
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a high quantum yield dye for tumour imaging†
Dan
Yang‡
a,
Huasen
Wang‡
b,
Chengjie
Sun‡
a,
Hui
Zhao
a,
Kuan
Hu
 a,
Weirong
Qin
a,
Rui
Ma
b,
Feng
Yin
a,
Xuan
Qin
a,
Qianling
Zhang
c,
Yongye
Liang
*b and
Zigang
Li
*a
a,
Weirong
Qin
a,
Rui
Ma
b,
Feng
Yin
a,
Xuan
Qin
a,
Qianling
Zhang
c,
Yongye
Liang
*b and
Zigang
Li
*a
aSchool of Chemical Biology and Biotechnology, Shenzhen Graduate School of Peking University, Shenzhen, 518055, China. E-mail: lizg@pkusz.edu.cn
bDepartment of Materials Science and Engineering, South University of Science and Technology of China, Shenzhen 518055, China. E-mail: liangyy@sustc.edu.cn
cShenzhen Key Laboratory of Functional Polymer, College of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen, Guangdong 518060, China
First published on 19th July 2017
Abstract
A fluorescent dye, FEB, with high fluorescence quantum yield for tumour imaging is reported. FEB dyes can be efficiently synthesized in three steps and then easily modified with either PEG or PEG-iRGD to yield FEB-2000 or FEB-2000-iRGD, respectively. Both modified dyes showed negligible toxicity and were thus able to be adopted for in vivo tumour imaging. PEG modification endowed the dye FEB-2000 with both long circulating times and good tumour targeting properties in a MDA-MB-231 xenograft model. Further conjugation with iRGD to generate FEB-2000-iRGD showed minimal targeting enhancement. These results provide a template for the efficient preparation of FEB dyes for use in tumour imaging, thus providing a foundation for future modifications.
Introduction
It has remained a high priority to develop sensitive and non-invasive diagnostic tools for cancer—a collection of the most life-threatening diseases.1 Conventional approaches such as positron emission tomography (PET), single-photon emission computed tomography (SPECT), magnetic resonance imaging (MRI), and optical imaging have already shown great promise for non-invasive, real-time, and high-resolution tumour imaging.2–4 Among these, fluorescence imaging in the far red and NIR window has several advantages including minimal photo-damage to biological samples, satisfying penetration depth, and negligible interference from background auto-fluorescence.5–7Continuous work is being invested in exploring better fluorescent dyes for use in imaging. To date, numerous dyes have been developed and marketed for various biomedical applications, including tumour diagnosis and molecular image-guided surgeries.8 However, lots of them suffer from low biocompatibility, high toxicity, and low quantum yields.9,10 Some dyes—such as Rhodamine B—have small Stokes shifts (typically less than 35 nm) that cause systematic errors due to excitation backscattering effects.11 In addition, most tumour-targeting fluorescent probes require a combination of targeting partners (such as metabolic substrates),12 cell-surface receptor targeting peptides,13 growth factors,7,14 antibodies,15 and cancer-specific cell-surface biomarkers.16,17
Recently, fluorene and 3,4-ethoxylene dioxythiophene (EDOT) were employed as the shielding and donor units, respectively, to construct a donor–acceptor–donor-based fluorophore that absorbs in the NIR-II window. It was found that using both fluorene and EDOT could reduce the intermolecular or intramolecular interactions in the fluorophore molecules, enhancing the fluorescence quantum yield. Recent work from our lab developed a molecular fluorophore (IR-E1) that contained this donor–acceptor–donor structure.18 Specifically, we used benzo[1,2-c:4,5-c′]bis([1,2,5] thiadiazole) (BBTD) as the acceptor and thiophene-based units as the donor. This fluorophore was used to characterize dynamic vascular changes in a mouse model of traumatic brain injury. Herein, we report a new fluorescent dye, FEB, that is easily synthesized and was found to be suitable for in vivo tumour imaging (Scheme 1). We noticed that benzo-2,1,3-thiadiazole (BTD) has been utilized as a core unit with good fluorescence in both solution and solid state.10,19 By replacing the acceptor unit BBTD with BTD, new fluorophores with an emission peak at 670 nm were synthesized. The non-planar, EDOT–BTD–EDOT configuration decreased inter- and intramolecular interactions.20 The introduction of long alkyl chains reduced intermolecular interactions and enhanced the dye’s quantum yield.21 These fluorophores demonstrated a high fluorescence quantum yield of 35% in aqueous solution and a large Stokes shift of 120 nm, which are promising for bioimaging applications. We intentionally retained a bromine atom in the alkyl chain, allowing for further modifications.22 We further explored the tumour imaging application with the PEGylated fluorophore and the one labelled with iRGD. Interestingly, PEGylated FEB itself was shown to have a tumour targeting effect.
 | ||
| Scheme 1 Schematic representation of the FEB molecule based on benzo-2,1,3-thiadiazole. | ||
Results and discussion
A non-planar EDOT–BBTD–EDOT configuration could avoid inter- and intramolecular interactions.18 The 3,4-ethylenedioxy groups on EDOT improve dye dispersity in water. Here, a non-planar EDOT–BTD–EDOT configuration was introduced. Fluorescent BTDs are usually efficient and stable, and show a good signal-to-noise ratio. The fluorene and long alkyl chains also served as donors, thereby reducing intermolecular interactions and avoiding fluorescence quenching. The FEB dye was synthesized in three steps using a palladium-catalyzed Stille coupling reaction as its key step (Scheme 2). Compound 4 was obtained by di-acylation of compound 3, which was derived from cross-coupling between compounds 1 and 2 (see ESI†). Compound 4 was then coupled with the core part of FEB, namely compound 5, to derive FEB. This coupling step is facile and has a high yield. FEB retained four alkyl bromide moieties for further modifications. Azidation was conducted to obtain FEB-N3, which can be modified via ‘click’ chemistry. | ||
| Scheme 2 The synthesis of FEB-N3, FEB-2000 and FEB-2000-iRGD. See synthesis details and compound characterization in the ESI.† | ||
FEB-N3 showed an absorbance peak at ∼550 nm and an emission peak at ∼675 nm in toluene (Fig. 1a). The absolute fluorescence quantum yield of FEB-N3 was 86% in toluene. To further protect the dye core and increase its aqueous solubility, FEB-N3 was allowed to react with 2 kDa alkyne-PEG-COOH via ‘click’ chemistry.23 Successful PEGylation was confirmed using GPC (Gel Permeation Chromatography, see GPC data in ESI†). Fluorescence spectroscopy data showed that FEB-2000 had an absorbance peak at ∼550 nm and an emission peak at ∼670 nm (Fig. 1b), which is significantly better than Rhodamine dyes with small Stokes shifts.
 | ||
| Fig. 1 (a) Optical characterization of FEB-N3, demonstrating an absorbance peak at ∼550 nm and an emission peak at ∼675 nm in toluene; (b and c) optical characterization of FEB-2000 and FEB-2000-iRGD, demonstrating an absorbance peak at ∼550 nm and an emission peak at ∼670 nm in water. | ||
Previous reports have shown that the iRGD peptide c(CRGDK/RGPDC) can interact with both integrin and neuropilin-1 receptors, thereby enabling better cellular and deep tissue penetration.24,25 To increase the cancer cell targeting ability, we prepared the cyclic iRGD peptide and conjugated it with alkyne-PEG-COOH by amide bond formation. The PEGylated peptide was then conjugated to FEB via ‘click’ chemistry, producing FEB-2000-iRGD. FEB-2000-iRGD showed an absorbance peak at ∼550 nm and an emission peak at ∼670 nm in water (Fig. 1c).
As shown in Fig. 1, both the PEG and iRGD modifications did not interfere with the absorption behaviour. The absorption coefficients of FEB-2000 and FEB-2000-iRGD in water were 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 530 M cm−1 and 27300 M cm−1, respectively (Fig. S1†). Both FEB-2000 and FEB-2000-iRGD showed good stability (Fig. S2†). As the BTD acceptor was weaker in comparison to the NIR-II fluorophore IR-E1, the emission spectrum of the FEB derivatives exhibited a blue shift. However, the reduction in conjugation resulted in a high fluorescence quantum yield in water. Using Rhodamine B (QY = 31%) as a reference,26 the quantum yields of FEB-2000 and FEB-2000-iRGD in water were determined to be 35% and 36%, respectively (Fig. S3†). The quantum yields of the FEB derivatives were therefore on par with most commercial deep red fluorescence dyes, making them suitable for in vivo imaging. Similar to typical benzothiadiazole derivatives, the designed compounds displayed large Stokes shifts (120–125 nm). In addition, they showed longer emission wavelengths.
530 M cm−1 and 27300 M cm−1, respectively (Fig. S1†). Both FEB-2000 and FEB-2000-iRGD showed good stability (Fig. S2†). As the BTD acceptor was weaker in comparison to the NIR-II fluorophore IR-E1, the emission spectrum of the FEB derivatives exhibited a blue shift. However, the reduction in conjugation resulted in a high fluorescence quantum yield in water. Using Rhodamine B (QY = 31%) as a reference,26 the quantum yields of FEB-2000 and FEB-2000-iRGD in water were determined to be 35% and 36%, respectively (Fig. S3†). The quantum yields of the FEB derivatives were therefore on par with most commercial deep red fluorescence dyes, making them suitable for in vivo imaging. Similar to typical benzothiadiazole derivatives, the designed compounds displayed large Stokes shifts (120–125 nm). In addition, they showed longer emission wavelengths.
Fluorescent molecule uptake was first examined using in vitro systems.27 MDA-MB-231 cells were incubated with 22 μM FEB-2000 and FEB-2000-iRGD in serum-free media for 6 h, washed, and then imaged using confocal microscopy (Fig. 2a). Both FEB-2000 and FEB-2000-iRGD showed high cellular uptake. We compared the FEB derivatives with the commercially available near-infrared dye IR-783,28,29 which preferentially targets tumour cells (Fig. S4†). Although the FEB derivatives showed a poorer ability to label living cells than IR-783, they could be easily prepared in three steps and conveniently modified via ‘click’ chemistry. Notably, both FEB-2000 and FEB-2000-iRGD showed negligible cytotoxicity in the MDA-MB-231 and HeLa cell lines at a concentration of 100 μM (Fig. 2b and S5†).
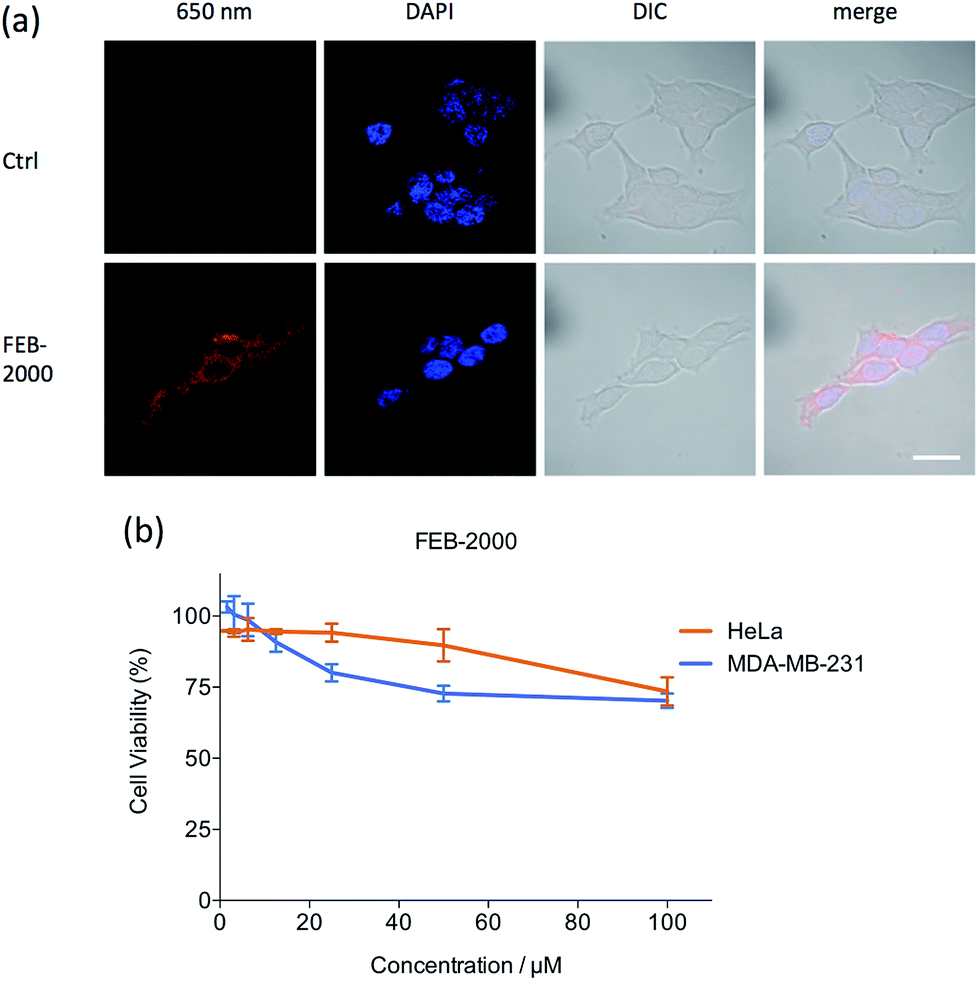 | ||
| Fig. 2 (a) Confocal laser-scanning microscopy images of MDA-MB-231 cells incubated with PBS, FEB-2000 at 310 K for 6 h. Scale bar = 20 μm; (b) cytotoxicity of FEB-2000 on the MDA-MB-231 cell line and HeLa cell line at 310 K. | ||
To evaluate FEB-2000 distribution in vivo, we next performed fluorescence imaging using a MDA-MB-231 xenograft model. With intravenous injection of 100 μL FEB-2000 (270 μM), specific tumour uptake of the dye was observed within 6 h. The tumour was clearly visible with a tumour/muscle (T/M) ratio of 2.0 (see Fig. 3a). After 24 h, the T/M ratio increased to 2.5. Tumour accumulation was still observed after 96 h. At 48 h post-injection, we quantified the dye accumulation in different critical organs, including the tumour, heart, liver, spleen, lung, and kidney. As shown in Fig. 3b, there was little dye accumulation in organs other than the liver. It has been reported that the PEG chain itself showed noticeable tumour accumulation through EPR effects.30,31 FEB-2000 accumulation in the tumour could be attributed to EPR effects caused by PEGylation.
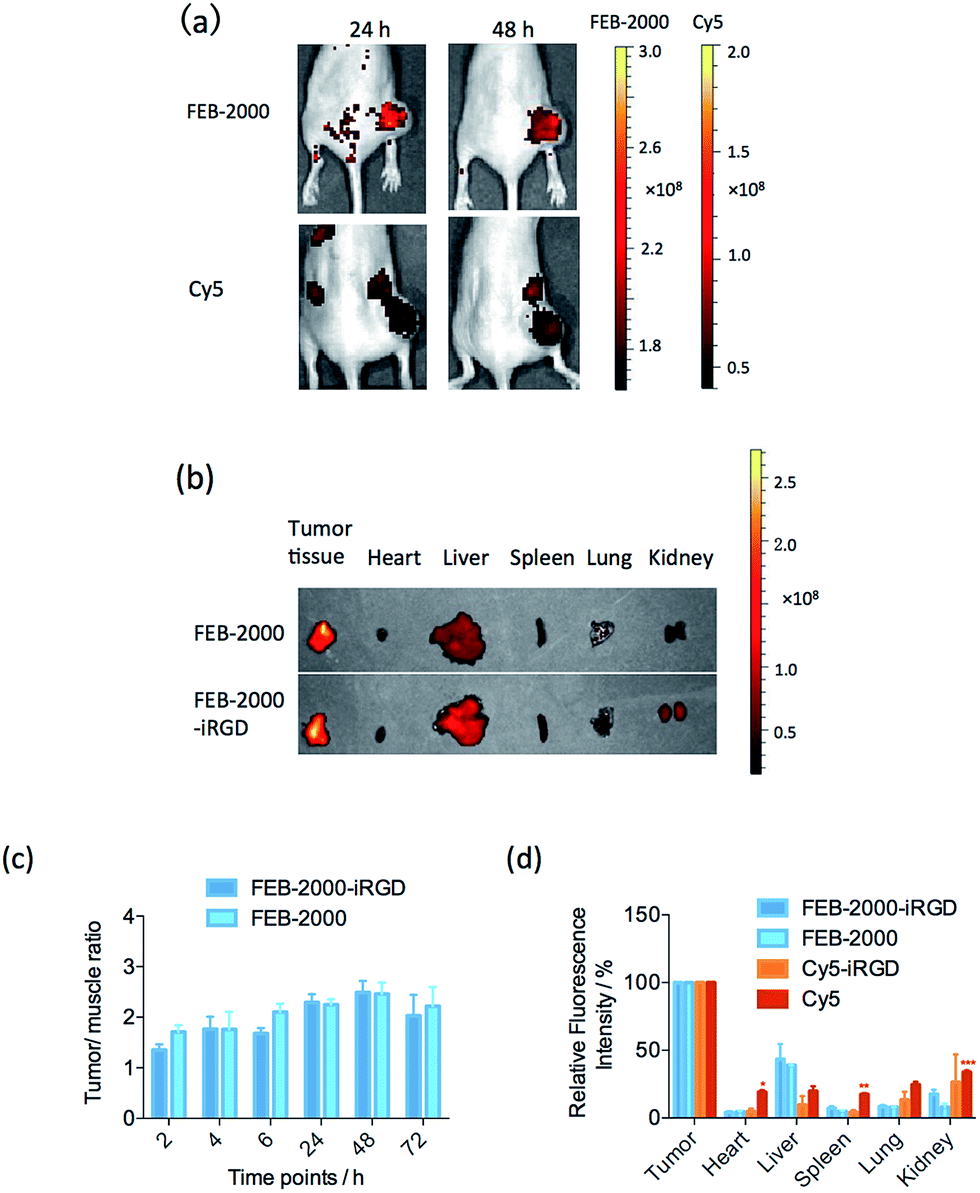 | ||
| Fig. 3 Optical imaging of MDA-MB-231 xenograft models. (a) In vivo optical tumour imaging with FEB-2000; (b) organ and tumour samples were examined at 48 h post-injection; (c) time-dependent tumour/muscle ratio of FEB-2000 and FEB-2000-iRGD (n = 5). Error bars represent the standard error of mean (SEM) from five independent experiments; (d) semi-quantitative results obtained using fluorescence imaging for ex vivo organ and tumour samples 48 h post-injection. Error bars represent the standard error of mean (SEM) from more than three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared with FEB-2000 treated mice. | ||
As previously reported, some dyes require targeting partner conjugation for efficient tumour imaging.27 We explored the influence of the iRGD peptide on tumour-targeted optical imaging of the FEB derivatives. The in vivo performance of FEB-2000-iRGD was evaluated to determine any enhanced targeting abilities. However, no statistically significant targeting enhancement was observed when comparing FEB-2000 with FEB-2000-iRGD (Fig. 3c). As a positive control, 100 μL of Cy5 (50 μM) was also injected. According to the optical imaging results, Cy5 showed a stronger background than that of FEB-2000. We also prepared Cy5-iRGD by conjugating Cy5-NHS and the iRGD peptide (confirmed by LC-MS, presented in ESI†). Except for the liver, direct imaging of dissected tissues and organs revealed that the FEB derivatives showed lower absorption in major organs when compared with either Cy5-iRGD or Cy5 (Fig. 3d and S6†). In contrast to some earlier reports, recent work has shown that co-administration of iRGD has little effect on the permeability of the chemotherapeutic agent doxorubicin (DOX).32,33 Our results also suggest that iRGD peptides have little effect on FEB tumour accumulation.
Taken together, our results indicate that PEGylated FEB alone has high tumour accumulation and long-lasting properties, while further attachment of iRGD provides no increase in FEB tumour accumulation. This indicates that FEB-2000 is a powerful dye for use in tumour imaging. Pathological data using H&E staining from heart, liver, spleen, lung, and kidney also showed its good biocompatibility properties (Fig. 4).
 | ||
| Fig. 4 Pathological data using H&E staining of different organs. No obvious toxicity was observed. 200× magnification. | ||
NIR fluorescence agents generally have low background autofluorescence, which helps give better imaging contrast.34,35 Near-infrared dyes including MH-148 and IR-783 showed a higher cell uptake and tumour/muscle ratio than the FEB derivatives.29 Therefore, future work will focus on developing high-quantum-yield NIR-II fluorophores based on the core structure of FEB.36
Conclusions
Fluorescence imaging is a useful tool for cancer diagnosis and tumour image-guided surgery. Our study introduces an easily accessible fluorescent dye, FEB, which is suitable for in vivo tumour imaging. The PEG modified FEB-2000 is water soluble and stable, showing high tumour accumulation and long-circulating properties. In vitro studies demonstrated that FEB dye derivatives penetrated MDA-MB-231 cells without conjugation of targeting peptides in addition to showing minimal cellular toxicity above the requisite imaging doses. Notably, targeting modifications showed negligible enhancement of FEB-2000, suggesting that further development of FEB could potentially lead to an efficient, small molecule tumour imaging agent.Acknowledgements
This work is supported by the Natural Science Foundation of China Grants 21372023 and 81572198; MOST 2015DFA31590, the Shenzhen Science and Technology Innovation Committee JSGG20140519105550503, JCYJ20150331100849958, JCYJ20150403101146313, JCYJ20160301111338144, JCYJ20160331115853521 and JSGG20160301095829250; the Shenzhen Peacock Program KQTD201103. Animal experiments were performed at the Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. We thank Prof. Olaf Wiest for helpful discussions.Notes and references
- A. Jemal, R. Siegel, J. Xu and E. Ward, Ca-Cancer J. Clin., 2010, 60, 277–300 CrossRef PubMed
.
- Y. Ye, L. Zhu, Y. Ma, G. Niu and X. Chen, Bioorg. Med. Chem. Lett., 2011, 21, 1146–1150 CrossRef CAS PubMed
- N. Kosaka, M. Mitsunaga, M. R. Longmire, P. L. Choyke and H. Kobayashi, Int. J. Cancer, 2011, 129, 1671–1677 CrossRef CAS PubMed
- L. Fass, Mol. Oncol., 2008, 2, 115–152 CrossRef PubMed
- M. Zhang, X. Liu, M. Yang, S. Zheng, Y. Bai and B.-Q. Yang, Tetrahedron Lett., 2015, 56, 5681–5688 CrossRef CAS
- L. Y. Niu, Y. S. Guan, Y. Z. Chen, L. Z. Wu, C. H. Tung and Q. Z. Yang, J. Am. Chem. Soc., 2012, 134, 18928–18931 CrossRef CAS PubMed
- K. E. Adams, S. Ke, S. Kwon, F. Liang, Z. Fan, Y. Lu, K. Hirschi, M. E. Mawad, M. A. Barry and E. M. Sevick-Muraca, J. Biomed. Opt., 2007, 12, 024017 CrossRef PubMed
- T. Terai and T. Nagano, Curr. Opin. Chem. Biol., 2008, 12, 515–521 CrossRef CAS PubMed
- R. Alford, H. M. Simpson, J. Duberman, G. C. Hill, M. Ogawa, C. Regino, H. Kobayashi and P. L. Choyke, Mol. Imaging, 2009, 8, 341–354 CAS
- B. A. Neto, P. H. Carvalho and J. R. Correa, Acc. Chem. Res., 2015, 48, 1560–1569 CrossRef CAS PubMed
- Y. Q. Sun, J. Liu, X. Lv, Y. Liu, Y. Zhao and W. Guo, Angew. Chem., Int. Ed. Engl., 2012, 51, 7634–7636 CrossRef CAS PubMed
- J. A. Yang, W. H. Kong, D. K. Sung, H. Kim, T. H. Kim, K. C. Lee and S. K. Hahn, Acta Biomater., 2015, 12, 174–182 CrossRef CAS PubMed
- O. A. Andreev, A. D. Dupuy, M. Segala, S. Sandugu, D. A. Serra, C. O. Chichester, D. M. Engelman and Y. K. Reshetnyak, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7893–7898 CrossRef CAS PubMed
- F. Danhier, A. L. Breton and V. Préat, Mol. Pharm., 2012, 9, 2961–2973 CrossRef CAS PubMed
- B. Ballou, G. W. Fisher, A. S. Waggoner, D. L. Farkas, J. M. Reiland, R. Jaffe, R. B. Mujumdar, S. R. Mujumdar and T. R. Hakala, Cancer Immunol. Immunother., 1995, 41, 257–263 CrossRef CAS PubMed
- C. Shi, J. B. Wu and D. Pan, J. Biomed. Opt., 2016, 21, 50901 CrossRef PubMed
- J. Gao, K. Chen, Z. Miao, G. Ren, X. Chen, S. S. Gambhir and Z. Cheng, Biomaterials, 2011, 32, 2141–2148 CrossRef CAS PubMed
- X. D. Zhang, H. Wang, A. L. Antaris, L. Li, S. Diao, R. Ma, A. Nguyen, G. Hong, Z. Ma, J. Wang, S. Zhu, J. M. Castellano, T. Wyss-Coray, Y. Liang, J. Luo and H. Dai, Adv. Mater., 2016, 28, 6872–6879 CrossRef CAS PubMed
- X. Zhang, H. Gorohmaru, M. Kadowaki, T. Kobayashi, T. Ishi-i, T. Thiemann and S. Mataka, J. Mater. Chem., 2004, 14, 1901–1904 RSC
- L. L. Li and E. W. Diau, Chem. Soc. Rev., 2013, 42, 291–304 RSC
- X. Zhang, L. Chen, X. Li, J. Mao, W. Wu, H. Ågren and J. Hua, J. Mater. Chem. C, 2014, 2, 4063 RSC
- A. L. Antaris, H. Chen, K. Cheng, Y. Sun, G. Hong, C. Qu, S. Diao, Z. Deng, X. Hu, B. Zhang, X. Zhang, O. K. Yaghi, Z. R. Alamparambil, X. Hong, Z. Cheng and H. Dai, Nat. Mater., 2015, 15, 235–242 CrossRef PubMed
- L. Cheng, W. He, H. Gong, C. Wang, Q. Chen, Z. Cheng and Z. Liu, Adv. Funct. Mater., 2013, 23, 5893–5902 CrossRef CAS
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, O. M. Girard, D. Hanahan, R. F. Mattrey and E. Ruoslahti, Cancer Cell, 2009, 16, 510–520 CrossRef CAS PubMed
- J. P. Houston, S. Ke, W. Wang, C. Li and E. M. Sevick-Muraca, J. Biomed. Opt., 2005, 10, 054010 CrossRef PubMed
- A. T. R. Williams, S. A. Winfield and J. N. Miller, Analyst, 1983, 108, 1067–1071 RSC
- J. L. Crisp, E. N. Savariar, H. L. Glasgow, L. G. Ellies, M. A. Whitney and R. Y. Tsien, Mol. Cancer Ther., 2014, 13, 1514–1525 CrossRef CAS PubMed
- X. Yang, C. Shi, R. Tong, W. Qian, H. E. Zhau, R. Wang, G. Zhu, J. Cheng, V. W. Yang and T. Cheng, Clin. Cancer Res., 2010, 16, 2833 CrossRef CAS PubMed
- X. Yang, C. Shao, R. Wang, C.-Y. Chu, P. Hu, V. Master, A. O. Osunkoya, H. L. Kim, H. E. Zhau and L. W. K. Chung, J. Urol., 2013, 189, 702–710 CrossRef CAS PubMed
- A. A. Maawy, Y. Hiroshima, G. A. Luiken, Y. Zhang, R. M. Hoffman and M. Bouvet, Cancer Res., 2014, 74, 4306 CrossRef PubMed
- K. Kanazaki, K. Sano, A. Makino, F. Yamauchi, A. Takahashi, T. Homma, M. Ono and H. Saji, J. Controlled Release, 2016, 226, 115–123 CrossRef CAS PubMed
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, D. R. Greenwald and E. Ruoslahti, Science, 2010, 328, 1031–1035 CrossRef CAS PubMed
- C. Mantis, I. Kandela and F. Aird, eLife, 2017, 6, pii: e17584 Search PubMed
- S. Luo, E. Zhang, Y. Su, T. Cheng and C. Shi, Biomaterials, 2011, 32, 7127–7138 CrossRef CAS PubMed
- J. B. Wu, C. Shi, G. C. Chu, Q. Xu, Y. Zhang, Q. Li, J. S. Yu, H. E. Zhau and L. W. Chung, Biomaterials, 2015, 67, 1–10 CrossRef CAS PubMed
- Y. Sun, C. Qu, H. Chen, M. He, C. Tang, K. Shou, S. Hong, M. Yang, Y. Jiang, B. Ding, Y. Xiao, L. Xing, X. Hong and Z. Cheng, Chem. Sci., 2016, 7, 6203–6207 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02698f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |