
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoredox mediated nickel catalyzed C(sp3)–H thiocarbonylation of ethers†
Byungjoon
Kang
and
Soon Hyeok
Hong
 *
*
Department of Chemistry, College of Natural Sciences, Seoul National University, 1 Gwanak-ro, Seoul 08826, South Korea. E-mail: soonhong@snu.ac.kr
First published on 24th July 2017
Abstract
The first direct C(sp3)–H thiocarbonylation reaction is achieved by visible light photoredox/Ni dual catalysis. The thioester group of thiobenzoate is transferred to the α-oxy carbon of various cyclic/acyclic ethers, which is the opposite to the commonly expected chemical reactivity involving acyl group transfer via the weaker C(acyl)–S activation. Through mechanistic studies, we proposed that the reaction is initiated by photocatalytic reduction and fragmentation of the thioester into an acyl radical and a thiolate. A nickel complex binds to the thiolate and induces the decarbonylation of the acyl radical to form an aryl radical, which abstracts hydrogen from the α-oxy carbon of the ether. The resulting α-oxy C(sp3) centered radical re-binds to the (RS)(CO)Ni complex, which undergoes CO migratory insertion and reductive elimination to give the desired thioester product.
Introduction
Thioesters are versatile synthetic building blocks1 and convenient protecting groups for thiols in organic synthesis.2 As an activated analogue of an alcohol-derived ester, it can be easily transformed into an ester, amide or ketone. In Nature, the thioester group plays a critical role in metabolism3 and cellular function regulation.4 The thioester moiety also plays a central role in Native Chemical Ligation (NCL)5 and Expressed Protein Ligation (EPL)6 in the biological sciences.Due to its versatility, the direct and selective incorporation of a thioester group into the desired position of a molecule has high synthetic value. The most common method for thioester synthesis is a reaction between a thiol and an activated carboxylic acid, or the coupling of a thioacid and an electrophile (Scheme 1A).7 However, the requirements of high oxidation state carbon-based reactants, harsh reaction conditions, and the sensitivity of thiols towards oxidation, limit the utility of the reactions. Several alternative synthetic methods have been developed including the oxidative thioesterification of aldehydes8 and the thiocarbonylation of alkenes9 or organic halides10 with carbon monoxide gas. Most of these methodologies still require a thiol and a suitable coupling partner as the reactants, sharing similar limitations with classical syntheses.
 | ||
| Scheme 1 Thioester synthesis. | ||
Direct C–H thiocarbonylation can serve as an ideal approach for thioester synthesis. Indeed, Nature utilizes an inspiring C–H functionalization strategy for the synthesis of metabolically important thioesters. Methylmalonyl-CoA mutase (MCM) and coenzyme B12 catalyse the L-methylmalonyl-CoA to succinyl-CoA isomerization by generating a radical on an sp3 hybridized carbon followed by the intramolecular 1,2-migration of the thioester group (Scheme 1A).11 However, to the best of our knowledge, a direct thioester group transfer reaction to a C–H bond has never been achieved in organic synthesis. To address the challenge, we envisioned an unprecedented C(sp3)–H thiocarbonylation utilizing simple thioester molecules as the thioester group source.
The biggest hurdle for the thioester group transfer reaction is the relative weakness of C(acyl)–S bonds compared to the C(acyl)–C bond (Scheme 1B). Indeed, to the best of our knowledge, all literature examples concerning the oxidative addition of an organometallic complex into thioesters show that the reaction preferably occurs in the C(acyl)–S bond to give an acyl–metal complex.12 We could not find any example of selective C–C(acyl) bond activation in a thioester. Nevertheless, we initially postulated that the selective activation of the C–C(acyl) bond could be achieved by controlling the electronic and steric character of the thioesters, motivated by the examples of selectivity control in C(aryl)–O vs. C(acyl)–O bond activation in esters.13 The theory was eventually proven incorrect.
Recently, visible light photoredox/transition metal dual catalysis14 has been utilized for selective sp3 C–H functionalization. Examples have shown that even weakly reactive α-amino15 and α-oxy15d,16 C(sp3)–H bonds could be functionalized. We devised a strategy to merge photoredox driven C–H functionalization with transition metal mediated thioester activation. Specifically, we focused on the α-oxy C(sp3)–H thiocarbonylation reaction, since α-oxy carboxylic acid derivatives are key functional groups in some pharmaceuticals such as selexipag or cetirizine. Herein, we report the first chemo-selective α-oxy C(sp3)–H thiocarbonylation reaction with a newly proposed photoredox/Ni dual thioester activation strategy (Scheme 1C).
Results and discussion
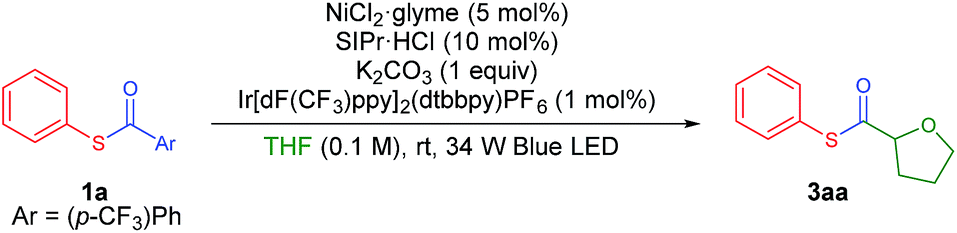
Recently, Doyle and co-workers succeeded in reacting an N-aryl amine with a 2-pyridylthioester under photoredox/Ni dual catalytic conditions (Scheme 1B).15a In this case, only the C–H acylation product via activation of the weaker C(acyl)–S bond was observed. To preferably activate the C(acyl)–C bond instead, we hypothesized that increasing the electron deficiency of the acyl group of the thioester could selectively weaken the C(acyl)–C bond. Indeed, an electron deficient thioester 1a showed reasonably high reactivity toward the target reaction (Tables 1 and S1†). After intensive optimizations (Table S2†), an excellent yield (90%) of 3aa was obtained using an N-heterocyclic carbene (NHC) based Ni(II) complex with Ir[dF(CF3)ppy]2(dtbbpy)PF6 as a visible light photoredox catalyst in THF (2a) (Table 1, entry 1). The only detected side product was 4-trifluoromethyl diphenyl sulfide (4a), which was possibly formed by the decarbonylation process.17 It should be emphasized that the thioester product was the solely formed product without generation of the α-oxy C–H acylation product.15a The reactivity highly depends on the electronic nature of the thioester. Among those tested, only thiobenzoate derivatives exhibited the desired activity (Table S1†). A dramatic decrease in yield was observed when relatively electron rich acyl groups were used (Table 1, entries 2 and 3), while the pentafluorophenyl group gave a moderate yield (entry 4). Control experiments showed that the Ni catalyst, NHC precursor, and photoredox catalyst are essential components of the reaction (entries 5–7). When the separately prepared N-heterocyclic carbene (NHC) SIPr was used as an additive, a moderate yield was obtained without the use of additional base, indicating that the NHC bound nickel complex acted as the catalytically active species (entry 8). In addition, a good yield with full conversion was also obtained in a shortened reaction time of 4 h (entry 10).|
|
|||
|---|---|---|---|
| Entry | Reaction conditions | t (h) | Yieldb of (%) |
| a Reaction conditions: thioester (0.25 mmol), NiCl2·glyme (5 mol%), SIPr·HCl (10 mol%), K2CO3 (1 equiv.), Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1 mol%), and THF (2.5 mL) in a 4 mL vial irradiated with a 34 W Blue LED. b GC yield using dodecane as an internal standard. c α,α,α-Trifluorotoluene (75%) was observed. d SIPr·HCl = 1,3-bis(2,6-diisopropylphenyl)imidazolinium chloride. e Isolated yield. | |||
| 1 | As shown | 12 | 90c |
| 2 | Ar = Ph (1u) | 12 | 7 |
| 3 | Ar = (p-Me)Ph (1v) | 12 | 2 |
| 4 | Ar = C6F5 (1w) | 12 | 49 |
| 5 | No NiCl2·glyme | 12 | 0 |
| 6 | No SIPr·HCld | 12 | 0 |
| 7 | No Ir[dF(CF3)ppy]2(dtbbpy)PF6 | 12 | 0 |
| 8 | SIPr instead of SIPr·HCl with no K2CO3 | 12 | 60 |
| 9 | Ni(COD)2 instead of NiCl2·glyme | 12 | <1 |
| 10 | As shown | 4 | 89 (83e) |
With the optimized conditions in hand, diverse aryl thioesters were applied for the thiocarbonylation reaction of THF (Table 2). A range of aryl thioesters with alkyl substituents in the p-, m- and o-positions gave good yields of the desired products (3ba–3ea). Alkoxy groups in the substrate did not mediate any side reactions, presumably due to the higher amount of the THF radical (3fa–3ha). A thioether group was also tolerated (3ia). Fluorine-containing thioesters showed excellent reactivity (3ja–3la). Encouragingly, C–N and C–O unsaturated bonds did not induce any side reactions or interfere with the catalytic reactivity (3ma–3oa). Naphthyl thioesters reacted smoothly, although 1-naphthylthioester gave a decreased yield (3pa, 3qa). Notably, an aryl boronate, which can be further functionalized, was also tolerated under the reaction conditions (3ra). Finally, benzyl thioesters were successfully transferred to the α-oxy carbon of THF (3sa, 3ta). However, aliphatic thioesters did not exhibit any reactivity under the described reaction conditions.


Next, the scope of ethers was investigated (Table 3). Selective and efficient reactions occurred in the α-oxy position of common cyclic ether solvents such as tetrahydropyran (3ab) and 1,4-dioxane (3ac). In the case of ethers in which the solubility of the Ni complex is poor, acetonitrile was adopted as a co-solvent. Diethylether (2d) underwent moderate thiocarbonylation (3ad). The use of anisole (2e) or tert-butyl methyl ether (MTBE, 2f) gave the desired products in low yields (3ae, 3af), possibly due to the relatively low stability of the primary alkyl radical compared to its secondary analogue. Other heterocycles such as thiophene and N-protected pyrrolidines (N-methyl, N-phenyl, and N-Boc) were tested as reactants using acetonitrile or DMA as the co-solvent due to solubility issues, and no conversion was observed.


From the mechanistic investigations, we realized that the initial hypothesis on preferable C–C(acyl) bond activation by Ni complexes should be reconsidered. To obtain insight into relative bond strengths, we compared the calculated bond dissociation energy (BDE) of the C–C(acyl) bonds with that of the C–S bonds as a preliminary tool, similarly to the Ni catalyzed C(aryl)–O vs. C(acyl)–O bond activation in esters.13d In contrast to our expectation of a significantly lowered BDE of the C–C(acyl) bond in the electron deficient thioester 1a, the calculated BDE of the C–C(acyl) bond in thioester 1a is still much higher than that of the C–S bonds (Table S3†). Also, the BDE cannot explain the unique reactivity of 1a among various thioesters since no significant difference among thioesters was found. In pursuit of an alternative pathway, a mechanism initiated by the oxidative addition of Ni(0) into the C(acyl)–S bond was considered. If this is followed by decarbonylation and migratory insertion of CO into the Ni–S bond, a thiocarbonyl Ni intermediate might be generated. However, Holm and Tucci reported that CO migratory insertion preferably occurred into the Ni–C bond over a Ni–S bond in [Ni(bpy)(R′)(SR)] complexes.18 Moreover, we found that Ni(COD)2, a commonly used Ni(0) complex which has high oxidative addition activity, did not mediate the desired reaction. Instead, a significant amount of decarbonylation product 4a was formed (Table 4). To identify the active catalytic intermediates, syntheses of monomeric NHC–Ni(I) and Ni(II) complexes were attempted, but were unsuccessful. Instead, the catalytic activities of the reported NHC–Ni dimer complexes [(SIPr)NiCl]2 ([Ni-I])19 and [(SIPr)NiCl]2(μ-Cl)2 ([Ni-II])20 were investigated to get insight into the oxidation state of Ni during the reaction (Table 4).21 With [Ni-I], the decarbonylation product 4a was observed as the major product (29%) with 5% of 3aa. In contrast, the well-defined NHC–Ni(II) dimer complex [(SIPr)NiCl]2(μ-Cl)2 ([Ni-II]) produced more of the C–H thiocarbonylation product 3aa (11%) than 4a (8%).
|
|
||||
|---|---|---|---|---|
| Entry | [Ni] | Conversion | 3aa | 4a |
| a With SIPr·HCl (10 mol%) and K2CO3 (1 equiv.). | ||||
| 1 | Ni(COD)2 (5 mol%) + SIPr (10 mol%) | 11% | <1% | 10% |
| 2 | [Ni-I] (2.5 mol%) | 48% | 5% | 29% |
| 3 | [Ni-II] (2.5 mol%) | 21% | 11% | 8% |
| 4 | NiCl2·glyme (1 mol%)a | 36% | 21% | 12% |
| 5 | Ni(COD)2 (5 mol%) + NiCl2·glyme (1 mol)a | 45% | 18% | 27% |
| 6 | [Ni-I] (2.5 mol%) + NiCl2·glyme (1 mol%)a | 52% | 21% | 28% |
| 7 | [Ni-II] (2.5 mol%) + NiCl2·glyme (1 mol%)a | 63% | 42% | 11% |

Because the tested Ni complexes exhibited significantly lower conversions, we re-tested the catalytic activities in the presence of 1 mol% of NiCl2·glyme (Table 4, entries 4–7). Under these conditions, the difference in the catalytic ability of Ni(0), Ni(I), and Ni(II) becomes more significant. While the addition of 5 mol% of Ni(0) or Ni(I) does not facilitate the formation of 3aa, the presence of the NHC–Ni(II) species gave meaningful improvements in reactivity, implying that Ni(II) could be the major catalytically active species.
The results indicate that oxidative addition type C–C(acyl) or C(acyl)–S bond activation by electron rich Ni(0) or Ni(I) did not lead to the formation of the desired product in our reaction. Another possible thioester activation pathway is the single electron reduction of the thioester. It is known that thioesters can be electrochemically reduced and fragmented into acyl radicals and thiolates.22 The same process could be mediated by photoredox catalysis in our system. To check the validity of this hypothesis, cyclic voltammetry (CV) measurements of 1a (Fig. S5†) were carried out. The results show an irreversible reduction peak [1a/1a−] at EredP = −1.96 V vs. the Ag/AgNO3 (0.01 M) electrode in CH3CN (correlated to −1.65 V vs. the saturated calomel electrode (SCE) in CH3CN). The significantly lower reduction potential of 1a compared to aliphatic thioesters and other relatively electron rich aryl thioesters could be the reason for the unique reactivity of 1a.23 Although the reduction potential [1a/1a−] is higher than that of the possible reductant Ir(II) (Ered1/2 [Ir(III)/Ir(II)] = −1.37 V vs. the SCE in CH3CN),24 a small portion of reduction wave overlap may induce single electron transfer at a meaningful rate considering that an irreversible fragmentation may occur after reduction of the thioester. Also, it has been proposed that the cationic character of the photoredox catalyst may electrostatically stabilize the generated radical anion, facilitating the reduction process.25 Additionally, in the optimized reaction conditions, there is a chance that complexation of the thioester and nickel complex could facilitate the thioester reduction process. To prove this hypothesis, a CV experiment with the thioester was conducted in the presence of a stoichiometric amount of a Lewis acid. The Mg(II) cation was utilized because it has redox-inactive character within the scan range (Ered = −2.61 V vs. the SCE in H2O) and has a similar ionic radius (72 pm) compared to that of Ni(II) (69 pm).26 Indeed, the reduction peak shift from −1.65 V to −1.21 V (vs. the SCE) was observed (Fig. S6†). To further validate this reduction process by capturing the in situ generated acyl radical, methyl acrylate was added to the system in the absence of a Ni complex (Scheme 2A). The coupling product 5a was isolated in 26% yield, confirming that the proposed photocatalytic fragmentation of the thioester can happen.
 | ||
| Scheme 2 Mechanistic studies. | ||
Based on this rationale, a revised mechanism is proposed (Fig. 1). Initially, the photo-activated Ir[dF(CF3)ppy]2(dtbbpy)PF6 complex (Ered1/2 [*Ir(III)/Ir(II)] = +1.21 V vs. the SCE in CH3CN)24 oxidizes a model NHC–Ni(II) species (EredP [[Ni-II]+/[Ni-II]] = +1.01 V vs. the SCE in CH3CN) to Ni(III) (Fig. S7†). The resulting Ir(II) complex reduces the thioester and returns to Ir(III). The reduced thioester is then fragmented into an aryl acyl radical excluding the thiolate, which can be immediately captured by Ni(III). The resulting aryl acyl radical is proposed to undergo Ni mediated decarbonylation to give the aryl radical. There have been a few reports that transition metal catalysts can mediate this process,27 although the formation of an aryl radical from an aryl acyl radical with CO exclusion would be difficult.28 To prove the generation of a Ni–CO intermediate during the reaction, we performed a reaction under a 13CO atmosphere utilizing a two-chamber reactor developed by Skrydstrup et al. (Fig. S1†).29 A significant amount of 13C incorporation in the acyl carbon of 3aa was observed, indicating that a Ni–CO intermediate is formed during the course of the reaction (Scheme 2B). The aryl radical is highly reactive so it can abstract a hydrogen from the relatively weak α-oxy carbon of THF as the next step.16a The role of the aryl radical as a hydrogen abstraction reagent was confirmed by the generation of α,α,α-trifluorotoluene-d (4a–d) when the reaction was performed in THF-d8 (Fig. S3†). The resulting α-oxy alkyl radical adds to the Ni(III) complex to give Ni(IV). Migratory insertion of the CO ligand followed by reductive elimination can produce the desired thioester, regenerating the Ni(II) species. Although it is still difficult to rule out the possible more general Ni(I)–Ni(III) cycle, we prefer the proposed a Ni(II)–Ni(IV) cycle based on the experiments in Table 4.
 | ||
| Fig. 1 Proposed mechanism. | ||
Conclusions
The first selective C(sp3)–H thiocarbonylation reaction is achieved. It is the first example of utilizing simple thioester molecules as the thioester group source in the thiocarbonylation reaction, avoiding the use of CO gas. Diverse aryl and benzyl thioesters could be synthesized in high yields. An iridium photoredox catalyst mediated electron transfer between a nickel complex and the thioester comprises the key step of the reaction. The developed direct C–H thiocarbonylation reaction will serve as a novel strategy to synthesize biologically and synthetically important thioesters under mild conditions.Acknowledgements
We thank J. G. Lee in the Prof. T. D. Chung group for help with the cyclic voltammetry analysis. This work was supported by Samsung Science and the Technology Foundation under Project Number SSTF-BA1601-12. B. Kang was supported by the Global PhD Fellowship Program through the NRF (NRF-2012H1A2A1049157).Notes and references
- (a) L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260 CrossRef CAS; (b) H. Tokuyama, S. Yokoshima, S. C. Lin, L. P. Li and T. Fukuyama, Synthesis, 2002, 1121 CrossRef CAS; (c) S. G. Modha, V. P. Mehta and E. V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 5042 RSC; (d) F. Dénès, C. H. Schiesser and P. Renaud, Chem. Soc. Rev., 2013, 42, 7900 RSC.
- P. G. M. Wuts and T. W. Greene, Greene’s Protective Groups in Organic Synthesis, John Wiley & Sons, New Jersey, 4th edn, 2007 Search PubMed.
- A. L. Lehninger, D. L. Nelson and M. M. Cox, Lehninger principles of biochemistry, W.H. Freeman, New York, 5th edn, 2008 Search PubMed.
- (a) M. E. Linder, P. Middleton, J. R. Hepler, R. Taussig, A. G. Gilman and S. M. Mumby, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3675 CrossRef CAS PubMed; (b) J. E. Smotrys and M. E. Linder, Annu. Rev. Biochem., 2004, 73, 559 CrossRef CAS PubMed.
- (a) P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776 CAS; (b) P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923 CrossRef CAS PubMed; (c) S. B. H. Kent, Chem. Soc. Rev., 2009, 38, 338 RSC.
- (a) T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705 CrossRef CAS PubMed; (b) T. W. Muir, Annu. Rev. Biochem., 2003, 72, 249 CrossRef CAS PubMed.
- (a) M. Kazemi and L. Shiri, J. Sulfur Chem., 2015, 36, 613 CrossRef CAS; (b) T. C. Zheng, M. Burkart and D. E. Richardson, Tetrahedron Lett., 1999, 40, 603 CrossRef CAS.
- (a) C.-L. Yi, Y.-T. Huang and C.-F. Lee, Green Chem., 2013, 15, 2476 RSC; (b) J. Chung, U. R. Seo, S. Chun and Y. K. Chung, ChemCatChem, 2016, 8, 318 CrossRef CAS.
- C. F. Li, W. J. Xiao and H. Alper, J. Org. Chem., 2009, 74, 888 CrossRef CAS PubMed.
- (a) H. Cao, L. McNamee and H. Alper, J. Org. Chem., 2008, 73, 3530 CrossRef CAS PubMed; (b) M. N. Burhardt, A. Ahlburg and T. Skrydstrup, J. Org. Chem., 2014, 79, 11830 CrossRef CAS PubMed.
- (a) A. I. Scott and K. Kang, J. Am. Chem. Soc., 1977, 99, 1997 CrossRef CAS PubMed; (b) A. I. Scott, J. B. Hansen and S. K. Chung, J. Chem. Soc., Chem. Commun., 1980, 388 RSC; (c) S. Wollowitz and J. Halpern, J. Am. Chem. Soc., 1988, 110, 3112 CrossRef CAS.
- (a) A. Shaver, H. L. Uhm, E. Singleton and D. C. Liles, Inorg. Chem., 1989, 28, 847 CrossRef CAS; (b) P. W. G. Ariyananda, M. T. Kieber-Emmons, G. P. A. Yap and C. G. Riordan, Dalton Trans., 2009, 4359 RSC; (c) A. M. Royer, T. B. Rauchfuss and D. L. Gray, Organometallics, 2009, 28, 3618 CrossRef CAS; (d) S. Kundu, W. W. Brennessel and W. D. Jones, Organometallics, 2011, 30, 5147 CrossRef CAS; (e) A. N. Desnoyer, F. W. Friese, W. L. Chiu, M. W. Drover, B. O. Patrick and J. A. Love, Chem.–Eur. J., 2016, 22, 4070 CrossRef CAS PubMed.
- (a) D. G. Yu, B. J. Li and Z. J. Shi, Acc. Chem. Res., 2010, 43, 1486 CrossRef CAS PubMed; (b) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A. M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346 CrossRef CAS PubMed; (c) K. Amaike, K. Muto, J. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2012, 134, 13573 CrossRef CAS PubMed; (d) X. Hong, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 2017 CrossRef CAS PubMed.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed; (c) J. J. Douglas, M. J. Sevrin and C. R. J. Stephenson, Org. Process Res. Dev., 2016, 20, 1134 CrossRef CAS; (d) M. D. Levin, S. Kim and F. D. Toste, ACS Cent. Sci., 2016, 2, 293 CrossRef CAS PubMed; (e) M. N. Hopkinson, B. Sahoo, J. L. Li and F. Glorius, Chem.–Eur. J., 2014, 20, 3874 CrossRef CAS PubMed.
- (a) C. L. Joe and A. G. Doyle, Angew. Chem., Int. Ed., 2016, 55, 4040 CrossRef CAS PubMed; (b) Z. W. Zuo, D. T. Ahneman, L. L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed; (c) D. T. Ahneman and A. G. Doyle, Chem. Sci., 2016, 7, 7002 RSC; (d) M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304 CrossRef CAS PubMed.
- (a) B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719 CrossRef CAS PubMed; (b) D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715 CrossRef CAS PubMed.
- E. Wenkert and D. Chianelli, J. Chem. Soc., Chem. Commun., 1991, 627 RSC.
- G. C. Tucci and R. H. Holm, J. Am. Chem. Soc., 1995, 117, 6489 CrossRef CAS.
- B. R. Dible, M. S. Sigman and A. M. Arif, Inorg. Chem., 2005, 44, 3774 CrossRef CAS PubMed.
- C. A. Laskowski and G. L. Hillhouse, Organometallics, 2009, 28, 6114 CrossRef CAS.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed.
- (a) R. D. Webster, A. M. Bond and R. G. Compton, J. Phys. Chem., 1996, 100, 10288 CrossRef CAS; (b) R. D. Webster and A. M. Bond, J. Org. Chem., 1997, 62, 1779 CrossRef CAS; (c) S. Ozaki, H. Yoshinaga, E. Matsui and M. Adachi, J. Org. Chem., 2001, 66, 2503 CrossRef CAS PubMed; (d) S. Ozaki, M. Adachi, S. Sekiya and R. Kamikawa, J. Org. Chem., 2003, 68, 4586 CrossRef CAS PubMed; (e) M. Weïwer, S. Olivero and E. Duñach, Tetrahedron, 2005, 61, 1709 CrossRef.
- Cyclic voltammetry analysis of 1u and 1v was performed. EredP[1u/1u−] = –1.89 V vs. the SCE in CH3CN. EredP[1v/1v−] = –1.99 V vs. the SCE in CH3CN.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef CAS.
- (a) K. Okamoto, K. Ohkubo, K. M. Kadish and S. Fukuzumi, J. Chem. Phys., 2004, 108, 10405 CrossRef CAS; (b) A. Singh, J. J. Kubik and J. D. Weaver, Chem. Sci., 2015, 6, 7206 RSC.
- W. M. Haynes, D. R. Lide and T. J. Bruno, CRC handbook of chemistry and physics: a ready-reference book of chemical and physical data, CRC Press, Florida, 95th edn, 2014 Search PubMed.
- (a) G. Domazetis, B. Tarpey, D. Dolphin and B. R. James, J. Chem. Soc., Chem. Commun., 1980, 939 RSC; (b) R. M. Belani, B. R. James, D. Dolphin and S. J. Rettig, Can. J. Chem., 1988, 66, 2072 CrossRef CAS.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS PubMed.
- S. D. Friis, A. T. Lindhardt and T. Skrydstrup, Acc. Chem. Res., 2016, 49, 594 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and full characterization of substrates and products. Crystallographic data for compound [Ni-II]. CCDC 1516709. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02516e |
| This journal is © The Royal Society of Chemistry 2017 |