
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simplified characterization of S-adenosyl-L-methionine-consuming enzymes with 1-Step EZ-MTase: a universal and straightforward coupled-assay for in vitro and in vivo setting†
Emmanuel S.
Burgos
 *a,
Ryan O.
Walters
bcd,
Derek M.
Huffman
bcd and
David
Shechter
*a
*a,
Ryan O.
Walters
bcd,
Derek M.
Huffman
bcd and
David
Shechter
*a
aDepartment of Biochemistry, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, New York 10461, USA. E-mail: emmanuel.burgos@einstein.yu.edu; david.shechter@einstein.yu.edu; Fax: +1-718-430-8565; Tel: +1-718-430-4120 Tel: +1-718-430-4128
bDepartment of Molecular Pharmacology, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, New York 10461, USA
cDepartment of Medicine, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, New York 10461, USA
dDepartment of Institute for Aging Research, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, New York 10461, USA
First published on 27th July 2017
Abstract
Methyltransferases use S-adenosyl-L-methionine (SAM) to deposit methyl marks. Many of these epigenetic ‘writers’ are associated with gene regulation. As cancer etiology is highly correlated with misregulated methylation patterns, methyltransferases are emerging therapeutic targets. Successful assignment of methyltransferases' roles within intricate biological networks relies on (1) the access to enzyme mechanistic insights and (2) the efficient screening of chemical probes against these targets. To characterize methyltransferases in vitro and in vivo, we report a highly-sensitive one-step deaminase-linked continuous assay where the S-adenosyl-L-homocysteine (SAH) enzyme-product is rapidly and quantitatively catabolized to S-inosyl-L-homocysteine (SIH). To highlight the broad capabilities of this assay, we established enzymatic characteristics of two protein arginine methyltransferases (PRMT5 and PRMT7), a histone-lysine N-methyltransferase (DIM-5) and a sarcosine/dimethylglycine N-methyltransferase (SDMT). Since the coupling deaminase TM0936 displays robust activity over a broad pH-range we determined the pH dependence of SDMT reaction rates. TM0936 reactions are monitored at 263 nm, so a drawback may arise when methyl acceptor substrates absorb within this UV-range. To overcome this limitation, we used an isosteric fluorescent SAM-analog: S-8-aza-adenosyl-L-methionine. Most enzymes tolerated this probe and sustained methyltransfers were efficiently monitored through loss of fluorescence at 360 nm. Unlike discontinuous radioactive- and antibody-based assays, our assay provides a simple, versatile and affordable approach towards the characterization of methyltransferases. Supported by three logs of linear dynamic range, the 1-Step EZ-MTase can detect methylation rates as low as 2 μM h−1, thus making it possible to quantify low nanomolar concentrations of glycine N-methyltransferase within crude biological samples. With Z′-factors above 0.75, this assay is well suited to high-throughput screening and may promote the identification of novel therapeutics.
Introduction
Protein post-translational modifications (PTM) regulate many biochemical processes.1–3 For instance, the deposition and removal of histone PTMs, can govern cell fate. Methyl marks are written by methyltransferases (MTases) and fueled by a universal methyl-donor: S-adenosyl-L-methionine (SAM).4 Small molecule methyltransferases (SMMT) were the first discovered.5,6 Further studies identified DNA methyltransferases (DNMT) as key catalysts to edit cytosine at certain CpG sequences, thus modulating cellular differentiation and transcriptional silencing.7 Later, protein lysine and arginine methyltransferases (PKMT and PRMT, respectively) emerged as crucial enzymes responsible for histone tail modifications.8,9 While methylation of lysine 4 on histone H3 (H3K4me3) is undeniably responsible for activation of transcription, establishing an universal code to translate PTM's and their cross talk is still at the early stage of development.10,11 Nonetheless, it is evident that erratic methylation patterns are implicated in oncogenesis and tumor progression.9,12,13 Overexpression of PKMTs and PRMTs in tumors is correlated with poor clinical prognosis.14–18 MTases are emerging cancer targets and they provide a new horizon for biological chemists to enhance the clinical use of personalized therapies.19–21Current MTase assays rely on the detection of either product of the transferase reactions, methyl marks or S-adenosyl-L-homocysteine (SAH; Fig. S1 and Table S1†). The use of radiolabeled SAM makes it possible to quantify the radioactivity incorporated within the acceptor target. Whether on a DNA strand or a peptide, the methylated product can be separated from methyl-donor via specifically charged filters, solid phase extraction sorbents or liquid chromatography (Fig. S1,† arrows 1 and 2).22–25 Methylation can also be detected via antibody-specific recognition combined with fluorescence resonance energy transfer (FRET, AlphaLISA; Fig. S1,† arrow 3).26,27 Although low detection limits may be reached with these assays, there are major drawbacks to these methods, including: (1) a discontinuous approach limiting the analysis throughput, (2) the elevated costs of radioactive waste treatment along with unstable radioactive SAM and (3) the highly specific immuno-detection may limit the analysis to one single MTase.
On the over hand, SAH detection is well suited to the characterization of a wider range of MTases as SAH is the universal by-product of all transferase reactions. Therefore, multiple assays are based on this detection, either directly or using recombinant coupling enzymes to catabolize SAH and channel it into a metabolite easily detectable (Fig. S1;† arrows 4–12). An additional experimental benefit is that this approach relieves the MTases from product-inhibition.
Many assays have been developed to detect SAH. For instance, bacterial S-adenosyl-L-homocysteine nucleosidase (MTAN, E.C. 3.2.29) generates adenine and S-(5-deoxy-D-ribos-5-yl)-L-homocysteine (SRH). Adenine can either be detected continuously by (1) luminescence at 570 nm through efficient conversion into AMP and ATP using adenine phosphoribosyltransferase (APRT, E.C. 2.4.2.7), pyruvate phosphate dikinase (PPDK, E.C. 2.7.9.11) and firefly luciferase (FLUC, E.C. 1.13.12.7; Fig. S1,† arrow 4)28,29 or (2) decrease of absorbance at 265 nm following deamination into hypoxanthine (Hx; Fig. S1,† arrow 5) using adenosine deaminase (ADA, E.C. 3.5.4.2).30,31 Meanwhile, S-ribosylhomocysteinase (LuxS, E.C. 4.4.1.21) catabolizes SRH into L-homocysteine (Hcy-SH) for further detection of free-thiol with Ellman's reagent at 412 nm (Fig. S1,† arrow 6).32 Likewise, recombinant S-adenosyl-L-homocysteine hydrolase (SAHH, E.C. 3.3.1.1) may utilize SAH to generate Hcy-SH, later detected with thiol-sensitive reagents (e.g. ThioGlo®-1; Fig. S1,† arrow 7).33,34 In presence of ATP and adenosine kinase (AK, E.C. 2.7.1.20), the adenosine product of the SAHH reaction, is phosphorylated to AMP and further detected with a specific antibody (Fig. S1,† arrow 8).35 The remaining ATP from this kinase reaction can also be quantified by KinaseGlo® reagent (luminescence; Fig. S1,† arrow 9).36 Another approach, involving PPDK and FLUC, allows for continuous monitoring of SAH through recording of light output (Fig. S1,† arrow 10).37 A universal, yet discontinuous detection of SAH, based on competitive fluorescence polarization immunoassay (FPIA), was also achieved.38
Finally, a very early report described a continuous assay involving a single coupling enzyme.39 The conversion of SAH into S-inosyl-L-homocysteine (SIH) was implemented to characterize the rat liver catechol O-methyltransferase through monitoring of absorbance at 265 nm (Fig. 1A and B). Isolation of the deaminase from Aspergillus oryzae is likely the major drawback in using the assay,40 so the method has not been used since 1973. However, recent efforts towards the annotation of enzyme function have predicted the SAH-deaminase activity of TM0936 from Thermotoga maritima (Fig. 1A).41 Further reports have described additional members of this enzyme family, none of which are able to catabolize SAM.42,43
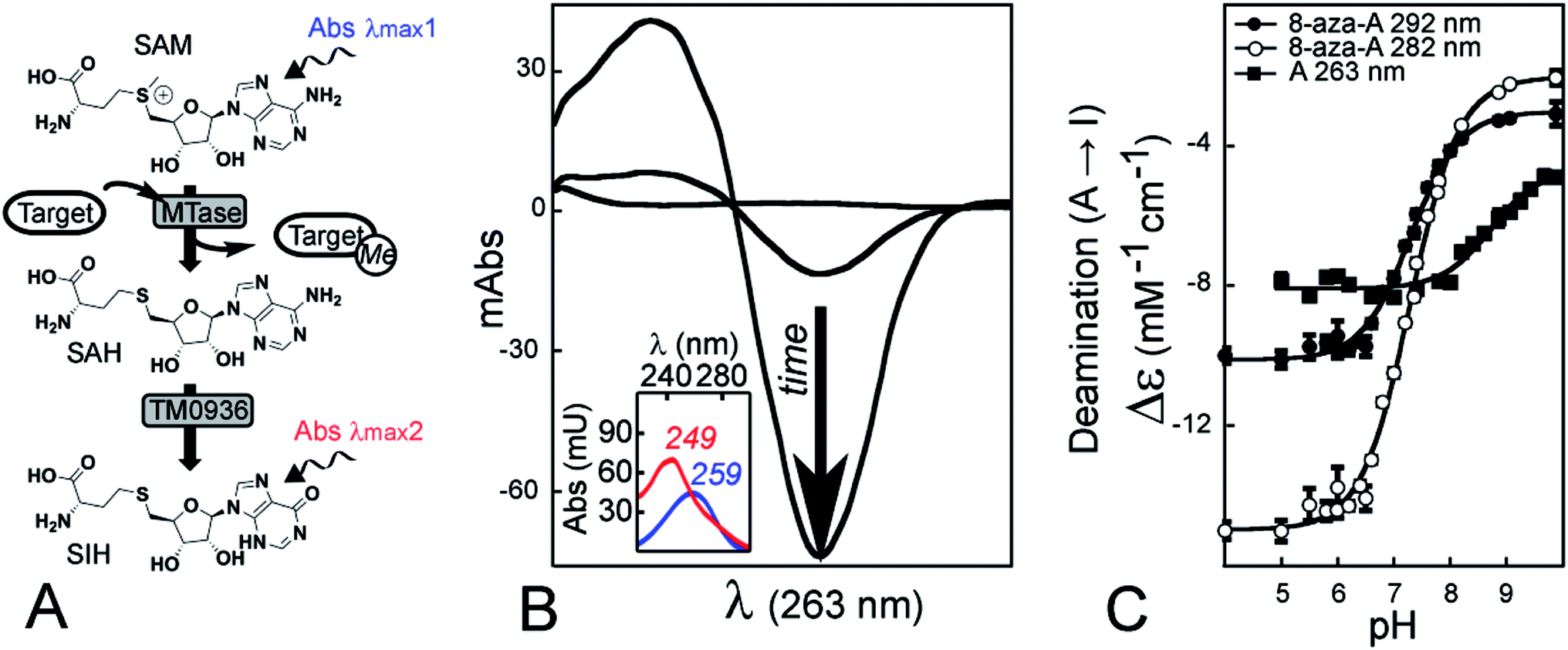 | ||
| Fig. 1 The reaction catalyzed by deaminase TM0936 and its use as a coupling enzyme for assay development. (A) The SAH by-product of methyltransferase (MTase) reactions is efficiently converted into its inosyl-derivative (SIH) by the deaminase TM0936. (B) The different spectral signatures between SAH and SIH (λmax1 = 259 nm in blue and λmax2 = 249 nm in red, respectively) allow continuous monitoring of methyltransfer through UV-detection. The reactions are characterized by a decrease of absorbance at 263 nm (black arrow). (C) The pH-dependence of differential extinction coefficients for the deamination reaction Δε = f(pH). UV-spectroscopy (scans 220–320 nm) was used to monitor changes in absorbance during the reaction catalyzed by TM0936. The adenosyl (A) to inosyl (I) conversion was monitored at 263 nm (black squares) while the homologous reaction using 8-aza-adenosyl (8-aza-A) was monitored at both 292 and 282 nm (black and white circles, respectively). | ||
A coupled assay relying on one single enzyme is a clear asset, thus we took advantage of TM0936. This SAH-deaminase is a robust and efficient catalyst.41,43 By converting SAH into SIH, the enzyme relieves MTAses from product inhibition.39,44 We coupled this catalyst to several MTase families, including two PRMTs (PRMT5 and PRMT7), a PKMT (DIM-5) and a SMMT (i.e. sarcosine/dimethylglycine N-methyltransferase; SDMT). Here, we demonstrate that this assay can provide efficient measurement of kinetic parameters well suited to high-throughput screening (HTS). Furthermore, we measured the inhibition value of sinefungin against SDMT, thus establishing the compatibility between sinefungin and TM0936 and demonstrating its use for inhibitor screening.
TM0936 is a strong catalyst and pH variations only affected the deaminase activity to a small extent; the decrease of absorbance at 263 nm was monitored accurately across a 5-unit pH range (5.0–10.0). Thus, in a technical tour de force, we established the pH dependence of SDMT reaction rates. Likewise, variations in salt concentration had no effect onto TM0936 activity, and we quantified the impact of ionic strength onto the affinity between histone tails and their MTase target. Conscious that a UV-mode of detection may limit the applications of this assay, we synthesized a fluorescent SAM analog. In most cases, the S-8-aza-adenosine-L-methionine (8-aza-SAM) was a good substrate for MTases. As TM0936 efficiently converted 8-aza-SAH into the non-fluorescent product 8-aza-SIH, PRMT7 activity was monitored through a decrease of fluorescence emission at 360 nm.
Finally, taking advantage of a low limit of SAH-detection (2 μM h−1), we successfully quantified glycine N-methyltransferase (GNMT) activity within rat liver extracts, demonstrating the in vivo applicability of this assay. Unlike other techniques, the continuous detection of methyltransferase with 1-Step EZ-MTase is compatible with adenine, phosphorylated adenosines and reactive thiol species (e.g. glutathione, homocysteine and cysteine) often present in crude biological samples. Thus, this novel assay allows for fast, simple and accurate measurement of GNMT activity, while overcoming limitations of interference observed in previous formats. Importantly, measurements can be detected in as little as 30 μg of tissue protein, suggesting applicability even for biologic samples where protein yield is limited.
Results and discussion
The deaminase TM0936 is a prime-choice candidate for MTase assays
We used a two-step purification of TM0936 as a polyhistidine-tagged protein expressed in bacteria. The high-purity enzyme from the hyperthermophile Thermotoga maritima can sustain multiple freeze/thaw cycles, it remains unable to catabolize SAM and it displays an excellent reactivity towards SAH at room temperature (Fig. S2A;†Km = 106 ± 18 μM, kcat = 2.2 ± 0.1 s−1. These parameters are in good agreement with previous reports describing Km and kcat values of 210 ± 40 μM and 12.2 ± 0.8 s−1, respectively).41,43Coupled-enzyme assays for MTase reactions are based on the same principal of rapid channeling of SAH to a signal output, such that SAH is virtually absent and coupling enzymes are not rate-limiting. Thus, the signal output reflects solely on the MTase reaction. In our hands, commercial kits for detection of methyltransfer (Fig. 2A; Cayman Chemical, #700150) suffered from poor performances with a slow and incomplete processing of SAH. It took 10 min for a 200 nM standard concentration of SAH to be digested by coupling enzymes (Fig. 2B). Furthermore, a comparison between SAH and resorufin standard curves supports that channeling of the MTase product is incomplete, thus resulting in a 50% loss of sensitivity (Fig. 2C).
 | ||
| Fig. 2 The drawbacks from a commercial kit. (A) The coupled-assay for MTase detection (Cayman Chemical, #700150). Through two enzymatic reactions, SAH is channeled to hypoxanthine; further oxidation by xanthine oxidase (XO) will produce uric acid and two molecules of hydrogen peroxide. The peroxide fuels horse radish peroxidase (HRP) to convert 10-acetyl-3,7-dihydroxyphenoxazine (ADHP) into fluorescent resorufin. (B) Slow channeling of SAH molecule. The coupling enzymes from the kit do not convert SAH fast enough and a 10 min lag phase is observed. (C) Channeling of SAH is not quantitative. A comparison between resorufin and SAH standard curves (squares and circles, respectively), highlighted the incomplete coupling between enzymes; nearly 50% of SAH-equivalent was lost before fluorescence emission. | ||
We previously developed methods for detection of SAH and its derivatives (e.g. adenosine, adenine).37,45 Although we routinely use these highly sensitive luciferase-based coupled assays,46 we encountered limitations (Table S1†). The preparation of four highly-purified recombinant enzymes (i.e. MTAN, APRT, PPDK, FLUC) is a tedious process and batch-to-batch reproducibility may be an issue. Furthermore, the coupling buffer requires 5-phospho-α-D-ribosyl-1-pyrophosphate (PRPP), a highly unstable and expensive substrate to fuel APRT. Likewise, the presence of adenine and phosphorylated adenosine species within crude biological samples preclude the use of luciferase-based assays for the in vivo settings. Therefore, based on the TM0936 performance, we developed the 1-Step EZ-MTase assay to provide a simple and robust method for measuring SAM consumption.
Spectral signature of the SAH deamination reaction catalyzed by TM0936
To determine the precise relationship between absorbance and concentration for the adenosyl to inosyl conversion, we measured the differential extinction coefficient from the reaction catalyzed by TM0936. A nucleoside absorbance spectrum (i.e. maximum absorption wavelength λmax, extinction coefficient ε) is pH-dependent, thus reflects the ionization state of this molecule. The adenosyl UV-signature remains unchanged in water under most conditions (neutral species at pH > 3.6; Fig. S2B†); however, the inosyl group may behave differently as it becomes deprotonated in alkaline solutions (pKa = 8.9).47,48We established the Δε = f(pH) relationship for the deamination (Fig. 1C). This data-set is a key component of the assay so that MTase reaction rates may be determined accurately across a broad pH-range. Data were fitted to ESI eqn (S2)† where Δεhigh pH (−4655 ± 186 M−1 cm−1) and Δεlow pH (−8076 ± 80 M−1 cm−1) are the extinction coefficient measured at 263 nm for the deaminase reaction at high and low pH, respectively, and −pKa (−8.72 ± 0.09) is the logarithm of acid dissociation constant for inosine (lit.,49 −3890 M−1 cm−1, −8270 M−1 cm−1 and 8.85, respectively).
In addition, similar Δε = f(pH) relationships were established at 282 and 292 nm for the 8-aza-adenosine (8-aza-A) to 8-aza-inosine (8-aza-I) reaction (Fig. 1C). The 8-aza analogs of SAM/SAH are fluorescent while their inosyl counterparts are not.50,51 Thus, 8-aza-SAM may be a valuable substrate for monitoring MTase activity using a fluorescence mode. Our experiments provided: Δεhigh pH (−2026 ± 174 and −3016 ± 119 M−1 cm−1), Δεlow pH (−14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 975 ± 129 and −10117 ± 87 M−1 cm−1) and pKa (7.29 ± 0.03 and 7.30 ± 0.03), at 282 and 292 nm, respectively.
975 ± 129 and −10117 ± 87 M−1 cm−1) and pKa (7.29 ± 0.03 and 7.30 ± 0.03), at 282 and 292 nm, respectively.
Kinetic parameters from four methyltransferases using 1-Step EZ-MTase
To establish a coupled assay that can sample the vast majority of MTase targets, all having diverse kinetic properties (range of SAM concentrations: 1–1000 μM; Fig. S3†), we performed experiments in 96-well plates compatible with UV detection. This format allows for higher throughput of data vs. a regular cell-changer spectrophotometer using 1/2–3 mL cuvettes. The wells accommodate 50–250 μL sample volumes, resulting in an adjustable optical path so that a linear relationship between absorbance and concentration is preserved (Fig. S3†). Furthermore, the assay displays a broad range of SAH-detection with a upper limit set at 10 × 103 μM h−1 when using a 4 μM concentration of TM0936 (i.e. 42 × 103 pmol min−1 in a 250 μL well at pH 8.00; Fig. S4A and B†). Although the cofactor slowly and partially degrades into SAH, our analytical tool senses methyltransfer rates as low as 2 μM h−1 (i.e. 8 pmol min−1 in a 250 μL well at pH 8.00 and 3.5 μM h−1 at pH 9.50; Fig. S4C and D†).We tested this coupled assay with four unique methyltransferases. TM0936 (4 μM) was coupled to these transferases to establish their kinetic behavior: determination of Michaelis constant (Km) and catalytic turnover (kcat) for either SAM or acceptor substrate. Using the protein arginine methyltransferase from Caenorhabditis elegans (CePRMT5) and saturating levels of SAM (25 μM), we determined the kinetic profile as a function of the peptide H4(1–20) concentration (6–180 μM, at 300 nM CePRMT5). The data fit to the Morrison equation52 gives a Km of 26 ± 2 μM and a kcat of 32.9 ± 0.8 h−1 (Fig. 3A), consistent with values we reported using the luciferase-based assay (lit.,46Km = 54 ± 4 μM, kcat = 28.6 ± 0.7 h−1). Another member of the PRMT family, PRMT7 from Trypanosoma brucei (400 nM), was also tested using saturating concentration of peptide H4(1–20) (200 μM). Varying SAM concentration (1.5–20.0 μM), reaction rates were determined and plotted against cofactor concentration. The data were fitted as for CePRMT5, giving a Km of 1.1 ± 0.2 μM and a turnover of 22.3 ± 0.6 h−1. Likewise, the activity from Neurospora crassa protein lysine N-methyltransferase (histone H3 lysine-9 specific; NcDIM-5) was successfully detected at pH = 9.50. Our data (Km = 0.9 ± 0.1 μM, kcat = 30 ± 1 min−1) and reported values (lit.,53Km = 7.4 μM, kcat = 138 h−1) display a 100-fold difference in the overall catalytic efficiencies (2000 μM−1 h−1vs. 19 μM−1 h−1, respectively). However, we used a H3(1–53) peptide while previous report mentioned a H3(1–20) peptide harboring a N-terminal biotinylation; such a modification, vicinal to the reactive lysine-9, may account for the discrepancy observed. Finally, we selected sarcosine/dimethylglycine N-methyltransferase from Galdieria sulphuraria (GsSDMT) to represent members of the SMMT family. We thought the 1-Step EZ-MTase assay would be challenged by this enzyme. Indeed, GsSDMT behaves differently from other MTases we tested: it is a fast enzyme and displays a high micromolar Km for SAM (lit.,54Km = 144 ± 44 μM, kcat = 52 ± 4 min−1). Yet, using a 60 μL reaction volume to accommodate the elevated SAM levels (50–750 μM), along with a low enzyme concentration (195 nM), we successfully determined the GsSDMT kinetic parameters. Both the Km of 95 ± 18 μM and the kcat of 42 ± 2 min−1 are in agreement with the data mentioned earlier.
 | ||
| Fig. 3 Monitoring methyltransfer activities using our 1-Step EZ-MTase assay and UV-mode of detection. (A) Application to the protein arginine methyltransferase from Caenorhabditis elegans (CePRMT5). Kinetic parameters using peptide H4(1–20) were determined at pH = 7.60 using 25 μM SAM and 300 nM CePRMT5: Km = 26 ± 2 μM, kcat = 32.9 ± 0.8 h−1. (B) Application to the protein arginine methyltransferase from Trypanosoma brucei (TbPRMT7). Kinetic parameters using SAM were determined at pH = 7.60 using 200 μM peptide H4(1–20) and 400 nM TbPRMT7: Km = 1.1 ± 0.2 μM, kcat = 22.3 ± 0.6 h−1. (C) Application to the protein lysine N-methyltransferase (histone H3 lysine-9 specific) from Neurospora crassa (NcDIM-5). Kinetic parameters using peptide H3(1–53) were determined at pH = 9.50 using 25 μM SAM and 7.6 nM NcDIM-5: Km = 0.9 ± 0.1 μM, kcat = 30 ± 1 min−1. (D) Application to the sarcosine/dimethylglycine N-methyltransferase from Galdieria sulphuraria (GsSDMT). Kinetic parameters using SAM were determined at pH = 7.80 using 100 μM MgCl2, 5 mM sarcosine and 195 nM GsSDMT: Km = 95 ± 18 μM, kcat = 42 ± 2 min−1. The experiments recorded at 263 nm were all performed using 4 μM final concentration of TM0936. Inserts are Z′-factors determined at multiple MTase concentrations; the lowest transferase concentration displays a Z′ ≈ 0.5, a characteristic for a good HTS assay.64 | ||
As we demonstrated with these four examples, the combination of a single coupling enzyme with a UV-mode of detection and a 96-well plate format makes the characterization of methyltransferases simple and straightforward. To further assist the user in the treatment of her/his experimental data-set, we developed two exhaustive template spreadsheets “My 1-Step EZ-MTase Assay (UV) Acceptor Km.xlsx” and “My 1-Step EZ-MTase Assay (UV) CoFactor Km.xlsx” (cf. ESI†). These files will help the users in (1) setting-up their experimental conditions (worksheet “Experiment conditions”), (2) importing their raw data (worksheet “Plate reader data”), (3) reporting their key experimental conditions e.g. pH, nature of methyl cofactor being used; worksheet “Coupling enzyme parameters” and (4) computing and displaying the kinetic parameters of their own experiment. The first template mentioned above is pre-loaded with experimental data pertinent to the kinetic behavior of H4(1–20) peptide with TbPRMT7, while the second template contains experimental results leading to the description of 8-aza-SAM substrate with CePRMT5. An overall summary of kinetic parameters from MTases we assayed is represented in Table 1.
| K m | k cat | |
|---|---|---|
| a From P. Rathert, X. Cheng and A. Jeltsch, BioTechniques, 2007, 43, 602, 604, 606. | ||
| CePRMT5 | ||
| SAM | 6.8 ± 0.3 μM | 31.9 ± 0.5 h−1 |
| H4(1–20) | 26 ± 2 μM | 32.9 ± 0.8 h−1 |
| 8-Aza-SAM | 35 ± 20 μM | 15 ± 6 h−1 |
|
||
| TbPRMT7 | ||
| SAM | 1.1 ± 0.2 μM | 22.3 ± 0.6 h−1 |
| H4(1–20) | 39 ± 3 μM | 28.2 ± 0.7 h−1 |
| 8-Aza-SAM | 15.3 ± 0.7 μM | 23.0 ± 0.4 h−1 |
|
||
| NcDIM-5 | ||
| SAMa | 0.68 ± 0.20 μM | 3.1 h−1 |
| H3(1–53) | 0.9 ± 0.1 μM | 30 ± 1 min−1 |
| 8-Aza-SAM | Not a substrate | Not a substrate |
|
||
| GsSDMT | ||
| SAM | 95 ± 18 μM | 42 ± 2 min−1 |
| Sarcosine | 1.7 ± 0.2 mM | 90 ± 5 min−1 |
| 8-Aza-SAM | 443 ± 33 μM | 81 ± 3 min−1 |
TM0936 activity is resilient to pH variations
Another key limitation to the luciferase-based assays is their sensitivity in different chemical environments, as the detection is optimum at a very narrow pH value (≈7.7) and luminescence output is drastically reduced upon pH variations (Fig. S5A and Table S1†). As the pH decreases, so does the green component of the luminescence (Fig. S5B†); light was no longer detected under acidic conditions (pH < 6.0; Fig. S5A†).55 Therefore, key mechanistic insights are undetectable with this assay as it is impossible to describe MTase enzymology over a wide pH-range.In contrast, TM0936 displays a sustained activity across a broad pH-range (Fig. 4A). With the 1-Step EZ-MTase assay, the transferase rates are the limiting ones at all pHs and absorbance recordings directly relate to methyltransfer. This is a characteristic to consider and take advantage of when developing a coupled enzymatic assay. As support of broad utility of this assay, we harnessed the deaminase activity of TM0936 to probe the enzymatic mechanism from a poorly characterized methyltransferase: sarcosine/dimethylglycine N-methyltransferase (SDMT). SDMT catalyzes a two-step methylation process leading to a key metabolite: betaine. Trimethylglycine is an effective methyl donor involved in the biosynthesis of L-methionine from L-homocysteine;56 furthermore, under extreme conditions (e.g. high salt concentrations or low temperatures), this molecule stabilizes proteins acting as an osmoprotectant.57 Little mechanistic information is available regarding this enzyme, with a handful of kinetic reports54,58,59 and one single crystal structure of the apo-form of SDMT from Galdieria sulphuraria.54,60 In an experimental tour de force, we established the pH-dependence of GsSDMT reaction rates for sarcosine (Dixon plots; Fig. 4B). By reporting Km and kcat for this substrate across a 4-unit range of pHs, we gleaned valuable information and proposed an inventory of residues potentially involved in sarcosine capture and processing by the SDMT enzyme.
 | ||
| Fig. 4 The 1-Step EZ-MTase assay is a simple tool to decipher enzymatic mechanisms. (A) The enzyme TM0936 remains a robust deaminase other a broad pH-range. Both substrates adenosine (A; black squares) and 8-aza-adenosine (8-aza-A; black circles) were assayed. Deamination rates (pmol min−1) were measured under different pH conditions, at 10 μM final substrate concentration and 1 nM TM0936. (B) The pH dependence for kcat/Km and kcat using the GsSDMT enzyme. Methyltransfer was monitored at 263 nm (pH 5.80–9.25) using sarcosine as the variable substrate (0.5–12.5 mM) and saturating levels of SAM (750 μM) with 976 nM GsSDMT and 4 μM coupling enzyme. Both log(kcat/Km) and log(kcat) pH functions are depicted. (C) Ionic strength does not affect TM0938 activity. Deaminase activity was monitored with 10 μM adenosine at both low and high sodium chloride concentrations (0–2 M). Relative activity was arbitrary set to 100% when no salt was used. (D) High ionic strength reduces affinity between peptide substrate and the Trypanosoma brucei PRMT7 target (TbPRMT7). The Km for H4(1–20) peptide was determined at saturating levels of SAM (25.8 μM) with 1 μM TbPRMT7, and 4 μM coupling enzyme at four different buffer concentrations (phosphate pH = 7.60; 25, 50, 75 and 100 mM, black squares, white circles, black circles and white squares, respectively). As the buffer concentration decreases, so does the Km for H4(1–20) peptide. | ||
Fitted to the ESI eqn (S8),† the logkcat/Kmvs. pH displays a symmetrical bell-shaped curve with an optimum enzymatic activity between pH 7.5–8.5; pKa values of 6.87 ± 0.05 and 9.2 ± 0.1 were assigned to the ascending and descending limbs, respectively (Fig. 4B, left). Since sarcosine was the varied substrate, the kcat/Km is the apparent second-order rate constant for the reaction between free sarcosine and GsSDMT·SAM complex. Thus, the effects of pH onto this rate constant likely describe the ionization states of these two entities. The protonation of sarcosine carboxylate (pKa ≈ 2.2) does not explain the loss of activity at acidic pHs. Indeed, the pKa for sarcosine carboxylate is much lower than the observed 6.87 pKa-value for the ascending limb. Such a pKa-value may be reminiscent of histidine residues (pKa 6.0–7.0) important for efficient sequestration of sarcosine by the GsSDMT·SAM complex. With respect to the descending limb (pKa = 9.2), the deprotonation of either the methyl-amine group from sarcosine (pKa ≈ 10.0) or a tyrosine residue (pKa ≈ 10.1) may account for the loss of enzymatic activity under alkaline conditions.
The logkcat plotted against pH (Fig. 4B, right; ESI eqn (S9)†) established that reaction rates increased with pH and reached a maximum value at pH = 6.82. This plot reports the ionization state of an important residue from the GsSDMT·SAM·sarcosine complex. Our results support our hypothesis that deprotonation of a crucial histidine may enhance the methyltransfer reaction.
One single structure of SDMT has been reported and the apo-form of this enzyme makes it difficult to support our experimental results.54 However, we successfully superimposed this structure (PDB: 2O57)54 with the SAH·sarcosine complex of the glycine sarcosine N-methyltransferase from Methanohalophilus portucalense (MpGSMT; PDB: 5HIL).61 Both structures display a good alignment of their N-terminus (Fig. S6;† light shades) and the cofactor binding site was easily identified within this conserved domain. Indeed, SAM interacts with key conserved amino-acids: π-stacking between adenosine and W115, stabilization of the ribosyl through hydrogen bond with D88 (F141 and N112 from GsSDMT are predicted to be homologous). Likewise, the homocysteyl binding-mode depicted additional groups involved in stabilization of the cofactor. Residues R60, A91 and Q157 from GsSDMT are structurally homologous to R43, A67 and L132 from MpGSMT, respectively, thus we hypothesize their interaction with the homocysteyl moiety from SAM/SAH (Fig. S6†). Although the sarcosine binding site is located within the most divergent region of the structures (dark shades; C-terminus), we identified Y242 (Y206 structural homolog in MpGSMT) and H241 from GsSDMT as potential candidates involved in sarcosine stabilization via hydrogen bond with the carboxylate tail. Furthermore, the histidine H162 from GsSDMT, also present in MpGSMT (H138), may influence methyltransfer rate since it is equidistant from both nitrogen and sulfur reactive centers from sarcosine and SAH, respectively (Fig. S6†).
The conclusive assignment of our experimental pKa values to definite residues or chemical groups requires further work, such as site-directed mutagenesis of GsSDMT. Yet, establishing this data set is a first step towards the elucidation of a more elaborate enzymatic mechanism.
TM0936 activity is resilient to variations of ionic strength
We previously established the role of the MEP50 WD-repeat protein in presenting histone substrates to the PRMT5 active site.46 To measure the sub-micromolar affinity between MEP50 and H4(1–20) peptide, we relieved methyltransfer activity through titration of exogenous MEP50. Many WD-repeat proteins are highly hydrophobic and require high salt concentrations to promote their solubility.62 Although we had monitored methyltransfer with the highly sensitive luciferase coupled assay, it was critical to maintain ionic strength constant throughout MEP50 titration.46 Indeed, a slight increasing in salt concentration drastically decreases FLUC light output (Fig. S5C and Table S1†), making FLUC-based assays more difficult. Importantly, TM0936 was not affected by increasing levels of salt. In our hands, the deaminase steadily catabolized adenosine (10 μM), even at sodium chloride concentrations as high as 2 M (Fig. 4C). This property, specific to TM0936, may facilitate future experimental set-up.To test the utility of this new assay in varied ionic conditions we measured histone methyltransferase activity. Histone tails are positively charged under physiological conditions, as they are enriched in lysine and arginine residues; this electrostatic property may partially account for binding of these substrates onto MTase target. Using TbPRMT7, we determined the kinetic behavior for H4(1–20) peptide at four phosphate concentrations (pH = 7.60, 25–100 mM). The initial rates were plotted against peptide concentrations (Fig. 4D, left). The data fit to the Michaelis–Menten equation (ESI eqn (S5)†) gives four Km values (μM) of 39 ± 3, 93 ± 5, 212 ± 7, 413 ± 8 at 25, 50, 75 and 100 mM phosphate concentrations, respectively (Fig. 4D, right). As the buffer concentration increased, so did the Km value for the histone peptide, thus reflecting a loss of affinity between substrate and PRMT7. Our data are in good agreement with the isothermal titration calorimetry experiments performed with the same PRMT7/peptide pair. While a 20-fold decrease in affinity between peptide and PRMT7 was observed when varying salt concentration from 20 to 150 mM, a 300 mM salt concentration precluded binding event (lit.,63Kd ≈ 400000 μM).
1-Step EZ-MTase is well suited for high-throughput screening
Our analytical tool provides an alternative to overcome major drawbacks from previous MTase assays (Fig. S1†); methods involved as much as four coupling enzymes, and the single deaminase activity from 1-Step EZ-MTase may decrease the risk for off-target inhibition and apparition of false positives. A high sensitivity and wide dynamic range of detection make this assay very competitive. Methyltransfer rates as low as 2 μM h−1 are detectable and the use of nanomolar MTase concentrations is also achieved, important as many eukaryotic MTases are difficult to purify in quantity sufficient for other assays (Fig. 5A and B). Very good values of screening window coefficient (Z′-factor) are obtained and always above 0.50, reaching maximum values of 0.87, 0.93, 0.80 and 0.92 for CePRMT5, TbPRMT7, NcDIM-5 and GsSDMT, respectively (Fig. 3; inserts).64 Such high values reflect the overall quality of the 1-Step EZ-MTase assay. Our analytical tool is well suited for HTS, with a signal displaying low variability and a good separation from background (e.g. SAM decomposition; Fig. S4E†). | ||
| Fig. 5 The methyltransfer reaction catalyzed by Caenorhabditis elegans PRMT5 is sustained by the 8-aza analog of SAM. (A). A typical kinetic experiment. Transferase rates were monitored at 263 nm in phosphate buffer (pH = 7.60) using various SAM concentrations (2.35–46.96 μM), saturating levels of H4(1–20) peptide (104 μM), 302 nM CePRMT5 and 4 μM coupling enzyme. Depicted rates are corrected for SAM decomposition and all adjusted to the same start absorbance. (B) Graphic representation of a kinetic experiment. Initial rates from panel A were plotted against SAM concentration and fitted to the Michaelis–Menten equation. Best fit provides both Km and kcat for this substrate (6.8 ± 0.3 μM and 31.9 ± 0.5 h−1, respectively). (C) HPLC traces of a transfer reaction using 8-aza-SAM. Fueled by 8-aza-SAM (grey trace; tR = 4.75, λmax = 280), CePRMT5 methylates H4(1–20) peptide and release 8-aza-SAH. This metabolite is instantaneously catabolized into 8-aza-SIH by TM0936 (black trace; tR = 11.60, λmax = 254). The tR retention times are in minutes, while λmax are in nm; both were determined following HPLC Method D (cf. SEI). (D) Kinetic parameters for 8-aza-SAM against CePRMT5. Reactions were monitored at 282 nm in phosphate buffer (pH = 7.60) using various 8-aza-SAM concentrations (4.16–124.8 μM), saturating levels of H4(1–20) peptide (104 μM), 1.5 μM CePRMT5 and 4 μM coupling enzyme. Initial rates were corrected for 8-aza-SAM decomposition and plotted against substrate concentration. Best fit provides Km, Ks and kcat for this substrate (35 ± 19 μM, 67 ± 42 μM and 15 ± 6 h−1, respectively). | ||
Sinefungin is a potent, yet non-selective inhibitor of MTasees, and frequently is used as a positive control during inhibitor screening. Sinefungin is a known inhibitor of the SAHH enzyme,36,65 while MTAN catabolizes sinefungin into adenine (Fig. 6A and Table S1†). Thus, most enzyme-coupled assays for MTase analysis are incompatible with this molecule (Fig. S1,† arrows 4–10). We evaluated the possibility to use sinefungin within the 1-Step EZ-MTase assay and established the reactivity profile between TM0936 and this chemical. Unlike other coupling enzymes, TM0936 displayed moderate reactivity towards this inhibitor and deamination of sinefungin was slower at pH = 6.80 than at pH = 8.00 (Fig. 6B). Indeed, the coupling enzyme deaminates the inhibitor very slowly under acidic conditions (pH ≤ 6.80, 125 nM min−1; Fig. 6C). As pH increases, so does the rate of deamination. The –NH3+ group from sinefungin is deprotonated into –NH2, thus becoming a lesser mimic of the CH3–S+ group from SAM (pH = 8.00, 760 nM min−1; Fig. 6C). This observation is in good agreement with TM0936 inability to catabolize SAM.
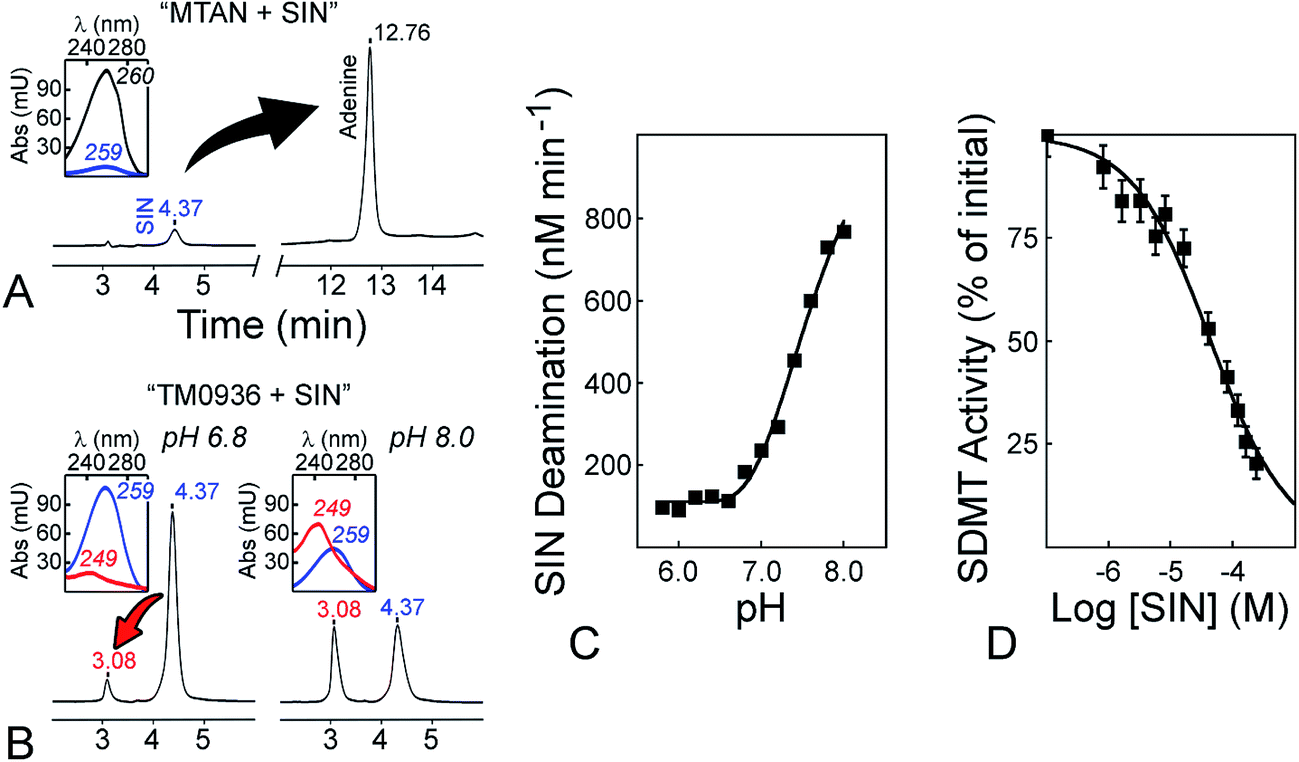 | ||
| Fig. 6 Sinefungin is a positive control compatible with the 1-Step EZ-MTase assay. (A) MTAN-based assays catabolize the methyltransferase inhibitor sinefungin (SIN). After one hour incubation with bacterial MTAN (1 μM, pH = 6.80), the universal MTase inhibitor (120 μM; tR = 4.37, λmax = 259) was quantitatively converted into adenine (tR = 12.76, λmax = 260). The tR retention times are in minutes, while λmax are in nm; both were determined following HPLC Method D (cf. ESI†). (B) Sinefungin is a poor substrate for TM0936. In comparison to MTAN, TM0936 (4 μM, one hour incubation) was a poor catalyst of the sinefungin deamination (tR = 3.08, λmax = 249). (C) pH-dependence of the reaction between sinefungin and TM0936. The coupling enzyme deaminates the inhibitor very slowly under acidic conditions (pH ≤ 6.80, 125 nM min−1; pH = 8.00, 760 nM min−1). Reaction rates were monitored at 263 nm and carried out in 50 mM phosphate buffer with 120 μM sinefungin and 4 μM TM0936. (D) Inhibition of the sarcosine/dimethylglycine methyltransferase by sinefungin. The inhibitor (0–244 μM) along with saturating levels of SAM and sarcosine (763 μM and 5 mM, respectively) were incubated with GsSDMT (195 nM) and TM0936 (4 μM) at pH = 6.80. The initial methyltransfer rates were recorded at 263 nm. Further analysis provided the inhibition constant Ki (1.8 ± 0.4 μM). | ||
Taking advantage of this substrate selectivity, we measured (pH = 6.80) the inhibition constant (Ki) for sinefungin against GsSDMT (Fig. 6D). Under our experimental conditions, sinefungin was a potent inhibitor of the reaction between sarcosine and SAM catalyzed by GsSDMT (Ki = 1.8 ± 0.4 μM). This result demonstrated the compatibility of our enzyme-coupled assay with sinefungin.
8-Aza-SAM and its application to the 1-Step EZ-MTase assay
A second mode of detection, complementary to the current UV-based assay, may present advantages. The use of a fluorescent cofactor analog may improve detection specificity compared with UV absorption. 8-Aza-adenosine is an isosteric and fluorescent analog of the nucleoside. When excited at 282 nm, this probe exhibits a strong fluorescence signature with a maximum at 360 nm.50 This characteristic is not shared with the 8-aza-inosine and this deaminated product is a weak fluorophore.50 Several SAM analogs, including the 8-aza modification, are biologically active and display affinity with the SAM-III riboswitch, the EcoRI methyltransferase and other MTases.44,66 Therefore, we anticipated this probe may be used in place of the natural cofactor.We successfully resynthesized 8-aza-SAM through reaction between 8-aza-ATP and L-methionine.66 SAM isomerizes readily at the sulfonium center and only the S(S)-epimer is biologically active. Thus, we performed a short reaction at 35 °C (enzyme:triphosphate, 1:400) to limit such epimerization and yield 8-aza-SAM (65% yield, 5% S(R)-epimer; cf. ESI† NMR analysis). Although 8-aza-ATP is commercially available (TriLink Biotechnologies, #N-1004), this molecule is cost prohibitive for this assay. Therefore, we synthesized this chemical through multiple phosphorylation of the more affordable 8-aza-adenosine. This approach is reminiscent of a strategy we previously applied towards the preparation of a C–P lyase inhibitor.67 Performed on a 50 mg scale using a 2 mL tube, the one-pot synthesis provides an easy access to high quantities of pure 8-aza-ATP (95%; cf. ESI† Enzymatic syntheses).
We further assayed the reactivity of 8-aza-SAM towards our methyltransferases. This analog was well tolerated by our candidates since only NcDIM-5 was unable to process this cofactor. Likewise, the 8-aza-SAH product of methyltransfer reactions (λmax = 280 nm) was successfully converted into the deaminated 8-aza-SIH (λmax = 254 nm; Fig. 5C). In fact, the deamination of adenosine by TM0936 is 10-fold slower than that of its 8-aza analog (Fig. 4A). Using the UV-based assay, we determined the 8-aza-SAM kinetic parameters with CePRMT5 at saturating concentration of H4 peptide (Fig. 5D). Turnover was moderately affected and the enzyme processed SAM twice as fast as its 8-aza-analog (kcat = 31.9 ± 0.5 h−1vs. 15 ± 6 h−1, respectively; Fig. 5B and D). The catalytic efficiency for 8-aza-SAM was one order of magnitude lower than that of the natural cofactor (120 M−1 s−1 and 1300 M−1 s−1, respectively). Unlike SAM, the 8-aza analog displayed significant substrate inhibition (Ks = 67 ± 42 μM; Fig. 5D). When assayed against either TbPRMT7 or GsSDMT, this inhibitory behavior of the analog was not observed (Table 1).
With the relevance of 8-aza-SAM validated, we then calibrated the fluorescence signal at 360 nm for the 8-aza-adenosine (8-aza-A) to 8-aza-inosine (8-aza-I) reaction using two excitation wavelengths (i.e. 282 and 292 nm). The 282 nm excitation provided a higher limit of detection for 8-aza-A and fluorescence signal was most intense (≈2-fold; Fig. 7A). Plotted against 8-aza-A concentrations (0–25 μM), the variation of fluorescence (ΔFLUO) observed over deaminase reaction fits a polynomial equation (ESI eqn (S10) and Table S2†). Furthermore, experiments performed at various pHs (5.00–9.50) support the weak fluorescence properties of 8-aza-I. The deaminated product is a poor fluorophore, it does not emit light under acidic conditions and its fluorescence is 10-fold weaker than that of 8-aza-A at pH = 9.50 (35 RLU μM−1vs. 380 RLU μM−1; Fig. 7A, insert). Thus, at the early stage of methyltransfer reaction (i.e. less than 10% of 8-aza-SAM consumed), the 8-aza-SIH contribution to the fluorescence is negligible and accounts for less than 1% of the signal.
 | ||
| Fig. 7 The use of 8-aza-SAM and a fluorescence-mode of detection within the 1-Step EZ-MTase assay. (A) Calibration curves for the deamination of 8-aza-adenosine by TM0936. The deamination reactions using 8-aza-A (0–25 μM) were monitored through light emission at 360 nm with both 282 and 292 nm excitation wavelengths (white and black circles, respectively). Experiments were carried out at pH = 5.00 (black curve fit) and pH = 9.50 (red curve fit). The reaction product, 8-aza-inosine (8-aza-I), is a poor fluorophore: it does not emit light under acidic conditions and its fluorescence is 10-fold weaker than that of the 8-aza-A substrate at pH = 9.50 (insert). (B) Kinetic behavior for H4(1–20) peptide against TbPRMT7 using 8-aza-SAM cofactor. The Km and kcat for H4 peptide were determined using the fluorescence-mode of detection. | ||
To establish the utility of a fluorescence mode of detection (Table S1†), we measured kinetic parameters for the transfer reaction catalyzed by TbPRMT7 using 8-aza-SAM and various concentration of H4(1–20) peptide (Fig. 7B). The data fit to the Michaelis–Menten equation (ESI eqn (S5)†) gives Km and kcat values of 71 ± 8 μM and 39 ± 2 h−1, respectively (Fig. 7B). Similar to the UV-mode of detection and to facilitate data analysis, we developed a third template spreadsheet “My 1-Step EZ-MTase Assay (FLUO) Acceptor Km.xlsx” (cf. ESI†). This file contains the calibration curves for the 8-aza-adenosyl to 8-aza-inosyl reaction at various pHs and is pre-loaded with experimental data pertinent to the kinetic behavior of H4(1–20) peptide with TbPRMT7 using 8-aza-SAM cofactor.
Applicability to biological samples: detection of GNMT activity
Glycine N-methyltransferase (GNMT) is a key component of SAM homeostasis. As a methionine-rich diet replenishes the SAM pool, the increasing concentration of methyl donor inhibits the 5,10-methylene-tetrahydrofolate reductase, thus impairing the 5-methyltetrahydrofolate (5-CH3-THF) synthesis. GNMT is a folate-binding protein and 5-CH3-THF is deleterious to its activity.68,69 Within this feedback mechanism, the alleviating GNMT inhibition promotes SAM consumption through sarcosine synthesis.70 GNMT establishes the cross talk between the one carbon folate pathway and the methionine cycle, thereby maintaining a healthy SAM/SAH ratio, which is indicative of methylator potential.71,72Development of improved assays for probing GNMT activity within biological samples is important to accelerate understanding of the interplay among metabolic pathways, energetics, epigenetics and cancer metabolism.73–75 However, classical methods using tritiated SAM, and the detection of radioactive sarcosine within crude protein extracts is a tedious and discontinuous approach. Furthermore, valuable tissue samples from animal studies may be limited and the requirement for large amounts of extract (e.g. 250 μg total protein) may be challenging when using this method.73 To overcome these drawbacks, we optimized the 1-Step EZ-MTase platform and made it compatible with biological samples. Using protein extracts from rat liver, we successfully quantitated GNMT activity within these crude biological samples (Fig. 8).
 | ||
| Fig. 8 The 1-Step EZ-MTase detects glycine N-methyltransferase activity within biological samples. (A) Experimental set-up to monitor the 263 nm signal during the reaction catalyzed by human glycine N-methyltransferase (HsGNMT). Various enzyme concentrations (0–100 nM) were mixed with SAM (75 μM) and glycine (20 mM) at pH 8.00 in presence of 4 μM TM0936. (B) Compatibility between lysis buffer and the 1-Step EZ-MTase. Calibration curve were established with and without 10% v/v of buffer used for lysis of liver samples (white cross and black square, respectively). Both curves are superimposable (i.e. 619 ± 9 nM h−1 nM−1vs. 618 ± 8 nM h−1 nM−1). (C) Detection of GNMT activity within rat liver extracts. Increasing volumes of extract (6–24 μL) were mixed at pH 8.00 with SAM, glycine and TM0936 (75 μM, 20 mM and 4 μM, respectively). The methyltransfer (MT) catalyzed by rat GNMT is detected through loss of absorbance at 263 nm. The MT rates (μM h−1) are proportional to liver extract volumes (insert). (D) The traditional discontinuous and radioactive assay for detection of GNMT activity. Using radioactive SAM, the tritiated sarcosine product of the GNMT catalyzed reaction is isolated through solid phase extraction with charcoal. (E) GNMT activities measured within rat liver extracts. Two tissue samples from a same liver were prepared (1 and 2). GNMT activity was measured with both the radioactive (R; black bars) and the 1-Step EZ-MTase coupled assay (UV; grey bars). The effect of three freeze thaw (FTa–FTc) onto GNMT activity was evaluated. | ||
The deamination reaction catalyzed by TM0936 only occurs with SAH, methylthioadenosine and adenosine.41,43 The substrate specificity of our coupling enzyme makes it compatible with the highly complex content of biological samples. Luciferase-based assays (Fig. S1,† arrows 4, 9 and 10) are not suited for such a type of sample where adenine and phosphorylated adenosine species generate high background signal. Likewise, endogenous thiol species (e.g. glutathione, homocysteine, cysteine residues from proteins) preclude the continuous detection of GNMT activity through efficient derivatization of homocysteine (e.g. Ellman's reagent, ThioGlo®-1; Fig. S1,† arrows 6 and 7). In addition to reacting with these thiol species, the 5,5′-dithio-bis-(2-nitrobenzoic acid) completely inactivates the methyltransferase upon reaction with its cysteine residues.76
In our hand, GNMT activity was not affected by the tissue lysis buffer (150 mM NaCl, 20 mM Tris–HCl pH = 7.4, 1% Triton X-100, 1 mM orthovanadate, 1 mM EDTA, 10 mM NaF, 1× protease inhibitor cocktail, and 1 mM PMSF), thus establishing the compatibility between this universal reagent and the 1-Step EZ-MTase (Fig. 8A and B). With its low 2 μM h−1 SAH-detection limit (Fig. S4D†), our analytical tool successfully senses methyltransfer catalyzed by GNMT. At 75 μM SAM concentration, endogenous glycine was not sufficient for sarcosine synthesis catalyzed by rat GNMT (Fig. S7†). Upon addition of saturating glycine (20 mM), a 2.28-fold increase of methyltransfer rate was observed (Fig. S7A;† 2.38-fold increase as monitored with the radioactive assay, Fig. S7B†). Our observations are in good agreement with the estimated 50–100 μM endogenous glycine concentration (2.37 mM in rat liver)77 and a Km value for glycine of 130 μM.78
The decrease of absorbance at 263 nm is concentration dependent and levels as low as 30 μg of total protein were sufficient to monitor sarcosine synthesis over an extended period (Fig. 8C). Our platform surpasses the performance of the tedious radioactive assay (Fig. 8D) by only requiring a fraction of the material necessary for the radioactive assay, and yet remains capable of continuously detecting the SAH methyltransferase product. Values obtained with both methods were comparable (radio assay and spectro-based assay, R and UV, respectively; Fig. 8E) and GNMT displayed similar activity across liver samples (162 ± 14 and 139 ± 21 pmol min−1 mg−1 for UV1 and UV2, respectively; 245 ± 7 and 261 ± 11 pmol min−1 mg−1 for R1 and R2, respectively). To evaluate the GNMT stability within liver extracts, we measured GNMT activity after three freeze–thaw cycles (FTa–FTc; Fig. 8E). Our results display overlapping measurements (162 ± 14, 183 ± 26 and 149 ± 21 pmol min−1 mg−1 for FTa, FTb and FTc, respectively; Fig. 8E), confirming that GNMT enzyme activity is very stable against multiple freeze–thaw cycles.
To facilitate the quantitation of GNMT activity within crude biological samples, we developed a customized template spreadsheet “MY 1-Step EZ-MTase Assay (UV) GNMT Activity BioSample.xlsx” (cf. ESI†). This file provides a framework for the user to determine GNMT activity expressed in μM h−1 and pmol min−1 mg−1 after importing raw data from a plate reader.
Conclusion
Epigenetic modifications catalyzed by methyltransferases play a central role in gene transcription and parental imprinting. A correlation between dysregulation of methylation patterns and occurrence of human diseases (e.g. cancer, diabetes) is becoming more obvious. Several methyltransferases are now validated therapeutic targets. The regulation of these enzymatic activities by specific and potent inhibitors may offer new opportunities for patients. To promote our understanding of methyltransferases and the discovery of new chemotherapeutics, we have developed a simple and straightforward assay to study this class of enzymes. Unlike anything else available, this analytical tool harnesses the power of one single protein: the SAH-deaminase TM0936. The coupling enzyme is an easily accessible and robust catalyst that allows for quick and facile determination of enzymatic rates through monitoring of absorbance at 263 nm. We demonstrate its utility by coupling it to a panel of four enzymes, including lysine and arginine methyltransferases. A 96-well plate format allows high-throughput performance, and the high Z′-factors reflect the overall quality of this assay. Furthermore, sinefungin is compatible with this coupled assay, suggesting this platform may have a significant impact on the identification of new inhibitors using high-throughput screening. We implemented a second mode of detection, providing users with the option to detect methyltransfer through monitoring the loss of fluorescence at 360 nm. This approach relies on 8-aza-SAM, a fluorescent analog of the universal methyl donor. We established the relevance of this alternative cofactor and determine kinetic properties of the PRMT7 from Trypanosoma brucei using H4(1–20) peptide. Our analytical tool detects methyltransfer rates as low as 2 μM h−1 and is able to sense low nanomolar concentrations of GNMT within crude biological samples. Overall, the 1-Step EZ-MTase surpasses the performances of other techniques while retaining a compact and simple format with broad applicability (Table S1†). Finally, to further reduce data processing, we provide the users with worksheets in which absorbance/fluorescence recordings can be imported and the templates calculate key kinetic parameters of methyltransferases (Km, kcat, activity and curve fit).Acknowledgements
We thank Dr Bruce R. Branchini (Connecticut College, New London, USA) for providing his original construct for expression of the wild-type Photinus pyralis luciferase (FLUC); Dr Vern L. Schramm (Albert Einstein College of Medicine) for sharing the pDEST14-AgAK, and pDEST14-SeMTAN vectors encoding the respective adenosine kinase from Anopheles gambiae, and S-adenosyl-L-homocysteine nucleosidase from Salmonella enterica; Dr María A. Pajares (Instituto de Investigaciones Biomedicas “Alberto Sols”, Madrid, Spain) for forwarding the pET19b (pMj1208-1) vector encoding the His-tagged methionine adenosyltransferase from Methanococcus jannaschii; Dr Rui-Ming Xu (Institute of Biophysics, Chinese Academy of Sciences, Beijing, China) for his gift of the pET21a-CePRMT5; Dr Erik W. Debler (Laboratory of Cell Biology, The Rockefeller University, New-York, USA) for providing the pET28a-TbPRMT7 encoding wild-type PRMT7 from Trypanosoma brucei; Dr Erik U. Selker (Institute of Molecular Biology, University of Oregon, Eugene, USA) for the gift of pGST-DIM5 encoding wild-type histone H3 lysine MTase from Neurospora crassa (19-318; DIM-5); the DNASU Plasmid Repository for access to clones TmCD00084735 and GsCD00383580 for expression of deaminase TM0936 and GsSDMT, respectively; Dr Luka and Wagner (Vanderbilt University Medical Center, Nashville, USA) for their gift of pET-17b-HsGNMT vector encoding wild-type human glycine N-methyltransferase. Finally, we are thankful to Dr Sean Cahill and Edward Nieves (Albert Einstein College of Medicine) for their assistance in NMR and MS analyses. This work was supported by the American Cancer Society (Robbie Sue Mudd Kidney Cancer Research Scholar Grant 124891-RSG-13-396-01-DMC) and the National Institutes of Health (GM108646), both to David Shechter. Derek M. Huffman was supported by the National Institute on Aging (R00AG037574, R56AG052981 and P30AG038072), and the American Federation for Aging Research (AFAR). Ryan O. Walters was supported by a T32 training grant from the National Institute on Aging (T32AG23475). While the Bruker Avance IIIHD 300 MHz system from the Einstein Structural NMR Resource was purchased and is supported by the Albert Einstein College of Medicine, the Bruker Avance IIIHD 600 MHz system was purchased using funds from the National Institutes of Health (1S10OD016305).References
- F. Wold, Annu. Rev. Biochem., 1981, 50, 783–814 CrossRef CAS PubMed.
- T. Hitosugi and J. Chen, Oncogene, 2014, 33, 4279–4285 CrossRef CAS PubMed.
- A. P. Lothrop, M. P. Torres and S. M. Fuchs, FEBS Lett., 2013, 587, 1247–1257 CrossRef CAS PubMed.
- G. L. Cantoni, J. Am. Chem. Soc., 1952, 74, 2942–2943 CrossRef CAS.
- J. Axelrod and R. Tomchick, J. Biol. Chem., 1958, 233, 702–705 CAS.
- G. L. Cantoni, J. Biol. Chem., 1951, 189, 203–216 CAS.
- J. Song, M. Teplova, S. Ishibe-Murakami and D. J. Patel, Science, 2012, 335, 709–712 CrossRef CAS PubMed.
- S. S. Wolf, Cell. Mol. Life Sci., 2009, 66, 2109–2121 CrossRef CAS PubMed.
- Y. Yang and M. T. Bedford, Nat. Rev. Cancer, 2013, 13, 37–50 CrossRef CAS PubMed.
- A. J. Ruthenburg, C. D. Allis and J. Wysocka, Mol. Cell, 2007, 25, 15–30 CrossRef CAS PubMed.
- H. Chen, B. Lorton, V. Gupta and D. Shechter, Oncogene, 2017, 36, 373–386 CrossRef CAS PubMed.
- P. Chi, C. D. Allis and G. G. Wang, Nat. Rev. Cancer, 2010, 10, 457–469 CrossRef CAS PubMed.
- C. Sawan and Z. Herceg, Adv. Genet., 2010, 70, 57–85 CAS.
- X. Bao, S. Zhao, T. Liu, Y. Liu, Y. Liu and X. Yang, J. Histochem. Cytochem., 2013, 61, 206–217 CrossRef CAS PubMed.
- K. Mathioudaki, A. Scorilas, A. Ardavanis, P. Lymberi, E. Tsiambas, M. Devetzi, A. Apostolaki and M. Talieri, Tumour Biol., 2011, 32, 575–582 CrossRef CAS PubMed.
- C. Milite, A. Feoli, M. Viviano, D. Rescigno, A. Cianciulli, A. L. Balzano, A. Mai, S. Castellano and G. Sbardella, Clin. Epigenet., 2016, 8, 102 CrossRef PubMed.
- A. T. Nguyen and Y. Zhang, Genes Dev., 2011, 25, 1345–1358 CrossRef CAS PubMed.
- L. Wang, Z. Zhao, M. B. Meyer, S. Saha, M. Yu, A. Guo, K. B. Wisinski, W. Huang, W. Cai, J. W. Pike, M. Yuan, P. Ahlquist and W. Xu, Cancer Cell, 2014, 25, 21–36 CrossRef CAS PubMed.
- S. R. Daigle, E. J. Olhava, C. A. Therkelsen, A. Basavapathruni, L. Jin, P. A. Boriack-Sjodin, C. J. Allain, C. R. Klaus, A. Raimondi, M. P. Scott, N. J. Waters, R. Chesworth, M. P. Moyer, R. A. Copeland, V. M. Richon and R. M. Pollock, Blood, 2013, 122, 1017–1025 CrossRef CAS PubMed.
- E. Chan-Penebre, K. G. Kuplast, C. R. Majer, P. A. Boriack-Sjodin, T. J. Wigle, L. D. Johnston, N. Rioux, M. J. Munchhof, L. Jin, S. L. Jacques, K. A. West, T. Lingaraj, K. Stickland, S. A. Ribich, A. Raimondi, M. P. Scott, N. J. Waters, R. M. Pollock, J. J. Smith, O. Barbash, M. Pappalardi, T. F. Ho, K. Nurse, K. P. Oza, K. T. Gallagher, R. Kruger, M. P. Moyer, R. A. Copeland, R. Chesworth and K. W. Duncan, Nat. Chem. Biol., 2015, 11, 432–437 CrossRef CAS PubMed.
- A. Finley and R. A. Copeland, Chem. Biol., 2014, 21, 1196–1210 CrossRef CAS PubMed.
- H. Gowher and A. Jeltsch, J. Mol. Biol., 2001, 309, 1201–1208 CrossRef CAS PubMed.
- M. S. Kareta, Z. M. Botello, J. J. Ennis, C. Chou and F. Chedin, J. Biol. Chem., 2006, 281, 25893–25902 CrossRef CAS PubMed.
- B. B. Suh-Lailam and J. M. Hevel, Anal. Biochem., 2010, 398, 218–224 CrossRef CAS PubMed.
- C. Wilczek, R. Chitta, E. Woo, J. Shabanowitz, B. T. Chait, D. F. Hunt and D. Shechter, J. Biol. Chem., 2011, 286, 42221–42231 CrossRef CAS PubMed.
- N. Gauthier, M. Caron, L. Pedro, M. Arcand, J. Blouin, A. Labonte, C. Normand, V. Paquet, A. Rodenbrock, M. Roy, N. Rouleau, L. Beaudet, J. Padros and R. Rodriguez-Suarez, J. Biomol. Screening, 2012, 17, 49–58 CrossRef CAS PubMed.
- K. Devkota, B. Lohse, C. N. Jakobsen, J. Berthelsen and R. P. Clausen, Anal. Biochem., 2015, 476, 78–80 CrossRef CAS PubMed.
- G. Ibanez, J. L. McBean, Y. M. Astudillo and M. Luo, Anal. Biochem., 2010, 401, 203–210 CrossRef CAS PubMed.
- I. Hemeon, J. A. Gutierrez, M. C. Ho and V. L. Schramm, Anal. Chem., 2011, 83, 4996–5004 CrossRef CAS PubMed.
- K. M. Dorgan, W. L. Wooderchak, D. P. Wynn, E. L. Karschner, J. F. Alfaro, Y. Cui, Z. S. Zhou and J. M. Hevel, Anal. Biochem., 2006, 350, 249–255 CrossRef CAS PubMed.
- S. Duchin, Z. Vershinin, D. Levy and A. Aharoni, Epigenet. Chromatin, 2015, 8, 56 CrossRef PubMed.
- C. L. Hendricks, J. R. Ross, E. Pichersky, J. P. Noel and Z. S. Zhou, Anal. Biochem., 2004, 326, 100–105 CrossRef CAS PubMed.
- E. Collazo, J. F. Couture, S. Bulfer and R. C. Trievel, Anal. Biochem., 2005, 342, 86–92 CrossRef CAS PubMed.
- C. Wang, S. Leffler, D. H. Thompson and C. A. Hrycyna, Biochem. Biophys. Res. Commun., 2005, 331, 351–356 CrossRef CAS PubMed.
- T. A. Klink, M. Staeben, K. Twesten, A. L. Kopp, M. Kumar, R. S. Dunn, C. A. Pinchard, K. M. Kleman-Leyer, M. Klumpp and R. G. Lowery, J. Biomol. Screening, 2012, 17, 59–70 CrossRef CAS PubMed.
- K. M. Drake, V. G. Watson, A. Kisielewski, R. Glynn and A. D. Napper, Assay Drug Dev. Technol., 2014, 12, 258–271 CrossRef CAS PubMed.
- E. S. Burgos, S. A. Gulab, M. B. Cassera and V. L. Schramm, Anal. Chem., 2012, 84, 3593–3598 CrossRef CAS PubMed.
- T. L. Graves, Y. Zhang and J. E. Scott, Anal. Biochem., 2008, 373, 296–306 CrossRef CAS PubMed.
- J. K. Coward and F. Y. Wu, Anal. Biochem., 1973, 55, 406–410 CrossRef CAS PubMed.
- F. Schlenk, C. R. Zydek-Cwick and N. K. Hutson, Arch. Biochem. Biophys., 1971, 142, 144–149 CrossRef CAS PubMed.
- J. C. Hermann, R. Marti-Arbona, A. A. Fedorov, E. Fedorov, S. C. Almo, B. K. Shoichet and F. M. Raushel, Nature, 2007, 448, 775–779 CrossRef CAS PubMed.
- R. Guan, M. C. Ho, R. F. Frohlich, P. C. Tyler, S. C. Almo and V. L. Schramm, Biochemistry, 2012, 51, 9094–9103 CrossRef CAS PubMed.
- D. S. Hitchcock, H. Fan, J. Kim, M. Vetting, B. Hillerich, R. D. Seidel, S. C. Almo, B. K. Shoichet, A. Sali and F. M. Raushel, J. Am. Chem. Soc., 2013, 135, 13927–13933 CrossRef CAS PubMed.
- R. T. Borchardt, J. A. Huber and Y. S. Wu, J. Med. Chem., 1974, 17, 868–873 CrossRef CAS PubMed.
- K. Thomas, A. M. Haapalainen, E. S. Burgos, G. B. Evans, P. C. Tyler, S. Gulab, R. Guan and V. L. Schramm, Biochemistry, 2012, 51, 7541–7550 CrossRef CAS PubMed.
- E. S. Burgos, C. Wilczek, T. Onikubo, J. B. Bonanno, J. Jansong, U. Reimer and D. Shechter, J. Biol. Chem., 2015, 290, 9674–9689 CrossRef CAS PubMed.
- J. J. Christensen, J. H. Rytting and R. M. Izatt, Biochemistry, 1970, 9, 4907–4913 CrossRef CAS PubMed.
- J. Saevels, A. Zanoletty Perez, A. Salvat Jauma, A. Van Schepdael and J. Hoogmartens, Chromatographia, 1998, 47, 225–229 CAS.
- G. Cercignani, Anal. Biochem., 1987, 166, 418–423 CrossRef CAS PubMed.
- J. Wierzchowski, B. Wielgus-Kutrowska and D. Shugar, Biochim. Biophys. Acta, 1996, 1290, 9–17 CrossRef.
- J. Wierzchowski, J. M. Antosiewicz and D. Shugar, Mol. BioSyst., 2014, 10, 2756–2774 RSC.
- J. F. Morrison and C. T. Walsh, Adv. Enzymol. Relat. Areas Mol. Biol., 1988, 61, 201–301 CAS.
- H. Gowher, X. Zhang, X. Cheng and A. Jeltsch, Anal. Biochem., 2005, 342, 287–291 CrossRef CAS PubMed.
- J. G. McCoy, L. J. Bailey, Y. H. Ng, C. A. Bingman, R. Wrobel, A. P. Weber, B. G. Fox and G. N. Phillips Jr, Proteins, 2009, 74, 368–377 CrossRef CAS PubMed.
- Y. Wang, H. Akiyama, K. Terakado and T. Nakatsu, Sci. Rep., 2013, 3, 2490 CrossRef PubMed.
- V. Vanek, M. Budesinsky, P. Kabeleova, M. Sanda, M. Kozisek, I. Hanclova, J. Mladkova, J. Brynda, I. Rosenberg, M. Koutmos, T. A. Garrow and J. Jiracek, J. Med. Chem., 2009, 52, 3652–3665 CrossRef CAS PubMed.
- A. Sakamoto and N. Murata, Plant, Cell Environ., 2002, 25, 163–171 CrossRef CAS.
- A. Nyyssola, T. Reinikainen and M. Leisola, Appl. Environ. Microbiol., 2001, 67, 2044–2050 CrossRef CAS PubMed.
- R. Waditee, Y. Tanaka, K. Aoki, T. Hibino, H. Jikuya, J. Takano, T. Takabe and T. Takabe, J. Biol. Chem., 2003, 278, 4932–4942 CrossRef CAS PubMed.
- J. P. Kallio, J. Janis, A. Nyyssola, N. Hakulinen and J. Rouvinen, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2009, 65, 805–808 CrossRef CAS PubMed.
- Y. R. Lee, T. S. Lin, S. J. Lai, M. S. Liu, M. C. Lai and N. L. Chan, Sci. Rep., 2016, 6, 38071 CrossRef CAS PubMed.
- T. F. Smith, C. Gaitatzes, K. Saxena and E. J. Neer, Trends Biochem. Sci., 1999, 24, 181–185 CrossRef CAS PubMed.
- E. W. Debler, K. Jain, R. A. Warmack, Y. Feng, S. G. Clarke, G. Blobel and P. Stavropoulos, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2068–2073 CrossRef CAS PubMed.
- J. H. Zhang, T. D. Chung and K. R. Oldenburg, J. Biomol. Screening, 1999, 4, 67–73 CrossRef PubMed.
- K. Fabianowska-Majewska, J. A. Duley and H. A. Simmonds, Biochem. Pharmacol., 1994, 48, 897–903 CrossRef CAS PubMed.
- O. M. Ottink, F. H. Nelissen, Y. Derks, S. S. Wijmenga and H. A. Heus, Anal. Biochem., 2010, 396, 280–283 CrossRef CAS PubMed.
- S. S. Kamat, E. S. Burgos and F. M. Raushel, Biochemistry, 2013, 52, 7366–7368 CrossRef CAS PubMed.
- R. J. Cook and C. Wagner, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 3631–3634 CrossRef CAS.
- C. Wagner, W. T. Briggs and R. J. Cook, Biochem. Biophys. Res. Commun., 1985, 127, 746–752 CrossRef CAS PubMed.
- Z. Luka, S. H. Mudd and C. Wagner, J. Biol. Chem., 2009, 284, 22507–22511 CrossRef CAS PubMed.
- C. Huidobro, A. F. Fernandez and M. F. Fraga, Cell. Mol. Life Sci., 2013, 70, 1543–1573 CrossRef CAS PubMed.
- S. C. Lu and J. M. Mato, Physiol. Rev., 2012, 92, 1515–1542 CrossRef CAS PubMed.
- M. J. Rowling, M. H. McMullen and K. L. Schalinske, J. Nutr., 2002, 132, 365–369 CAS.
- Y. J. Cha, D. H. Kim, W. H. Jung and J. S. Koo, Int. J. Clin. Exp. Pathol., 2014, 7, 7824–7833 Search PubMed.
- Z. Heger, M. A. Merlos Rodrigo, P. Michalek, H. Polanska, M. Masarik, V. Vit, M. Plevova, D. Pacik, T. Eckschlager, M. Stiborova and V. Adam, PLoS One, 2016, 11, e0165830 Search PubMed.
- M. Fujioka, Y. Takata, K. Konishi and H. Ogawa, Biochemistry, 1987, 26, 5696–5702 CrossRef CAS PubMed.
- D. L. Bloxam, Br. J. Nutr., 1972, 27, 233–247 CrossRef CAS PubMed.
- H. Ogawa and M. Fujioka, J. Biol. Chem., 1982, 257, 3447–3452 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental materials and methods, characterization of all compounds (1H–1H COSY NMR, 1H–13C edited HSQC NMR, MS analysis), supplementary figures and tables, worksheets for the 1-Step EZ-MTase assay using both UV- and fluorescence-detection mode, a worksheet for the determination of glycine N-methyltransferase activity within biological samples. Samples of purified TM0936 will be distributed upon request. See DOI: 10.1039/c7sc02830j |
| This journal is © The Royal Society of Chemistry 2017 |