
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Full color palette of fluorescent D-amino acids for in situ labeling of bacterial cell walls†
Yen-Pang
Hsu‡
 a,
Jonathan
Rittichier‡§
b,
Erkin
Kuru‡§
a,
Jacob
Yablonowski
a,
Erick
Pasciak
b,
Srinivas
Tekkam¶
b,
Edward
Hall||
b,
Brennan
Murphy
b,
Timothy K.
Lee
c,
Ethan C.
Garner
d,
Kerwyn Casey
Huang
ce,
Yves V.
Brun
*f and
Michael S.
VanNieuwenhze
*ab
a,
Jonathan
Rittichier‡§
b,
Erkin
Kuru‡§
a,
Jacob
Yablonowski
a,
Erick
Pasciak
b,
Srinivas
Tekkam¶
b,
Edward
Hall||
b,
Brennan
Murphy
b,
Timothy K.
Lee
c,
Ethan C.
Garner
d,
Kerwyn Casey
Huang
ce,
Yves V.
Brun
*f and
Michael S.
VanNieuwenhze
*ab
aDepartment of Molecular and Cellular Biochemistry, Indiana University, Bloomington, IN 47405, USA. E-mail: mvannieu@indiana.edu
bDepartment of Chemistry, Indiana University, Bloomington, IN 47405, USA
cDepartment of Bioengineering, Stanford University, Stanford, CA 94305, USA
dMolecular and Cellular Biology (FAS) Center for Systems Biology, Harvard University, Cambridge, Massachusetts 02138, USA
eDepartment of Microbiology and Immunology, Stanford University School of Medicine, Stanford, CA 94305, USA
fDepartment of Biology, Indiana University, Bloomington, IN 47405, USA. E-mail: ybrun@indiana.edu
First published on 7th July 2017
Abstract
Fluorescent D-amino acids (FDAAs) enable efficient in situ labeling of peptidoglycan in diverse bacterial species. Conducted by enzymes involved in peptidoglycan biosynthesis, FDAA labeling allows specific probing of cell wall formation/remodeling activity, bacterial growth and cell morphology. Their broad application and high biocompatibility have made FDAAs an important and effective tool for studies of peptidoglycan synthesis and dynamics, which, in turn, has created a demand for the development of new FDAA probes. Here, we report the synthesis of new FDAAs, with emission wavelengths that span the entire visible spectrum. We also provide data to characterize their photochemical and physical properties, and we demonstrate their utility for visualizing peptidoglycan synthesis in Gram-negative and Gram-positive bacterial species. Finally, we show the permeability of FDAAs toward the outer-membrane of Gram-negative organisms, pinpointing the probes available for effective labeling in these species. This improved FDAA toolkit will enable numerous applications for the study of peptidoglycan biosynthesis and dynamics.
Introduction
The peptidoglycan (PG) cell wall is a macromolecular polymer of glycan strands crosslinked by short peptides that surrounds the cytoplasmic membrane of most bacteria.1,2 The cell wall serves as a rigid layer that protects bacteria from environmental stresses and dictates cell shape throughout their life cycle. Elucidating the mechanisms by which PG biosynthesis results in robust morphogenesis is a major challenge in microbiology, and inhibition of PG synthesis has proven a valuable strategy for the identification and development of antibiotics.Prior efforts directed at visualizing the sites of nascent PG growth have relied on fluorescently modified antibiotics or fluorescent lectins such as wheat germ agglutinin.3–6 We recently reported a new strategy that utilized a novel set of metabolic probes, fluorescent D-amino acids (FDAAs), for in situ PG labeling/monitoring.7–9 This approach relies on the inherent promiscuity of the PG biosynthetic pathway to incorporate small molecules conjugated to a D-amino acid backbone into the sites of new PG synthesis. This promiscuity is thought to be the result of a D-amino acid exchange reaction conducted by ubiquitous transpeptidases, penicillin-binding proteins (PBPs) and/or L,D-transpeptidases (Ldts) (Fig. 1A).10–12 The FDAA technology has demonstrated efficacy in diverse bacterial species, enabling spatiotemporal tracking of PG synthesis and modification in real-time and in live bacterial cells.13–24
 | ||
| Fig. 1 PBP-facilitated FDAA incorporation mechanism and novel FDAA structures. (A) Comparison of the transpeptidation reaction performed by penicillin-binding proteins (PBPs, top) and proposed mechanism of FDAA incorporation (bottom). The peptide-PBP intermediate (acyl donor) is attacked by the free amine of lysine in another peptide chain (acyl acceptor), which leads to cross-linking. FDAAs mimic the acyl acceptor to interact with the peptide-PBP intermediate. (B) Structures of FDAAs reported in this study. | ||
To further enhance the utility of FDAAs in a variety of labeling applications, we report the design and synthesis of a new set of FDAA probes that cover five spectrally separable colors of the optical spectrum ranging from violet to far-red (Fig. 1B). Additionally, we provide photochemical, physical, and biological characterizations that will serve as a valuable resource when choosing the optimal FDAA for a labeling experiment of interest.
Results and discussion
Design and synthesis of improved FDAAs
A new FDAA should follow the basic design principles for biomolecular fluorescent probes: a “carrier” moiety to specifically bind to the target of interest (i.e., a D-amino acid backbone), a fluorophore that emits light upon excitation, and a linker to join the two together.25 Parameters defining an ideal fluorescent probe can be largely separated into two categories: (1) photochemical properties that determine the performance of probes in microscopy, such as excitation/emission spectra and brightness, and (2) physical properties that are related to molecular interactions between probes, their targets, and their surroundings, such as solubility and stability. Optimal labeling with probes such as FDAAs relies on these properties, as well as appropriate labeling and imaging conditions required for a specific use/application.FDAAs are biocompatible, and their labeling provides a fluorescent readout of PG synthesis and remodeling activity.8 Combining FDAAs with protein labels and/or fluorescent antibiotics and with super-resolution imaging technologies provides a powerful experimental approach to elucidate the spatiotemporal regulation necessary for robust bacterial growth and division.14,24 Sequential pulses with differently colored FDAAs enable sub-cellular tracking of PG growth via “virtual time-lapse” microscopy that reveals the history of PG synthesis and remodeling sites during an experiment as reported by the emission and detection of each color.9,14 Given the demonstrated utility of FDAA probes, we sought to expand the available FDAA color palette with probes possessing improved photochemical and/or physical properties, thereby further enhancing the utility of this valuable class of probes in various experimental applications.
The general synthetic strategy for FDAAs is composed of three straightforward steps (Fig. 2A): (1) coupling of an Nα-protected-D-amino acid to an activated fluorophore, purchased in its pre-activated form or synthesized in-house (Table 1), (2) removal of the Nα protecting group, and (3) purification of the FDAA. The coupling reaction is usually carried out with an activated fluorophore and a D-amino acid with Boc/Fmoc-protected α-amine under basic conditions and at ambient temperature. Boc-deprotection is achieved by dissolving the coupling product in a mixture of dichloromethane (DCM) and trifluoroacetic acid (TFA) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and stirring at ambient temperature for approximately 2 h. An Fmoc-protecting group can be removed by suspension of the coupling product in DCM followed by treatment with 1,8-diazabicycl0(5.4.0)undec-7-ene (DBU). The crude FDAA product(s) can be purified using reverse-phase high-performance liquid chromatography (HPLC, 1:1 acetonitrile/water).
:1 ratio and stirring at ambient temperature for approximately 2 h. An Fmoc-protecting group can be removed by suspension of the coupling product in DCM followed by treatment with 1,8-diazabicycl0(5.4.0)undec-7-ene (DBU). The crude FDAA product(s) can be purified using reverse-phase high-performance liquid chromatography (HPLC, 1:1 acetonitrile/water).
 | ||
| Fig. 2 Synthesis scheme and spectra of FDAAs. (A) FDAA synthesis is achieved by coupling conjugation-activated fluorophores with 3-amino-D-alanine (3-aminopropionic acid) or D-lysine. (B and C) Excitation (B) and emission (C) spectra of FDAAs in phosphate-buffer saline (1× PBS) at pH 7.4. Absorbance and fluorescence intensity were normalized to their maximum values. | ||
| a Values for unsalted FDAAs. The molecular weight of conjugated salt was not included. b FDAA spectra were measured in PBS buffer at 1× pH 7.4 (containing 0.1% DMSO). c Extinction coefficient (ε) or quantum yield (Φ) of fluorophores used for FDAA synthesis. Values are from the dye manufacturer, except as noted from ref. 47. NA: not available. d Synthesis of Atto610ADA using commercially available Atto610 NHS ester resulted in a less stable product (data not shown). We modified the structure of the linker moiety, producing stable Atto610ADA. Please note that the commercially available Atto610 NHS ester contains a butyric linker. See ESI for detailed synthesis scheme. |
|---|
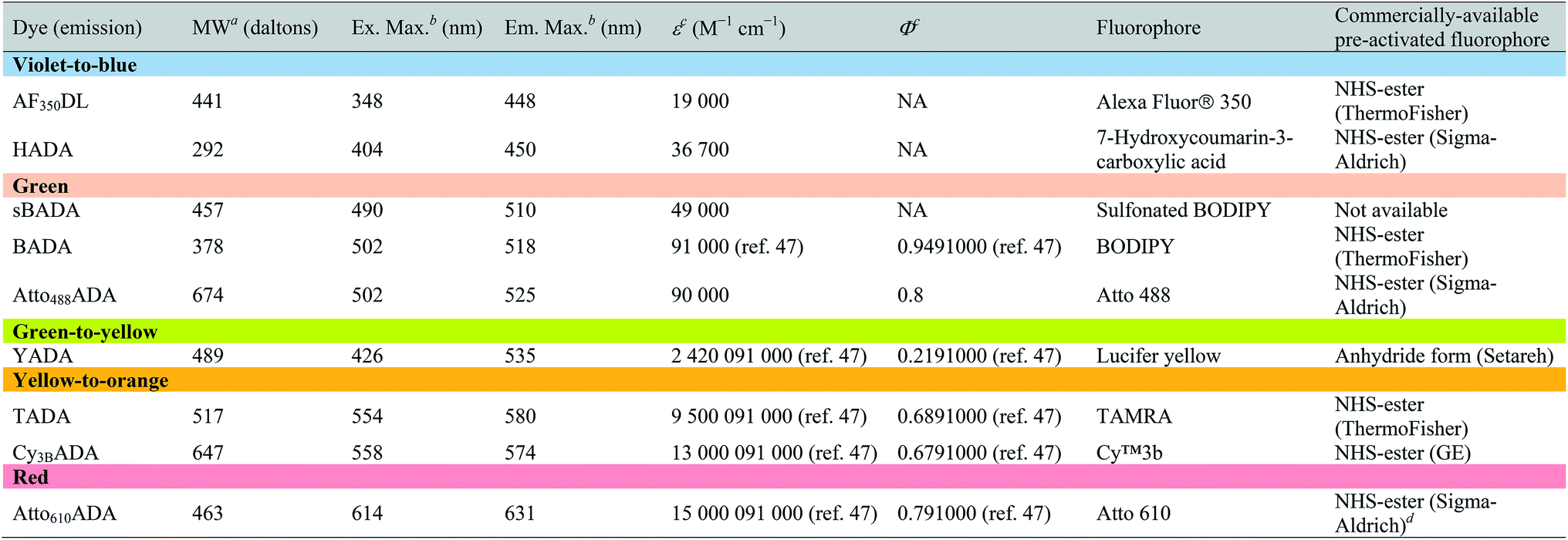
|
In an effort to develop a probe to expand FDAAs into the violet-to-blue region of the visible spectrum, we first attempted to couple an activated Alexa Flour 350 dye with 3-Amino-D-alanine. The chemical instability of the product obtained after the deprotection reaction (data not shown) prompted us to use D-lysine instead, which resulted in the stable AF350DL (Alexa Flour® 350 D-Lysine) (Table 2 and Fig. 2B). For green-emission probes, in addition to previously reported BADA (BODIPY-FL 3-amino-D-alanine), we further prepared sBADA (sulfonated BODIPY-FL 3-amino-D-alanine) and Atto488ADA (Atto 488 3-amino-D-alanine) from sulfonated BODIPY-FL and Atto 488 dye, respectively.26,27 For a new green-to-yellow emitting probe that would be spectrally separable from green FDAAs, we used Lucifer yellow to construct YADA (Lucifer Yellow 3-amino-D-alanine). For yellow-to-orange emission probe, in addition to TADA/TDL (TAMRA 3-amino-D-alanine/D-Lysine),15 we prepared Cy3BADA (Cy3B 3-amino-D-alanine) from Cy™3B. Finally, we synthesized a red-to-far-red FDAA, Atto610ADA (Atto 610 3-amino-D-alanine), based on the reported Atto 610 dye.
|
a Data were measured in PBS buffer at pH 7.4 (containing 0.1% DMSO).
b FDAAs were assigned “+++”, “++”, or “+” if the measured distribution coefficient logD7.4 was <−1, between −1 and 0, or >0, respectively.
c FDAAs were assigned “+++”, “++”, or “+” if the measured exponential decay coefficient was <0.1, 0.1–1, or >1, respectively.
d The data represent signal retention of absorbance of dye solution incubated at 37 °C for 5 min, 2 h, or 24 h, compared to the corresponding initial value.
|
|---|

|
Photochemical properties and labeling with new FDAAs
In fluorescence microscopy, the separation of excitation and emission wavelengths of a probe is achieved by selecting filters to block or pass wavelengths specific for that probe. Under physiological conditions (in 1× phosphate-buffer saline, or PBS, at pH 7.4), AF350DL (maximum λex/em = 348/448 nm) is ideally excited by ultraviolet light (∼350 nm). However, AF350DL can be partially excited by a visible violet light source (395/25 nm) and imaged using a typical blue filter set (435/26 nm, Tables 2, S3 and Fig. S1†). Thus, AF350DL can be used as an alternative of HADA (maximum λex/em = 400/450 nm), a previously published blue FDAA. BADA (maximum λex/em = 502/518 nm), sBADA (maximum λex/em = 490/510 nm) and Atto488ADA (maximum λex/em = 502/525 nm) can be visualized using a blue light source (470/24 nm) and a green emission filter (510/40 nm, Table 2). By comparison, YADA (maximum λex/em = 426/535 nm) has a shorter excitation wavelength but a red-shifted and wider emission spectrum. The large Stokes shift and wider excitation/emission spectra of YADA enable it to be distinguished from other blue-, green-, and red-emitting FDAAs when appropriate microscopy settings are used. For example, YADA can be excited by a violet light source and detected by a green filter (or even by a red filter if necessary). Similar to the published yellow-to-orange-emitting TADA (maximum λex/em = 554/580 nm), Cy3BADA (maximum λex/em = 558/574 nm) can be excited by green light (550/15 nm) and visualized by a red filter (595/40 nm, Table 2). Finally, Atto610ADA (maximum λex/em = 614/631 nm) can be excited by a red light source (640/30 nm) and detected through use of a far-red filter (705/72 nm). The extinction coefficient (ε) and quantum yield (Φ) of fluorophores used for FDAA synthesis are shown in Table 1. Since no significant changes in the excitation and emission spectra were observed after these fluorophores were coupled to D-amino acids, we expect the corresponding FDAAs to retain similar extinction coefficients and quantum yields.Two commonly utilized FDAA labeling strategies differ from each other in their respective labeling times. Long-pulse labeling experiments usually result in labeling of the whole cell, while short-pulse labeling enables visualization of the sites of transpeptidation activity during the pulse period. We confirmed the incorporation of our new FDAAs into PG by growing cells of the Gram-negative Escherichia coli and Gram-positive Bacillus subtilis, two evolutionarily distant model organisms, in the presence of these molecules for several generations (Fig. S2†). No toxic effects were observed. In addition, pre-fixed cells were not labeled, suggestive of metabolic incorporation of these FDAAs (Fig. S3†). TADA and Atto610ADA labeling in pre-fixed E. coli showed non-specific signal inside the cell (Fig. S3†). This might result from the trapped probes inside the cells after fixation which changes outer-membrane permeability toward FDAAs. Indeed, we also observed increased background intensity of FDAAs upon ethanol fixation in other Gram-negative species, such as C. crescentus (data not shown). Thus, control experiments using live cells were highly recommended if one needs to do cell fixation. Labeling using the L-isomer of TADA (TALA) showed no signal in live E. coli cells (data not shown), confirming the metabolic incorporation of the probe.
The addition of more distinct colors allows for increased spatiotemporal resolution of a virtual time-lapse labeling experiment, as seen for labeling of the polar-growing and branching bacterium Streptomyces venezuelae with four FDAAs (Fig. 3A). S. venezuelae cells were successively pulsed with Atto488ADA (3 h), Cy3BADA (15 min), AF350DL (15 min), and Atto610ADA (15 min). In this experiment, while the long-pulse labeling with Atto488ADA is necessary to delineate the pattern of the oldest PG at the start of the experiment, the short pulses with three distinct FDAAs resolved the polar, newer PG synthesis activity (Fig. 3A, white arrows) over a 45 min interval with a temporal resolution of 15 min per color.9,14 With appropriate adjustment to labeling and microscopy conditions, our expanded FDAA toolkit now enables virtual time-lapse labeling with up to five colors, in which YADA can be distinguished from blue- and green-emission probes (Fig. 3B).
 | ||
| Fig. 3 Virtual time-lapse FDAA labeling. (A) Streptomyces venezuelae cells were sequentially labeled with Atto488ADA (3 h), Cy3BADA (15 min), AF350DL (15 min), and Atto610ADA (15 min), and then fixed and imaged. Left: Phase-contrast; right: fluorescence. Scale bar: 5 μm. White arrows point out new branches formed at different time points (oldest to newest: (a) to (c)). (B) Lactococcus lactis cells were sequentially labeled with Atto610ADA (8 min), HADA (5 min), YADA (8 min), sBADA (5 min), and TADA (5 min), and then fixed and imaged. Left to right: Phase channel, Atto610ADA, HADA, YADA, sBADA, TADA, and merged image. Scale bar: 1 μm. | ||
Improved physical properties of the new FDAAs
FDAAs decorate PG with high specificity that is endowed by the unique selectivity of the PG biosynthetic pathways for D-amino acids over their L-isomers. However, FDAAs with low water solubility might result in non-specific staining of the cell membrane. We determined the water solubility of all FDAAs in order to evaluate their potential for non-specific labeling. Specifically, with a given total FDAA concentration, we performed extractions of FDAAs in 1× PBS (pH 7.4) and 1-octanol and then measured the amount of FDAA distributed in each solvent layer. These measurements were used to calculate the distribution coefficient, logD7.4, where a smaller value represents greater hydrophilicity.28,29 Our previous experience with various probes suggest that FDAAs with logD7.4 values higher than 1 show a greater propensity for non-specific labeling. Each of the novel FDAAs reported here had logD7.4 values less than 1 (Tables 2 and S1†). Labeling experiments in pre-fixed B. subtilis showed no significant signal, confirming that non-specific membrane labeling by these FDAAs is minimal (Fig. S3†). The degree of non-specific labeling could also depend on the species-specific composition of membranes and experimental conditions. Of all the FDAAs, BADA has the lowest relative water solubility, which might compromise its overall utility due to non-specific labeling. This observation was the underlying motivation for preparation of the sulfonated form of BADA (sBADA), which displays significantly increased hydrophilicity. High hydrophilicity also simplifies the washing steps for cell labeling with aqueous buffers, which are required to minimize background fluorescence, and allows for use of higher FDAA concentrations in aqueous solutions (e.g. growth medium), which is known to enhance the signal-to-background ratio favoring the incorporation reaction.9
Depending on the bacterial species and the goals of the labeling experiment, the FDAA labeling duration and sample processing could range from minutes to days. Thus, in most cases, high FDAA stability during labeling at physiological conditions is desired, especially for quantitative fluorescence experiments. We examined thermal stability of the FDAAs by measuring the percentage of signal retention after an incubation in PBS (pH 7.4) at 37 °C for 5 min, 2 h, or 24 h. No significant signal reduction was observed for all the FDAAs in short incubation times between 5 min and 2 h (Table 2). After 24 h of incubation, we observed a minimal signal decrease of <10% for most of the FDAAs, suggesting that these probes exhibit high thermal stability for at least 1 day under our experimental conditions. However, Atto610ADA showed a significant signal reduction after 24 h of incubation, with 50% signal loss after 10 h of incubation in PBS (Fig. S4†). Thus, careful optimization and control experiments are required when using Atto610ADA for long-pulse labeling and quantification, especially if the experiment exceeds ∼10 h at 37 °C.
Because FDAAs are non-toxic, they can be used for time-lapse microscopy. In a typical time-lapse experiment, the high photostability of a fluorescent probe is important as the probe gets regularly exposed to the excitation light.30–32 To evaluate the photostability of FDAAs, we measured relative decay curves of fluorescence emission intensity for each FDAAs at their emission maximum (λem) with continuous excitation at their corresponding excitation maximum (λex) in B. subtilis samples labeled under long-pulse conditions. Fitting to an exponential curve enabled determination of the exponential decay coefficient (EDC) as a metric of FDAA photostability, where a smaller EDC represents greater photostability (Tables 2 and S1†). Of all the FDAAs, HADA is the most sensitive to prolonged light exposure (Fig. S5†). It has a relatively high EDC value of 1.186 and a fluorescence half-life of 0.5846 s under our experimental conditions. In contrast, the other blue-to-violet FDAA, AF350DL, has an EDC of 0.0497 and a fluorescence half-life of 13.950 s under the same conditions. Since HADA has also shown utility for acquiring three-dimensional structured illumination microscopy (3D-SIM) images,8,14 which require prolonged excitation from a powerful laser (Fig. S6†), we predict all the FDAAs reported here will have sufficient photostability for most uses and applications. Furthermore, excitation-based phototoxicity could be a major issue for cell viability, especially in time-lapse microscopy experiments. Use of red-shifted TADA, Cy3BADA, and Atto610ADA for such experiments addresses this problem, because cells are tolerant to low-energy excitation light.31,33 Therefore, we conclude that the new FDAAs reported here have improved photophysical properties for use in imaging applications that require prolonged excitation.
Outer membrane permeability of FDAAs
The outer membrane (OM) of Gram-negative bacteria is an effective barrier that protects cells from toxins in the environment. Many antibiotics show low toxicity to Gram-negative species because the OM provides a permeability barrier that inhibits access to the periplasm and cytoplasm.34 The OM also blocks the entry of larger fluorescent probes, resulting in lower labeling efficiency. Gram-negative cells import water-soluble materials through the OM via porins, a class of membrane proteins that serve as transport channels. Transport is limited by the size of porin channels as well as of the cargo: a molecular weight (MW) of ∼600 g mol−1 was reported as the cargo cutoff size.35,36 The MW of FDAAs reported here ranges from ∼300 to ∼700 g mol−1. To investigate the OM permeability of the FDAAs in a model Gram-negative species, we labeled wild type E. coli BW25113 with FDAAs and measured the signal-to-background ratio (S/B) of the signal. HADA and YADA showed high labeling efficiency with S/B values >4 (Table 3). BADA, sBADA, and Atto610ADA had S/B values between 2 and 4. AF350DL, Atto488ADA, TADA, and Cy3BADA had relatively low labeling efficiency in WT E. coli, with S/B values <2.| a FDAAs were assigned “+++”, ”++”, or “+” if the signal-to-background (S/B) ratio of E. coli BW25113 (WT) images was >4, 2–4, or <2, respectively. b Data represent the fluorescence intensity ratio of E. coli imp4213 BW25113 to E. coli BW25113 under the same labeling conditions and microscopy settings. |
|---|

|
These S/B values are relative and are dependent upon species/strain, labeling conditions, auto-fluorescence of cells, and microscopy settings. To quantify the barrier effect of the OM on each of these FDAAs in E. coli, we compared FDAA intensity between wild-type E. coli BW25113 and E. coli imp4213 BW25113, a mutant with increased outer membrane permeability.37 Under the same labeling and imaging conditions, FDAA labeling S/B values increased significantly in E. coli imp4213 relative to the parental strain (Fig. S7A†). We represented the ratio of FDAA intensity in E. coli imp4213 over wild-type as the “OM blocking factor” (Table 3). Consistent with displaying lower S/B values in wild-type E. coli relative to the other FDAAs, sBADA, TADA, and Atto488ADA revealed a high blocking factor of ∼20, indicating that the OM is a strong permeability barrier for each of these FDAAs. On the other hand, HADA and YADA had low blocking factors, suggesting that the OM is highly permeable to these FDAAs.
Despite the poor OM permeability to some of the FDAAs, optimization of microscope settings can effectively improve S/B of PG labeling to acceptable levels (Fig. S7B†). Moreover, the use of higher FDAA concentrations and/or minimal medium in cell labeling increases FDAA labeling efficiency. In Fig. S7C,† we show TADA and BADA labeling with different FDAA concentrations and culture medium in wild type E. coli BW25113. We observed a significant increase in S/B with the use of high FDAA concentrations (3 or 2 mM) compared a lower one (1 mM). An increase was also found when using M9 minimal medium compared to LB. However, additional optimization and control experiments for toxicity effects are strongly recommended when changing experimental conditions.
FDAAs for STORM
Currently, stochastic optical reconstruction microscopy (STORM) and related technologies offer the highest, sub-diffraction-limit, spatial resolution achievable with fluorescence microscopy.38 Atto 488 and Cy3B fluorophores have been reported to have outstanding applicability for direct STORM (dSTORM) and reductive caging STORM (rcSTORM), respectively.39,40 Therefore, Atto488ADA and Cy3BADA should enable STORM for cell-wall imaging. Indeed, we conducted rcSTORM visualization of E. coli cell wall using Cy3BADA and the outer-membrane permeable imp strain (Fig. 4A). In addition, we note that TADA/TDL also has the potential to be visualized via dSTORM41 and rcSTORM (Fig. 4B). Further optimization is highly recommended to acquire reliable reconstruction in STORM. | ||
| Fig. 4 Stochastic optical reconstruction microscopy (STORM) of E. coli imp4213 Δ6 BW25113 cells lacking all L,D-transpeptidases. Δ6 cells were labeled with Cy3BADA (A) and TDL (B). Left: Epifluorescence microscopy; right panel: STORM (right). E. coli imp4213 Δ6 cells were treated with cephalexin for 2 h, washed, and then pulsed with 1 mM Cy3BADA/TDL for 2 min. Insets in (A) and arrow head highlight a division septum. Scale bar: 2 μm. | ||
Conclusion and remarks
The synthesis and characterization of the improved FDAA probes described here should be useful when choosing an optimal probe for PG labeling. The new colors yellow (YADA) and red (Atto610ADA) that can be spectrally separated from the typical blue (e.g. HADA), green (e.g. BADA), and orange-to-red (e.g. TADA) FDAAs will add versatility to the toolset, especially when pairing multiple FDAAs with fluorescent protein fusions. The increased water solubility of the new FDAAs compared to older FDAAs (e.g. sBADA vs. BADA) will improve S/B ratios of PG labeling in several ways: (1) by allowing pulses with higher FDAA concentrations; (2) by simplifying washes, and (3) by decreasing the non-specific binding to membranes. Similarly, FDAAs with improved photostability (e.g. AF350DL vs. HADA) or with STORM capabilities (e.g. Cy3BADA, Atto488ADA and TADA) will facilitate PG imaging with increased spatial resolution when combined with super-resolution microscopy techniques.14Phototoxicity is a major issue when frequent light exposure is required in an experiment such as time-lapse microscopy. Here, we also introduce Atto610ADA, a small, photostable FDAA that is even more red-shifted than the orange-to-red FDAA, TADA. The outer membrane permeability of Atto610ADA will allow tracking of PG remodeling in Gram-negative bacteria via time-lapse microscopy experiments in far-red region. Finally, our effort to determine the FDAA size dependency for OM permeability (OM blocking factor) not only confirms that FDAAs larger than ∼500 Da (TADA and Cy3BADA) are excluded by the OM, but also reveals that other factors might be equally important as size for determining permeability. Specifically, although sBADA (MW: 457 Da) is smaller than the similarly sulfonated FDAA YADA (MW: 489 Da), sBADA is approximately 10 times less permeable than YADA. This discrepancy could result from alternative OM transport mechanisms such as lipid-mediated diffusion.42,43
The modular and simple design of our fluorescent amino acids allows convenient conjugation to a variety of bright and common commercially available organic probes. Such fluorescent amino acids have a variety of potential uses. Fluorescent L-amino acids have found use in peptide synthesis.44 More importantly, encoding brighter, red-shifted, yet still relatively small fluorescent amino acids (such as the L-enantiomer of Atto610ADA), into proteins could be possible with the availability of new technologies, which in turn would address current issues with utilizing fluorescent protein fusions such as size and maturation.45 As we have demonstrated, fluorescent D-amino acids serve as ideal PG probes to elucidate bacterial cell growth and division in high spatiotemporal resolution,14,24 or to simply label bacterial cell surfaces for a variety of other applications in diverse bacteria.13,16–19,21,22,46 We provide here an improved toolset of FDAAs to study in situ peptidoglycan synthesis for various purposes that range from identification of modes of cell growth, to peptidoglycan metabolism and turnover, to peptidoglycan–enzyme interactions.
Methods
See compiled ESI.†Acknowledgements
We thank Josh Vaughan for help on STORM imaging; Xin Meng for help with FDAA synthesis; and David Kysela for help on image processing and analysis. This work was supported by NIH grants 5R01GM113172 to MSV and YVB; R35GM122556 to YVB; a NSF CAREER Award MCB-1149423 to KCH; and Stanford Center for Systems Biology under Grant P50-GM107615 to KCH. SIM was performed in the Indiana University Light Microscopy Imaging Center supported by S10RR028697-01.References
- A. Typas, M. Banzhaf, C. A. Gross and W. Vollmer, Nat. Rev. Microbiol., 2011, 10, 123–136 Search PubMed.
- W. Vollmer, D. Blanot and M. A. de Pedro, FEMS Microbiol. Rev., 2008, 32, 149–167 CrossRef CAS PubMed.
- R. A. Daniel and J. Errington, Cell, 2003, 113, 767–776 CrossRef CAS PubMed.
- K. Tiyanont, T. Doan, M. B. Lazarus, X. Fang, D. Z. Rudner and S. Walker, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11033–11038 CrossRef CAS PubMed.
- T. S. Ursell, J. Nguyen, R. D. Monds, A. Colavin, G. Billings, N. Ouzounov, Z. Gitai, J. W. Shaevitz and K. C. Huang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1025–E1034 CrossRef CAS PubMed.
- O. Kocaoglu, R. A. Calvo, L.-T. Sham, L. M. Cozy, B. R. Lanning, S. Francis, M. E. Winkler, D. B. Kearns and E. E. Carlson, ACS Chem. Biol., 2012, 7, 1746–1753 CrossRef CAS PubMed.
- Y. P. Hsu, X. Meng and M. S. VanNieuwenhze, in Methods in Microbiology, ed. H. Colin and J. J. Grant, Academic Press, 2016, vol. 43, pp. 3–48 Search PubMed.
- E. Kuru, H. V. Hughes, P. J. Brown, E. Hall, S. Tekkam, F. Cava, M. A. de Pedro, Y. V. Brun and M. S. VanNieuwenhze, Angew. Chem., Int. Ed., 2012, 51, 12519–12523 CrossRef CAS PubMed.
- E. Kuru, S. Tekkam, E. Hall, Y. V. Brun and M. S. Van Nieuwenhze, Nat. Protoc., 2015, 10, 33–52 CrossRef CAS PubMed.
- T. J. Lupoli, H. Tsukamoto, E. H. Doud, T.-S. A. Wang, S. Walker and D. Kahne, J. Am. Chem. Soc., 2011, 133, 10748–10751 CrossRef CAS PubMed.
- Y. Qiao, M. D. Lebar, K. Schirner, K. Schaefer, H. Tsukamoto, D. Kahne and S. Walker, J. Am. Chem. Soc., 2014, 136, 14678–14681 CrossRef CAS PubMed.
- F. Cava, M. A. de Pedro, H. Lam, B. M. Davis and M. K. Waldor, EMBO J., 2011, 30, 3442–3453 CrossRef CAS PubMed.
- T. M. Bartlett, B. P. Bratton, A. Duvshani, A. Miguel, Y. Sheng, N. R. Martin, J. P. Nguyen, A. Persat, S. M. Desmarais, M. S. VanNieuwenhze, K. C. Huang, J. Zhu, J. W. Shaevitz and Z. Gitai, Cell, 2017, 168, 172–185 CrossRef CAS PubMed.
- A. W. Bisson Filho, Y.-P. Hsu, G. Squyres, E. Kuru, F. Wu, C. Jukes, Y. Sun, C. Dekker, S. Holden, M. VanNieuwenhze, Y. Brun and E. Garner, Science, 2017, 355, 739–743 CrossRef CAS PubMed.
- M. J. Boersma, E. Kuru, J. T. Rittichier, M. S. VanNieuwenhze, Y. V. Brun and M. E. Winkler, J. Bacteriol., 2015, 197, 3472–3485 CrossRef CAS PubMed.
- E. Cserti, S. Rosskopf, Y. W. Chang, S. Eisheuer, L. Selter, J. Shi, C. Regh, U. Koert, G. J. Jensen and M. Thanbichler, Mol. Microbiol., 2017, 103, 875–895 CrossRef CAS PubMed.
- A. K. Fenton, L. E. Mortaji, D. T. Lau, D. Z. Rudner and T. G. Bernhardt, Nat. Microbiol., 2016, 2, 16237 CrossRef PubMed.
- A. Fleurie, C. Lesterlin, S. Manuse, C. Zhao, C. Cluzel, J. P. Lavergne, M. Franz-Wachtel, B. Macek, C. Combet, E. Kuru, M. S. VanNieuwenhze, Y. V. Brun, D. Sherratt and C. Grangeasse, Nature, 2014, 516, 259–262 CrossRef CAS PubMed.
- M. D. Lebar, J. M. May, A. J. Meeske, S. A. Leiman, T. J. Lupoli, H. Tsukamoto, R. Losick, D. Z. Rudner, S. Walker and D. Kahne, J. Am. Chem. Soc., 2014, 136, 10874–10877 CrossRef CAS PubMed.
- G. Liechti, E. Kuru, M. Packiam, Y.-P. Hsu, S. Tekkam, E. Hall, J. T. Rittichier, M. VanNieuwenhze, Y. V. Brun and A. T. Maurelli, PLoS Pathog., 2016, 12, e1005590 Search PubMed.
- J. M. Monteiro, P. B. Fernandes, F. Vaz, A. R. Pereira, A. C. Tavares, M. T. Ferreira, P. M. Pereira, H. Veiga, E. Kuru, M. S. VanNieuwenhze, Y. V. Brun, S. R. Filipe and M. G. Pinho, Nat. Commun., 2015, 6, 8055 CrossRef CAS PubMed.
- D. Morales Angeles, Y. Liu, A. M. Hartman, M. Borisova, A. de Sousa Borges, N. de Kok, K. Beilharz, J. W. Veening, C. Mayer, A. K. Hirsch and D. J. Scheffers, Mol. Microbiol., 2017, 104, 319–333 CrossRef CAS PubMed.
- M. Pilhofer, K. Aistleitner, J. Biboy, J. Gray, E. Kuru, E. Hall, Y. V. Brun, M. S. VanNieuwenhze, W. Vollmer, M. Horn and G. J. Jensen, Nat. Commun., 2013, 4, 2856 Search PubMed.
- X. Yang, Z. Lyu, A. Miguel, R. McQuillen, K. K. C. Huang and J. Xiao, Science, 2017, 355, 744–747 CrossRef CAS PubMed.
- J. Zhou and H. Ma, Chem. Sci., 2016, 7, 6309–6315 RSC.
- L. Li, J. Han, B. Nguyen and K. Burgess, J. Org. Chem., 2008, 73, 1963–1970 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS.
- J. Sangster, J. Phys. Chem. Ref. Data, 1989, 18, 1111–1226 CrossRef CAS.
- R. A. Scherrer and S. M. Howard, J. Med. Chem., 1977, 20, 53–58 CrossRef CAS PubMed.
- T. Bernas, M. Zarebski, J. W. Dobrucki and P. R. Cook, J. Microsc., 2004, 215, 281–296 CrossRef CAS PubMed.
- R. A. Hoebe, C. H. Van Oven, T. W. J. Gadella, P. B. Dhonukshe, C. J. F. Van Noorden and E. M. M. Manders, Nat. Biotechnol., 2007, 25, 249–253 CrossRef CAS.
- L. Song, R. P. van Gijlswijk, I. T. Young and H. J. Tanke, Cytometry, 1997, 27, 213–223 CrossRef CAS PubMed.
- R. Dixit and R. Cyr, Plant J. Cell Mol. Biol., 2003, 36, 280–290 CrossRef CAS.
- A. H. Delcour, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 808–816 CrossRef CAS PubMed.
- H. Nikaido and M. Vaara, Microbiol. Rev., 1985, 49, 1–32 CAS.
- F. Yoshimura and H. Nikaido, Antimicrob. Agents Chemother., 1985, 27, 84–92 CrossRef CAS PubMed.
- B. A. Sampson, R. Misra and S. A. Benson, Genetics, 1989, 122, 491–501 CAS.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–796 CrossRef CAS PubMed.
- G. T. Dempsey, J. C. Vaughan, K. H. Chen, M. Bates and X. Zhuang, Nat. Methods, 2011, 8, 1027–1036 CrossRef CAS.
- J. C. Vaughan, S. Jia and X. Zhuang, Nat. Methods, 2012, 9, 1181–1184 CrossRef CAS PubMed.
- T. Klein, A. Loschberger, S. Proppert, S. Wolter, S. van de Linde and M. Sauer, Nat. Methods, 2011, 8, 7–9 CrossRef CAS PubMed.
- B. Alberts, A. Johnson and J. Lewis, in Molecular Biology of the Cell, Garland Science, New York, 4th edn, 2002 Search PubMed.
- G. Camenisch, J. Alsenz, H. van de Waterbeemd and G. Folkers, Eur. J. Pharm. Sci., 1998, 6, 313–319 CrossRef CAS.
- A. H. Harkiss and A. Sutherland, Org. Biomol. Chem., 2016, 14, 8911–8921 CAS.
- J. Luo, R. Uprety, Y. Naro, C. Chou, D. P. Nguyen, J. W. Chin and A. Deiters, J. Am. Chem. Soc., 2014, 136, 15551–15558 CrossRef CAS PubMed.
- J. M. Fura, D. Kearns and M. M. Pires, J. Biol. Chem., 2015, 290, 30540–30550 CrossRef CAS PubMed.
- G. McNamara, McNamara 2007 Fluorophore Data Tables, Online Source, 2007 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc01800b |
| ‡ These authors contributed equally. |
| § Current address: Department of Genetics, Harvard Medical School, Boston, MA 021157, USA. |
| ¶ Current address: School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA. |
| || Current address: Department of Chemistry, Hanover College, Hanover, IN 47243, USA. |
| This journal is © The Royal Society of Chemistry 2017 |