; ESI Fig. 6†). Indeed, EnSYNΔMt and BaSYNΔMt exclusively produced the non-methylated analogues desmethyl-enniatin and desmethyl-bassianolide, respectively (Fig. 2a). Notably, these compounds represent the first example of a directed in vivo production of CDPs lacking backbone methylation. The findings are reminiscent of natural enniatins produced by EnSYN with only one or two N-methylations (type B2, B3), while for BaSYN, no natural non-methylated species have been reported so far. Finally, we challenged the linear CDP elongation model similar to a recent BeSYN-based approach10 by constructing versions of EnSYN with mutations at the conserved serines bearing the phosphopantetheine groups Ppant2a (S2538A) or Ppant2b (S2632A). Like the BeSYN-variants, both mutants (EnSYNΔPpant2a, EnSYNΔPpant2b) were still able to produce their wild-type CDP enniatin in vivo in E. coli (Fig. 2b). Similar to recent findings,10 the production of enniatin B by EnSYNΔPpant2a dropped to 0.4%, whereas EnSYNΔPpant2b still produced 42% compared to the wild-type synthetase (Fig. 2c). This observation confirms that the use of T2a or T2b as a waiting position is very unlikely, thus strongly supporting the linear/looping model.
Active chain-length control for hybrid CDP production
The identification of the C3 domain as the ring size determining domain in fungal iterative NRPSs10 inspired us to design new-to-nature CDPs of tailored ring size. To this end, we generated a set of hybrid synthetases based on the hexa-CDP-producing EnSYN and BeSYN as well as the octa-CDP producing BaSYN by exchanging the C3 domain or the T2b–C3 bidomain. This rendered XSYN-YC3 and XSYN-YTC3 constructs with X and Y referring to the NRPS core and the complementary heterologous segment, respectively (Fig. 3). Based on the identified domain boundaries in multiple sequence and structure alignments (see Experimental section), the domain swapping was performed in the inter-domain linker regions (ESI Fig. 1†). The resulting GST-tagged hybrid synthetases (∼375 kDa) were produced in soluble form in E. coli co-expressing NpgA (ESI Fig. 8†). Indeed, the hybrid synthetases EnSYN–BaTC3 and EnSYN–BaC3 both efficiently produced octa-enniatin B (4× N-Me-L-Val, m/z 853.553 [M + H]+; Fig. 3a). In analogy, the hybrids BeSYN–BaTC3 and BeSYN–BaC3 produced octa-beauvericin (m/z 1045.553 [M + H]+; Fig. 3c). So far, octa-beauvericin was only known from total synthesis26 and the in vivo biosynthesis has been reported very recently.10 Closer inspection of the biosynthesis products revealed that the octa-CDP producing hybrid synthetases EnSYN–Ba(T)C3 and BeSYN–Ba(T)C3 still produced trace amounts of the natural smaller ring size (Fig. 3a and c). Notably, the wild-type BaSYN is also able to produce minor amounts of hexa-bassianolide (Fig. 5b). Therefore, we can conclude that the BaSYN-C3 domain also cyclizes the premature CDP chain, which is in contrast to recent statements.10 The four BaSYN-based hybrid constructs BaSYN–En(T)C3 and BaSYN–Be(T)C3 produced the respective hybrid CDP hexa-bassianolide (corresponding to enniatin C, 3× N-Me-L-Leu, m/z 682.463 [M + H]+; Fig. 3b). Apart from BaSYN, hexa-bassianolide is also a natural side product of the wild-type EnSYN.27 The results reinforce the control function of the C3 domain with respect to the encrypted chain length for macrocyclization as well as its substrate flexibility concerning aliphatic and aromatic N-Me-L-AAs. | ||
| Fig. 3 Structural diversification of fungal CDPs. In vivo CDP production of wild-type and hybrid synthetases with swapped C-terminal part (see color code) based on EnSYN (a), BaSYN (b) and BeSYN (c) synthesized in E. coli and monitored by MALDI-TOF-MS. | ||
A detailed view of the C3 domain
In analogy to conventional elongation C domains (Celong) and fungal Cterm domains,20,22,28 the C3 domain consists of an N- and C-terminal subdomain (CNTD, CCTD), which together form a V-shaped pseudo-dimer with the active site buried in the cleft between the two subdomains (ESI Fig. 3†). Whereas CNTD harbors the catalytically relevant Hiscat, CCTD provides most of the interface residues towards the donor T domain. In contrast to the Cterm domain of the fungal tryptoquialanine (TqaA) synthetase that is closed at its acceptor site,22 the C3 domain of fungal CDP synthetases also catalyzes condensation reactions.10 Hence, the C3 domain features a functional acceptor site competent to catalyze ester bond formation. In order to gain insights into the molecular determinants of macrocyclization and ring size control by the C3 domain, we started with a sequence-based shuffling approach. An alignment of the C3 domains revealed ten residues potentially involved in ring size determination (ESI Fig. 5†). When visualized in a superposition of the TqaA-Cterm structure22 with a structural model of EnSYN-C3 (ESI Fig. 9†), five from the originally ten conspicuous residues remained, which were swapped from the BaSYN to the EnSYN sequence (EnSYN-mutC3). Exchanging these five amino acids did not hamper CDP production, but did not lead to an altered ring size either (Fig. 4a), ruling out these residues in terms of ring size control.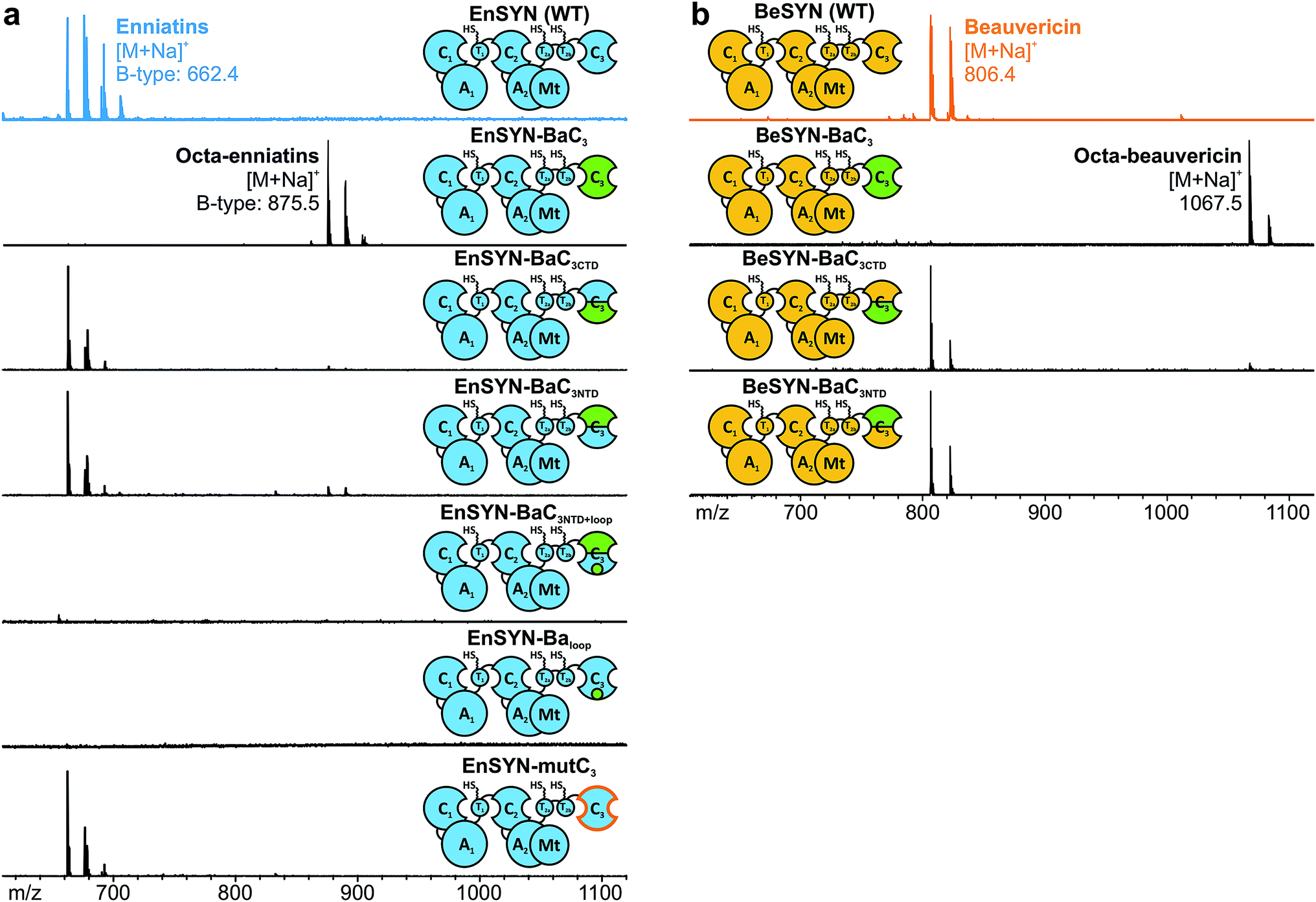 | ||
| Fig. 4 Swapping of C3 subdomains and elements thereof. In vivo CDP production of wild-type and hybrid synthetases based on EnSYN (a) and BeSYN (b) with swapped C3NTD, C3CTD, bridging loop region (small green circle) and five distinct AAs of BaSYN (orange line) in E. coli, respectively, as monitored by MALDI-TOF-MS. | ||
In an alternative approach, we swapped the subdomains C3NTD and C3CTD of BaSYN into the EnSYN and BeSYN system (ESI Fig. 4†). Expression of the four constructs EnSYN–BaC3NTD, EnSYN–BaC3CTD, BeSYN–BaC3NTD and BeSYN–BaC3CTD in E. coli co-expressing NpgA showed that CDP production was not hampered by the extensive heterologous intra-domain interfaces (Fig. 4). Remarkably, a swap of C3CTD gave rise to minor production of octa-CDPs. In the EnSYN-based system, a swap of C3NTD even slightly enhanced production of octa-enniatin B (Fig. 4a), whereas only trace amounts of octa-beauvericin could be observed for BeSYN–BaC3NTD (Fig. 4b). This data encouraged an additional swap of the bridging loop in C3CTD (EnSYN–BaC3NTD+loop, EnSYN–Baloop), the region which crosses over to C3NTD and forms a lid above the cyclization pocket (ESI Fig. 3†). However, swapping of the bridging loop completely abolished CDP production (Fig. 4a), suggesting that the conformation of the C3 domain has been massively disturbed. Thus, we hypothesize that CDP ring size is controlled not only by the size of the substrate pocket, but also by the acceptor- and donor-site interfaces that mediate the recruitment and correct positioning of the corresponding partner T domains. Such regulation via the T–C interfaces becomes in particular relevant in light of the recently postulated linear mode of CDP synthetases,10 which relies on a competition between T1 and T2a/b for binding to C3 leading either to chain elongation or macrocyclization.
Scale-up production in A. niger and bioactivities of hybrid CDPs
In previous engineering approaches, the use of E. coli as a bacterial heterologous host proved valuable for rapid proof-of-principle approaches.17 However, the fungal heterologous host A. niger appears to be more suitable for scale-up of fungal CDP production, as it renders significantly higher CDP yields.17,19 Therefore, we aimed for an enhanced production of the hybrid CDPs octa-enniatin B, octa-beauvericin and hexa-bassianolide by using A. niger under control of an inducible expression system.19 For that purpose, we performed polyethylene glycol (PEG)-mediated transformation of two A. niger strains differing in their protease profiles (strains AB1.13 and MA169.4) with the hybrid synthetase genes of EnSYN–BaTC3, BeSYN–BaTC3, BaSYN–EnTC3 and BaSYN–BeTC3. The genes were integrated into the fungal genome by homologous recombination (ESI Fig. 10†). From the two alternative constructs BaSYN–EnTC3 and BaSYN–BeTC3 synthesizing hexa-bassianolide, the former was chosen for scale-up in A. niger due to a higher production (ESI Fig. 11†).All hybrid CDPs were purified by extraction with ethyl acetate, flash chromatography and reversed phase HPLC. Purification of the hybrid octa-CDPs from A. niger yielded 4 mg L−1 of octa-enniatin B and 10.8 mg L−1 of octa-beauvericin, respectively. Intriguingly, purification of hexa-bassianolide produced by BaSYN–EnTC3 afforded very high titers of approximately 1.3 g L−1. To our knowledge, this is the first report of an artificial NRPS producing on the g L−1-scale, which underlines the great potential of fungal NRPS systems for biosynthetic engineering. Without understanding the catalytic assembly in detail, the higher titer in comparison to the hybrid octa-CDPs might be attributed to a more efficient interaction between the heterologous parts. For example, combining BaSYN with segments of EnSYN in E. coli rendered significantly higher titers than BaSYN–BeSYN combinations (Fig. 5b), which were also used for the production of octa-beauvericin (BeSYN–BaTC3). Since both wild-type BaSYN and EnSYN naturally produce minor amounts of hexa-bassianolide, the building blocks D-Hiv and L-Leu represent cognate substrates for both NRPS systems, thus ensuring efficient processing. To date, hexa-bassianolide could only be obtained in low amounts either from chemical synthesis,29 by precursor-directed biosynthesis (2.2 mg L−1)30 or from extraction of cultures of Verticillium hemipterigenum (BCC 1449; 0.3 mg L−1).27 In a very recent report on heterologous CDP production in S. cerevisiae, no yields were mentioned.10
 | ||
| Fig. 5 Multiple swapping of fungal CDP synthetases. (a) In vivo CDP production of the hybrid synthetases (see color code) monitored by MALDI-TOF-MS. (b) CDP titers of wild-type and hybrid NRPSs producing bassianolide and hexa-bassianolide in E. coli compared by LC-ESI-MS (n = 3 cultures, standard deviation). n. d.: not detected. | ||
Based on the sufficient amount of the hybrid CDPs for biological profiling and the broad activity range of CDPs,30–32 octa-enniatin B, octa-beauvericin and hexa-bassianolide were tested for antiparasitic, antibacterial and antifungal activity along with cytotoxicity measurements (ESI Tables 4–6†). As for hexa-bassianolide, antiplasmodial, antimycobacterial and cytotoxic properties were already shown.30,31 Additionally, hexa-bassianolide was found to be active against the parasites Trypanosoma b. rhodesiense, Trypanosoma cruzi and Leishmania donovani (ESI Table 4†). Most remarkably, both hybrid octa-CDPs showed a significantly increased antiparasitic activity (ESI Table 4†). Against T. cruzi, octa-enniatin B and octa-beauvericin showed up to 12-fold lower IC50 (1.76 μM and 0.54 μM, respectively) than the medical reference drug benznidazole (6.53 μM). Furthermore, they are superior to their corresponding natural hexa-CDPs enniatin B (2.31 μM) and beauvericin (0.76 μM), respectively. Against L. donovani, octa-enniatin B and octa-beauvericin showed up to >8-fold lower IC50 (0.07 μM and 0.14 μM, respectively) compared to the medically used reference drug miltefosine (0.58 μM). In comparison to enniatin B and beauvericin (IC50 0.72 μM and 0.31 μM, respectively), both compounds were also significantly more active and showed a similar cytotoxicity against rat-derived L6 cells (ESI Table 4†).
Multiple swapping of fungal CDP synthetases
With the assigned synthetase functions for precursor and ring size determination in hand, the fungal iterative NRPSs can be seen as a modular assembly platform for a tailored construction of new CDP structures. In order to test the flexibility of fungal CDP synthetases even further, we assessed the influence of multiple domain swaps on hybrid CDP synthesis. For this purpose, we constructed a set of synthetase chimeras for each of the three hybrid CDPs comprising parts from up to three CDP synthetases. All of these six multiple hybrids (EnSYN–BaM2, BaSYN–EnM2, BaSYN–BeM2, BeM1–BaM2–EnTC3, EnM1–BeM2–BaTC3, BeM1–EnM2–BaTC3) were capable of producing the predicted hexa- or octa-CDPs in E. coli co-expressing NpgA (Fig. 5a). These results demonstrate the first successful multiple cross-pathway assembly in fungal iterative NRPSs.Since bacterial NRPS-systems do not tolerate this type of swapping well,13 we further examined whether fungal CDP synthetases behave in a similar fashion. For that purpose, we determined the production titers of all wild-type and hybrid CDP synthetases producing hexa-bassianolide. The titers of all singly swapped hybrids (0.03–0.88 mg L−1) remained in the same range as that of wild-type EnSYN (0.15 mg L−1) and BaSYN (0.06 mg L−1), with the exception of BaSYN–EnC3 (24 mg L−1). The fact that BaSYN–EnC3 showed such a superior production in comparison to BaSYN–EnTC3 (0.88 mg L−1) highlights the influence of artificial T–C domain pairings in hybrid synthetases, which have a crucial role, i.e. in ester bond formation and macrocyclization. Surprisingly, the hybrid synthetases containing multiple swaps (EnSYN–BaM2, BeM1–BaM2–EnTC3) showed an enhanced production up to a factor of 520 (55–78 mg L−1) compared to the wild-type EnSYN (0.15 mg L−1), despite having more heterologous interfaces for domain–domain interactions. This is in stark contrast to virtually all previous studies on module and domain exchanges in bacterial NRPS systems suffering from a significant drop in product titers.32 Notably, the relatively high titer was achieved in the bacterial heterologous host E. coli, which previously rendered low production yields.17 Fungal NRPS systems might thus represent a viable alternative as an engineering platform with promising potential. Furthermore, the results suggest that the efficiency of the biosynthesis is influenced by the N-terminal part of the synthetase. This is supported by the fact that the hybrid EnSYN–BaM2 contains a homologous module 1 and T2bC3, thus pointing to highly important inter-modular interactions like the proposed C3–T1 interaction during ester bond formation.10 However, the highest production was achieved with a heterologous C3–T1 interface (BeM1–BaM2–EnTC3). The assumption of a readily exchangeable “bio-brick” system thus appears somewhat simplistic for a sustainable engineering approach.
Conclusions
Fungal CDP synthetases produce six- and eight-membered CDPs with a broad range of pharmacologically relevant bioactivities. Based on their modular structure, high homology and manageable size, these nonribosomal assembly lines bear good prospects to be an effective engineering platform for the production of novel CDP compounds. Still, a tailored engineering of CDP synthetases is somewhat limited by our poor understanding of the biosynthesis mechanisms. The systematic investigation of the individual contributions of domains to the catalytic activity and synthesis products in this study allow us to confirm the following model: the C-terminal C3 domain performs macrocyclization and determines the ring size independently from C1. In contrast to recent findings,10 truncated synthetases lacking the C1 domain are indeed functional in vivo. However, since the absence of C1 significantly affects the production yields of CDPs (drop of synthesis to 1–55%), a stabilizing and/or supportive role of C1 in the catalytic cycle is apparent. In accordance with a recent crystal structure comprising the C–A–T–Te domains of the bacterial NRPS AB3403,28 the C1 domain may serve as a resting site during precursor adenylation in the A1 domain. This stabilization may also apply to the A1 domain itself, since the C domains can affect the tertiary structure33 and substrate specificities of their downstream A domain partners.34With regard to substrate specificity, various functional hybrid synthetases with a heterologous C3 domain demonstrated that the C3 domain tolerates aliphatic as well as aromatic side chains (N-Me-L-Val/Leu/Ile/Phe). Apart from esterification, C3 also inherits the role of a gauge, measuring product chain length. In contrast to recent statements,10 this gauge also allows for cyclization of premature depsipeptide chains. Deletion of the Mt domain, affording the corresponding non-methylated peptides, was able to demonstrate that the absence of structure-modulating N-methylations is tolerated. As previous Mt domain deletions reportedly only affected β-methylation of Glu in daptomycin,13 our findings represent a rare example of complete in vivo NRP backbone engineering. The construction of a number of artificial CDP synthetases by means of domain swapping yielded hybrid hexa- and octa-CDPs under in vivo conditions. This way, we were able to produce the hybrid CDP hexa-bassianolide at very high titer of 1.3 g L−1 as well as new-to-nature octa-enniatin B (4 mg L−1) and octa-beauvericin (10.8 mg L−1) by using A. niger as a robust and cognate heterologous host. While previous approaches with the daptomycin synthetase have shown that multiple domain swaps lead to reduced product yields,13 we could significantly enhance CDP production in vivo by constructing functional hybrid synthetases from three different NRPSs. The results highlight the potential of fungal NRPS systems as engineering platforms as well as the crucial role of domain–domain and module–module interfaces in NRPS efficiency.
Swapping of parts of the C3 subdomain uncovered functional aspects of macrocyclization and ring size control. According to the bacterial Te mechanism, macrocyclization by the fungal C3 domain is performed when the free-standing hydroxy group at the tail of the linear depsipeptidyl chain is in close proximity to Hiscat of C3 and the substrate thioester. In consequence, CDP ring size probably depends on the size and composition of the cyclization pocket formed by C3NTD and C3CTD, along with a competitive process between the elongation and cyclization reaction. This assumption is supported by our observations that, unlike the octa-CDP-producing synthetases, all hexa-CDP-generating hybrids almost completely lost the capability of producing the cognate eight-membered ring. One reason might be the steric demand of a linear octa-depsipeptide, which may not fit well into the cyclization pocket of a hexa-CDP-producing synthetase. In addition to steric restrictions imposed by the (heterologous) cyclization pocket, ring size may also depend on intrinsic conformational properties of the growing depsipeptide chain. The nature of side chains, and in particular of backbone N-methylation, dictates the available conformational space of the depsipeptide backbone and thus affects cyclization efficiencies. For example, Ramachandran plots for peptides comprising N-Me-AAs demonstrate the restricted conformational freedom compared to their non-methylated analogs.35 However, the distinct mechanism of CDP ring size determination remains to be elucidated. By confirming recent findings of CDP production in synthetase variants lacking Ppant2a/b, our data also give strong support to the so-called “linear” CDP biosynthesis model. From a formal aspect, employing an alternating upstream and downstream elongation, this NRPS type might be reclassified as belonging to the “complex-type”32 rather than simply linear or iterative. We therefore suggest the term “looping mode”, implicating the substrate shuttling between two C domains in a consecutive manner. Still, since the role of the T2b domain remains unclear and odd-numbered T domain-bound intermediates are yet to be observed,8 neither of the two models can be completely ruled out. Hence, further information must be gathered with synthetase mutants, e.g. stalled at certain biosynthesis steps, as it has been applied to the bacterial balhimycin synthetase36 and rifamycin polyketide synthase.37
Finally, our experiments show that the design of bio-combinatorial swaps in NRPSs can serve well for the synthesis of compounds with enhanced bioactivities. Accordingly, the bioactivity profiling of octa-enniatin B and octa-beauvericin rendered a significantly stronger antiparasitic activity against T. cruzi (up to 12-fold) and L. donovani (up to 8.3-fold) compared to the reference drugs benznidazole and miltefosine, respectively, accompanied by similar cytotoxicity against L6 cells. Our findings give distinct clues for the active and applicable reprogramming of both iterative and linear NRPS assembly lines in further protein engineering approaches. In the future, we aim to expand the existing range of CDP ring sizes beyond the natural hexa- and octa-CDPs, giving rise to a whole class of new-to-nature cyclic fungal products with superior pharmacological activities and efficiencies.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank A. Mühlenweg and A. Poch for the construction of the PPTase-vectors and the Ppant knock-out mutants as well as S. Grätz for synthesizing the D-Hiv precursor. We also express our gratitude to C. Mudalungu for her help with NMR analysis as well as S. Böcker for the construction of pJET_BaSYN and for assisting the transformation of A. niger along with T. Schütze. We thank M. Cal, S. Keller-Märki and R. Rocchetti for their help with the parasite assays. KV wishes to thank C. Weigel for excellent technical help in the antimicrobial activity tests. We thank P. Durkin for proofreading of the manuscript.Notes and references
- R. D. Süssmuth, J. Müller, H. von Döhren and I. Molnár, Nat. Prod. Rep., 2011, 28, 99–124 RSC.
- S. C. Feifel, T. Schmiederer, T. Hornbogen, H. Berg, R. D. Süssmuth and R. Zocher, ChemBioChem, 2007, 8, 1767–1770 CrossRef CAS PubMed.
- J. Müller, S. C. Feifel, T. Schmiederer, R. Zocher and R. D. Süssmuth, ChemBioChem, 2009, 10, 323–328 CrossRef PubMed.
- D. Matthes, L. Richter, J. Müller, A. Denisiuk, S. C. Feifel, Y. Xu, P. Espinosa-Artiles, R. D. Süssmuth and I. Molnár, Chem. Commun., 2012, 48, 5674 RSC.
- M. Krause, A. Lindemann, M. Glinski, T. Hornbogen, G. Bonse, P. Jeschke, G. Thielking, W. Gau, H. Kleinkauf and R. Zocher, J. Antibiot., 2001, 54, 797–804 CrossRef CAS PubMed.
- N. Madry, R. Zocher, K. Grodzki and H. Kleinkauf, Appl. Microbiol. Biotechnol., 1984, 20, 83–86 CrossRef CAS.
- R. Zocher, U. Keller and H. Kleinkauf, Biochem. Biophys. Res. Commun., 1983, 110, 292–299 CrossRef CAS PubMed.
- W. Weckwerth, K. Miyamoto, K. Iinuma, M. Krause, M. Glinski, T. Storm, G. Bonse, H. Kleinkauf and R. Zocher, J. Biol. Chem., 2000, 275, 17909–17915 CrossRef CAS PubMed.
- M. Glinski, C. Urbanke, T. Hornbogen and R. Zocher, Arch. Microbiol., 2002, 178, 267–273 CrossRef CAS PubMed.
- D. Yu, F. Xu, S. Zhang and J. Zhan, Nat. Commun., 2017, 8, 15349 CrossRef CAS PubMed.
- X. Gao, S. W. Haynes, B. D. Ames, P. Wang, L. P. Vien, C. T. Walsh and Y. Tang, Nat. Chem. Biol., 2012, 8, 823–830 CrossRef CAS PubMed.
- H. D. Mootz, D. Schwarzer and M. A. Marahiel, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5848–5853 CrossRef CAS PubMed.
- K. T. Nguyen, D. Ritz, J.-Q. Gu, D. Alexander, M. Chu, V. Miao, P. Brian and R. H. Baltz, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17462–17467 CrossRef CAS PubMed.
- D. Butz, T. Schmiederer, B. Hadatsch, W. Wohlleben, T. Weber and R. D. Süssmuth, ChemBioChem, 2008, 9, 1195–1200 CrossRef CAS PubMed.
- H. D. Mootz, N. Kessler, U. Linne, K. Eppelmann, D. Schwarzer and M. A. Marahiel, J. Am. Chem. Soc., 2002, 124, 10980–10981 CrossRef CAS PubMed.
- D. Yu, F. Xu, D. Gage and J. Zhan, Chem. Commun., 2013, 49, 6176 RSC.
- S. Zobel, S. Boecker, D. Kulke, D. Heimbach, V. Meyer and R. D. Süssmuth, ChemBioChem, 2016, 17, 283–287 CrossRef CAS PubMed.
- J. W. A. van Dijk, C. Guo and C. C. C. Wang, Org. Lett., 2016, 18, 6236–6239 CrossRef CAS PubMed.
- L. Richter, F. Wanka, S. Boecker, D. Storm, T. Kurt, Ö. Vural, R. Süssmuth and V. Meyer, Fungal Biol. Biotechnol., 2014, 1, 4 CrossRef PubMed.
- S. A. Samel, G. Schoenafinger, T. A. Knappe, M. A. Marahiel and L.-O. Essen, Structure, 2007, 15, 781–792 CrossRef CAS PubMed.
- M. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T. G. Cassarino, M. Bertoni, L. Bordoli and T. Schwede, Nucleic Acids Res., 2014, 42, W252–W258 CrossRef CAS PubMed.
- J. Zhang, N. Liu, R. A. Cacho, Z. Gong, Z. Liu, W. Qin, C. Tang, Y. Tang and J. Zhou, Nat. Chem. Biol., 2016, 12, 1001–1003 CrossRef CAS PubMed.
- A. Tanovic, S. A. Samel, L.-O. Essen and M. A. Marahiel, Science, 2008, 321, 659–663 CrossRef CAS PubMed.
- E. Conti, T. Stachelhaus, M. A. Marahiel and P. Brick, EMBO J., 1997, 16, 4174–4183 CrossRef CAS PubMed.
- M. A. Marahiel, T. Stachelhaus and H. D. Mootz, Chem. Rev., 1997, 97, 2651–2674 CrossRef CAS PubMed.
- D. Lücke, T. Dalton, S. V. Ley and Z. E. Wilson, Chem.–Eur. J., 2016, 22, 4206–4217 CrossRef PubMed.
- S. Supothina, M. Isaka, K. Kirtikara, M. Tanticharoen and Y. Thebtaranonth, J. Antibiot., 2004, 57, 732–738 CrossRef CAS PubMed.
- E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov, C. Leigh Allen, G. Skiniotis, C. C. Aldrich and A. M. Gulick, Nature, 2016, 529, 235–238 CrossRef CAS PubMed.
- Y. A. Ovchinnikov, V. T. Ivanov, I. I. Mikhaleva and M. M. Shemyakin, Izv. Akad. Nauk SSSR, Ser. Khim., 1964, 581, 1912 Search PubMed.
- C. Nilanonta, M. Isaka, R. Chanphen, N. Thong-orn, M. Tanticharoen and Y. Thebtaranonth, Tetrahedron, 2003, 59, 1015–1020 CrossRef CAS.
- A. A. Sy-Cordero, C. J. Pearce and N. H. Oberlies, J. Antibiot., 2012, 65, 541–549 CrossRef CAS PubMed.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed.
- M. J. Tarry, A. S. Haque, K. H. Bui and T. M. Schmeing, Structure, 2017, 25, 783–793 CrossRef CAS PubMed.
- S. Meyer, J.-C. Kehr, A. Mainz, D. Dehm, D. Petras, R. D. Süssmuth and E. Dittmann, Cell Chem. Biol., 2016, 23, 462–471 CrossRef CAS PubMed.
- T. T. Tran, H. Treutlein and A. W. Burgess, Protein Eng., Des. Sel., 2006, 19, 401–408 CrossRef CAS PubMed.
- D. Bischoff, B. Bister, M. Bertazzo, V. Pfeifer, E. Stegmann, G. J. Nicholson, S. Keller, S. Pelzer, W. Wohlleben and R. D. Süssmuth, ChemBioChem, 2005, 6, 267–272 CrossRef CAS PubMed.
- A. Stratmann, C. Toupet, W. Schilling, R. Traber, L. Oberer and T. Schupp, Microbiology, 1999, 145, 3365–3375 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed methodology, alignments, cloning, purification, NMR, bioactivity. See DOI: 10.1039/c7sc03093b |
| This journal is © The Royal Society of Chemistry 2017 |