
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, crystal structure and atomic force microscopy study of thioether ligated D,L-cyclic antimicrobial peptides against multidrug resistant Pseudomonas aeruginosa†
Runze
He‡
a,
Ivan
Di Bonaventura‡
a,
Ricardo
Visini
a,
Bee-Ha
Gan
a,
Yongchun
Fu
a,
Daniel
Probst
a,
Alexandre
Lüscher
b,
Thilo
Köhler
b,
Christian
van Delden
b,
Achim
Stocker
a,
Wenjing
Hong
 ac,
Tamis
Darbre
a and
Jean-Louis
Reymond
*a
ac,
Tamis
Darbre
a and
Jean-Louis
Reymond
*a
aDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: jean-louis.reymond@dcb.unibe.ch
bDepartment of Microbiology and Molecular Medicine, University of Geneva, Service of Infectious Diseases, University Hospital of Geneva, Geneva, Switzerland
cState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
First published on 4th September 2017
Abstract
Here we report a new family of cyclic antimicrobial peptides (CAMPs) targeting MDR strains of Pseudomonas aeruginosa. These CAMPs are cyclized via a xylene double thioether bridge connecting two cysteines placed at the ends of a linear amphiphilic alternating D,L-sequence composed of lysines and tryptophans. Investigations by transmission electron microscopy (TEM), dynamic light scattering and atomic force microscopy (AFM) suggest that these peptide macrocycles interact with the membrane to form lipid–peptide aggregates. Amphiphilic conformations compatible with membrane disruption are observed in high resolution X-ray crystal structures of fucosylated derivatives in complex with lectin LecB. The potential for optimization is highlighted by N-methylation of backbone amides leading to derivatives with similar antimicrobial activity but lower hemolysis.
Introduction
There is an urgent need to develop new antibiotics to control multidrug resistant (MDR) bacteria. Here we report the design of new cyclic antimicrobial peptides (CAMPs, e.g.RH11, Scheme 1) against the Gram-negative opportunistic bacterium Pseudomonas aeruginosa, which represents a major cause of life threatening infections in hospitals today.1,2 CAMPs are examples of peptide macrocycles, one of the most important family of pharmacologically active natural products at sizes beyond small molecules.3–6 Peptide macrocycles are attractive because they can be easily prepared by ordered oligomerization of α-amino acids or related building blocks such as N-alkyl glycines7,8 using solid-phase peptide synthesis followed by cyclization,9 and their macrocyclic structure allows an organized positioning of pharmacophores favoring biological activity.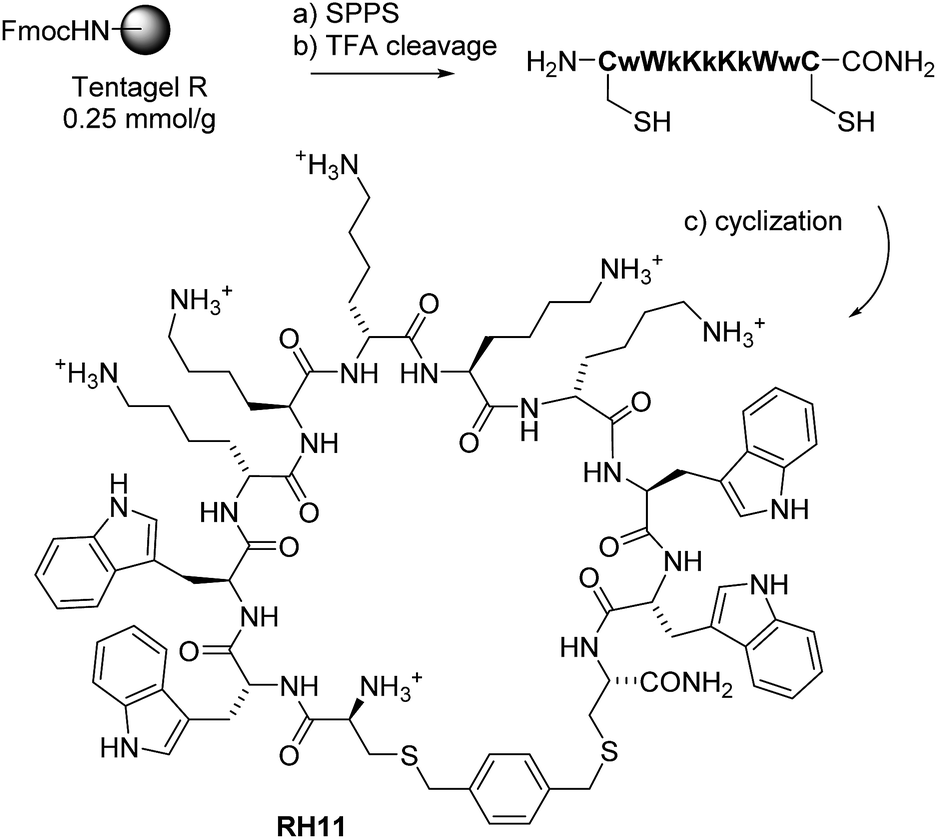 | ||
Scheme 1 Synthesis of RH11. Conditions: (a) SPPS: Fmoc deprotection: piperidine/NMP 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (v/v), 20 min; amino acid coupling: 3 eq. Fmoc-aa-OH, 3 eq. PyBOP, 5 eq. DIEA in NMP, 2–4 hours; (b) cleavage: TFA/TIS/H2O (95/4/1/); (c) cyclization: KI (20 eq.) DIPEA (5 eq.) α,α′-dichloro-p-xylene (3 eq.) in CH3CN/H2O (1:1, v/v), RT, 1–2 hours under Argon. :4 (v/v), 20 min; amino acid coupling: 3 eq. Fmoc-aa-OH, 3 eq. PyBOP, 5 eq. DIEA in NMP, 2–4 hours; (b) cleavage: TFA/TIS/H2O (95/4/1/); (c) cyclization: KI (20 eq.) DIPEA (5 eq.) α,α′-dichloro-p-xylene (3 eq.) in CH3CN/H2O (1:1, v/v), RT, 1–2 hours under Argon. | ||
Notable CAMPs against P. aeruginosa include the natural product polymyxin, currently a last resort antibiotic against MDR Gram-negative bacteria acting primarily by membrane disruption but against which resistance is appearing,10,11 the natural disulfide bridged cyclic dodecapeptide bactenecin and analogs,12 and POL7080, a recently developed synthetic CAMP blocking P. aeruginosa membrane assembly via the lipid transport protein lptD.13 Synthetic polymyxin analogs with fewer positive charges were recently shown to have reduced toxicity but also weaker antimicrobial activity.14,15
Here we decided to take a fresh start and search for entirely new CAMPs, keeping focused on membrane disruption because this represents the favored mechanism of action of CAMPs and their parent linear antimicrobial peptides (AMPs).16–20 In contrast to targeted protein binding by peptide macrocycles, which requires affinity selection from very large genetically encoded combinatorial libraries,21,22 membrane disruptive activities can be approached by design. Indeed, amphiphilic structures combining cationic residues potentially binding to the anionic phosphate groups of phospholipids with hydrophobic residues facilitating membrane penetration often disrupt biological membranes.
Amphiphilic CAMP structures can be observed directly in the crystal structures of the cyclic antimicrobial decapeptides Gramicidin S forming an intramolecular β-sheet23–25 and Tyrocidin A existing as a hydrogen-bonded amphiphilic dimer in the crystal.26 Many CAMPs have been discovered by modifying natural products such as Gramicidin S27–29 and bactenecin,12 as well as by cyclizing naturally occurring linear amphiphilic antimicrobial hexa- to octa-peptides featuring arginine–tryptophan as cationic–hydrophobic pair.30–33 Further examples of amphiphilic designs include cyclic peptoids with alternating aromatic and primary alkylamine side chains,34,35 and cyclic hexa- and octa-peptides featuring an alternating D,L-architecture forming supramolecular tubular aggregates.36–39 Strikingly however most of these CAMPs were developed against Gram-positive strains such as Staphylococcus aureus and some Gram-negative strains such as E. coli, but apart from bactenecin, polymyxin and POL7080 discussed above, only very few of them were reported to be active against P. aeruginosa.
In our attempt to discover new CAMPs against P. aeruginosa, we selected peptide macrocycles cyclized via a xylene double thioether bridge connecting a pair of cysteine residues placed at both ends of a linear sequence (Scheme 1). This xylene linker allows for a high-yielding cyclization40 and has been reported for synthetic epitope design41,42 and genetically encoded libraries,43 but has not been used previously in CAMPs. We adopted Ghadiri's alternating D,L-sequence design36–39 to promote aggregation and serum stability and used lysine/tryptophan as cationic/hydrophobic residue pair, an unprecedented combination for CAMPs.
As detailed below, reaching a size of 11 residues was essential to confer activity against P. aeruginosa including MDR clinical isolates. The optimal CAMP RH11 displays good stability in serum, moderate hemolysis, and the hallmarks of membrane disruptive activity. Imaging by transmission electron microscopy (TEM) and atomic force microscopy (AFM) shows that RH11 disrupts the bacterial membrane to form doughnut shaped structures suggesting the formation of lipid–peptide aggregates. Its amphiphilic structure is furthermore supported by high resolution X-ray crystal structures of fucosylated derivatives of close analogs in complex with lectin LecB of P. aeruginosa,44 a method which we have recently found to provide access to structural information on molecules otherwise difficult to crystallize.45,46 The structure also shows that backbone amides are not engaging in critical hydrogen bonds, leading to the exploration of backbone N-methylation as a route for property improvements.
Results and discussion
Design, synthesis and optimization
To build our cyclic peptides we focused on sequences containing an approximately even ratio of hydrophobic and cationic residues since this composition generally favors antimicrobial activity.16–20 As components of the amphiphilic sequence we selected tryptophan as hydrophobic residue because it is most often used in designed synthetic AMPs including CAMPs.30–33 On the other hand we chose lysine as cationic amino acid since it occurs most frequently in natural AMPs, and because lysine cationic side chains had served us well in obtaining antimicrobial peptide dendrimers and bicyclic peptides with high activities against P. aeruginosa.47–49 Despite of the fact that arginine is very prevalent in synthetic membrane active peptides, we did not want to use this residue because it also promotes cell penetration in eukaryotic cells and hemolysis.32,50–54 Finally we expected that the xylene bridge would play the role of a hydrophobic residue enhancing membrane interactions and possibly aggregation and might therefore positively contribute to antimicrobial effects.To probe the size requirement of our amphiphilic sequence design we initially prepared a small series of CAMPs between 6 and 12 residues (RH1–RH7, Table 1). We tested their possible antimicrobial activity by determining minimal inhibitory concentrations (MIC) in a broth dilution assay against our target Gram-negative bacterium P. aeruginosa PAO1, as well as against Bacillus subtilis BR151, a Gram-positive strain generally sensitive to membrane disruptive compounds. The shorter sequences RH1–RH5 showed activity against B. Subtilis BR151 but were inactive against PAO1. By contrast the larger cyclic undecapeptide RH6 showed significant activity against PAO1 (MIC = 16 μg mL−1), while retaining a robust activity against B. subtilis (MIC = 2 μg mL−1). Activity was preserved upon replacement of the para-xylene bridge with meta- or ortho-xylene bridges in RH6m and RH6o, but was strongly decreased in the disulfide-bridged analog RH6ss, indicating that the xylene bridge was necessary for activity, presumably due to its contribution to hydrophobicity. Activity was slightly reduced with the phenylalanine analog RH8 and lost with the leucine analog RH9, showing that tryptophan was the optimal hydrophobic residue for RH6. Activity was preserved when placing the p-xylene bridge at the center of the penta-lysine sequence in RH10, and slightly increased when placing the bridge at the center of the tetra-tryptophan sequence in RH11, which simultaneously reduced its hemolytic effect.
| Peptide | Hyd. | Pos.a | Linear sequenceb | MICc PA01 | MICc BR151 | MHCd |
|---|---|---|---|---|---|---|
| a Hyd. = number of hydrophobic residues including cysteines, Pos. = positive charges (from lysine side chains and N-terminus). b Sequences are cyclized with α,α′-dichloro-para-xylene. Upper case letters are used for L-amino acids and lower case letters for D-amino acids, the C-terminal cysteine is carboxamide (CONH2) from Rink amide synthesis. c The MIC (minimal inhibitory concentration) in μg mL−1 was measured by 1/2 serial dilutions in Müller–Hinton (MH) broth in 96 well plates after incubation for 16–18 hours at 37 °C. The MIC values were measured in independent triplicates with at least two experiments giving the same value. d MHC = minimal hemolytic concentration in μg mL−1, measured by 1/2 serial dilutions on human red blood cells (HRBCs) in 96 well plates. The MHC values were measured in duplicates giving the same value. n.d. = not determined. e Cyclized with α,α′-dichloro-meta-xylene. f Cyclized with α,α′-dichloro-ortho-xylene. g Cyclized with disulfide bridge. h Me for N-methylation of the peptide bond. | ||||||
| Pmx | 4 | 5 | Polymyxin B | 0.5 | 16 | >2000 |
| RH1 | 3 | 4 | cWkKkC | >64 | 64 | 1000 |
| RH2 | 4 | 4 | CwWkKkC | 64 | 8 | 125 |
| RH3 | 4 | 5 | cWwKkKkC | 64 | 16 | 500 |
| RH4 | 5 | 5 | CwWwKkKkC | 64 | 4 | 125 |
| RH5 | 5 | 6 | CwWwKkKkKC | >64 | 2 | 1000 |
| RH6 | 6 | 6 | CwWwWkKkKkC | 16 | 2 | 31 |
| RH7 | 6 | 7 | cWwWwKkKkKkC | 32 | 2 | 31 |
| RH6m | 6 | 6 | CwWwWkKkKkCe | 16 | 2 | 31 |
| RH6o | 6 | 6 | CwWwWkKkKkCf | 16 | 2 | 31 |
| RH6ss | 6 | 6 | CwWwWkKkKkCg | 64 | 8 | n.d. |
| RH8 | 6 | 6 | CfFfFkKkKkC | 32 | 2 | 500 |
| RH9 | 6 | 6 | ClLlLkKkKkC | >64 | 16 | >2000 |
| RH10 | 6 | 6 | CkKkWwWwKkC | 16 | 1 | 250 |
| RH11 | 6 | 6 | CwWkKkKkWwC | 8 | 1 | 125 |
| dRH11 | 6 | 6 | cWwKkKkKwWc | 8 | 1 | n.d. |
| RH11m | 6 | 6 | CwWkKkKkWwCe | 8 | 1 | n.d. |
| RH11o | 6 | 6 | CwWkKkKkWwCf | 8 | 1 | n.d. |
| RH12 | 6 | 6 | CkWkWkWkWkC | >64 | 16 | 1000 |
| RH13 | 7 | 6 | CWwKkKkKwWwC | 8 | 1 | 125 |
| dRH13 | 7 | 6 | cwWkKkKkWwWc | 8 | 1 | n.d. |
| RH14 | 7 | 5 | CwWkKkKwWwC | 32 | 2 | 8 |
| RH15 | 5 | 7 | CwKkKkKkWwC | 64 | 2 | 250 |
| RH16 | 6 | 6 | ClLkKkKkLlC | 64 | 4 | >2000 |
| RH17 | 6 | 6 | CWWKKKKKWWC | 16 | 1 | 250 |
| RH18 | 6 | 6 | CMewWkKkKkWMewCh | 16 | 4 | 8 |
| RH19 | 6 | 6 | CwMeWkKkKkWMewCh | 16 | 4 | 1000 |
| RH20 | 6 | 6 | CwWMekKkKMekWwCh | 16 | 2 | 1000 |
An optimum in sequence design was apparently reached with RH11. Sequence amphiphilicity was essential for its activity as indicated by the almost complete loss of activity in analog RH12 with alternating tryptophan and lysine residues. RH12 was non hemolytic, indicating that sequence amphiphilicity caused the hemolytic effect. Extending the hydrophobic part by one tryptophan preserved antimicrobial activity in RH13, while activity decreased when perturbing the lysine/tryptophan ratio in RH14 or RH15. Exchanging the four tryptophans against less hydrophobic leucines in RH16 also reduced antimicrobial activity, and completely abolished hemolysis. Interestingly RH17 containing only L-amino acids was as active as RH11, showing that the alternating D,L-architecture was not necessary for activity. Nevertheless RH17 was unstable in serum while RH11 was resistant to degradation, implying that the D,L-sequence design was required for stability (Fig. 1A). Interestingly the most active CAMPs RH11 and RH13 were comparably active as their enantiomers dRH11 and dRH13.
 | ||
| Fig. 1 (A) D,L-Peptides RH6 (red), RH11 (blue) and all L-peptide RH17 (black) (at c = 685 μg mL−1) were incubated in human serum over 24 hours and remaining peptide was quantified by LC/MS. (B) PAO1 membrane integrity was measured by monitoring DNA release using the SYTOX dye and fluorescence spectroscopy. PAO1 cells in minimal salts medium were treated with SYTOX and each of the antibiotics/AMPs (added at time = 0) at a concentration of 4 × MIC. The fluorescence change is shown during 60 min. (C) TEM images showing morphology of PAO1 cells incubated for 60 min without compound (left), or with 10 × MIC polymyxin B (center) and 10 × MIC RH11 (right). (D) Particle size distribution observed by DLS in solutions of 100 μg mL−1E. coli LPS without or with antibiotic compound at 100 μg mL−1. Each sample was measured in triplicate and the experiments were repeated three times. | ||
Activity against MDR strains
The three most active CAMPs RH6, RH11 and RH13 were further evaluated against clinical respiratory tract isolates of P. aeruginosa. The isolates ZEM9A, ZEM1A, PEJ2.6, and PEJ9.1 (provided by CHUV, Lausanne, Switzerland) were resistant against two to three different classes of antibiotics, including β-lactams (cephalosporins, carbapenems), aminoglycosides (amikacin, gentamicin, tobramycin), or quinolones (norfloxacin, ciprofloxacin). Because our CAMPs were quite active on the Gram-positive test strain B. subtilis, we also included the Gram-positive pathogens S. aureus Newman (methicillin sensitive) and S. aureus COL (methicillin-resistant), against which many CAMPs are active.Both optimized CAMPs RH11 and RH13 showed good activities (MIC ≤ 16 μg mL−1) against three of the four MDR strains of P. aeruginosa, although they were much less active than polymyxin B. Both compounds were also slightly active against S. aureus strains, further illustrating a significant activity against Gram-positive strains contrasting with polymyxin B, which is specific for Gram-negative bacteria. RH11 and RH13 also showed moderate activity against Acinetobacter baumannii, but not against Escherichia coli or Klebsiella pneumoniae (Table 2).
| RH6 | RH11 | RH13 | Polymyxin B | |
|---|---|---|---|---|
| a MICs were determined by serial two-fold dilutions in Mueller–Hinton (MH) broth in 96 well plates after incubation for 16–18 hours at 37 °C. Experiments were performed in triplicates with at least two independent experiments giving the same value. Resistance profiles in Table S1.† MSSA: methicillin-sensitive S. aureus, MRSA: methicillin-resistant S. aureus. | ||||
| ZEM1.A | 32 | 16–32 | 8–16 | 0.125 |
| ZEM9.A | 8 | 8–16 | 8 | 4 |
| PEJ2.6 | 16–32 | 4–8 | 8–16 | 0.5 |
| PEJ9.1 | >64 | 64 | 32 | 0.5 |
| S. aureus Newman (MSSA) | 64 | 16–32 | 32 | >64 |
| S. aureus COL (MRSA) | 32–64 | 16–32 | 32 | >64 |
| A. baumannii 19606 | n.d. | 16 | 16 | 0.125 |
| E. coli (W3110) | n.d. | 32 | 64 | 0.125 |
| K. pneumoniae s. | n.d. | >64 | >64 | 1 |
Membrane disruptive effects
RH11 behaved as a typical membrane disruptive compound. DNA was rapidly released from bacteria upon treatment with the peptide as measured by the SYTOX dye, an effect clearly linked to bacterial killing since the inactive analog RH12 did not induce DNA release (Fig. 1B).13,55 Furthermore RH11 induced fluorescein release from anionic phosphatidylglycerol vesicles mimicking the bacterial membrane (Fig. S1†). Transmission electron microscopy (TEM) images of P. aeruginosa cells exposed to RH11 were comparable to those obtained with polymyxin B,56 and showed the formation of membrane protrusions that suggest membrane disruption (Fig. 1C and S2†). The size of lipopolysaccharide (LPS) aggregates measured by dynamic light scattering (DLS) increased in the presence of RH11 as for polymyxin B, but were unaltered by tobramycin, a non-membrane active antibiotic, suggesting that the protrusions observed by TEM represented LPS–peptide aggregates (Fig. 1D and S3†).Atomic force microscopy
The TEM and DLS data suggested that RH11 interacts with the bacterial membrane to form lipid–peptide aggregates. To gain an additional insight into the action of RH11 on the bacterial membrane, we performed an atomic force microscopy (AFM) study. Unfortunately, no satisfactory AFM images could be obtained using P. aeruginosa. However, treatment of B. subtilis with RH11 induced the formation of highly homogeneous doughnut-like structures with a diameter around 80 nm and height around 0.8 nm (Fig. 2B–D). Control experiments showed that these doughnut-like structures were not formed with either B. subtilis or RH11 alone (Fig. S4†). Similar doughnut-like structures with 85–90 nm of diameter and 0.6 nm of height were also observed upon treatment of phosphatidylglycerol lipid vesicles with RH11 (Fig. 2E and F). Such regular nanostructures were not observed previously in AFM studies of bacterial and lipid vesicles with AMPs such as indolicidin,57 polymyxin B,58 magainin 2 (ref. 59) or endotoxin-binding Sushi peptides.60 On the other hand similar doughnut-like structures have been reported previously in AFM studies of designed amphiphilic peptide based surfactants in the absence of any membrane,61,62 however the objects were approximately double in size. In our studies the dimensions of the doughnut-like structures depend on the membrane, which suggests that they represent aggregates of membrane lipids and RH11. We propose that these structures are comparable to the protrusions observed by TEM which have a comparable size of 50–100 nm (Fig. 1C), and are formed by aggregation of the peptide at the bacterial membrane or lipid vesicle surface followed by detachment when a critical size has been reached, leading to membrane lysis and cell death in the case of bacteria (Fig. S5†).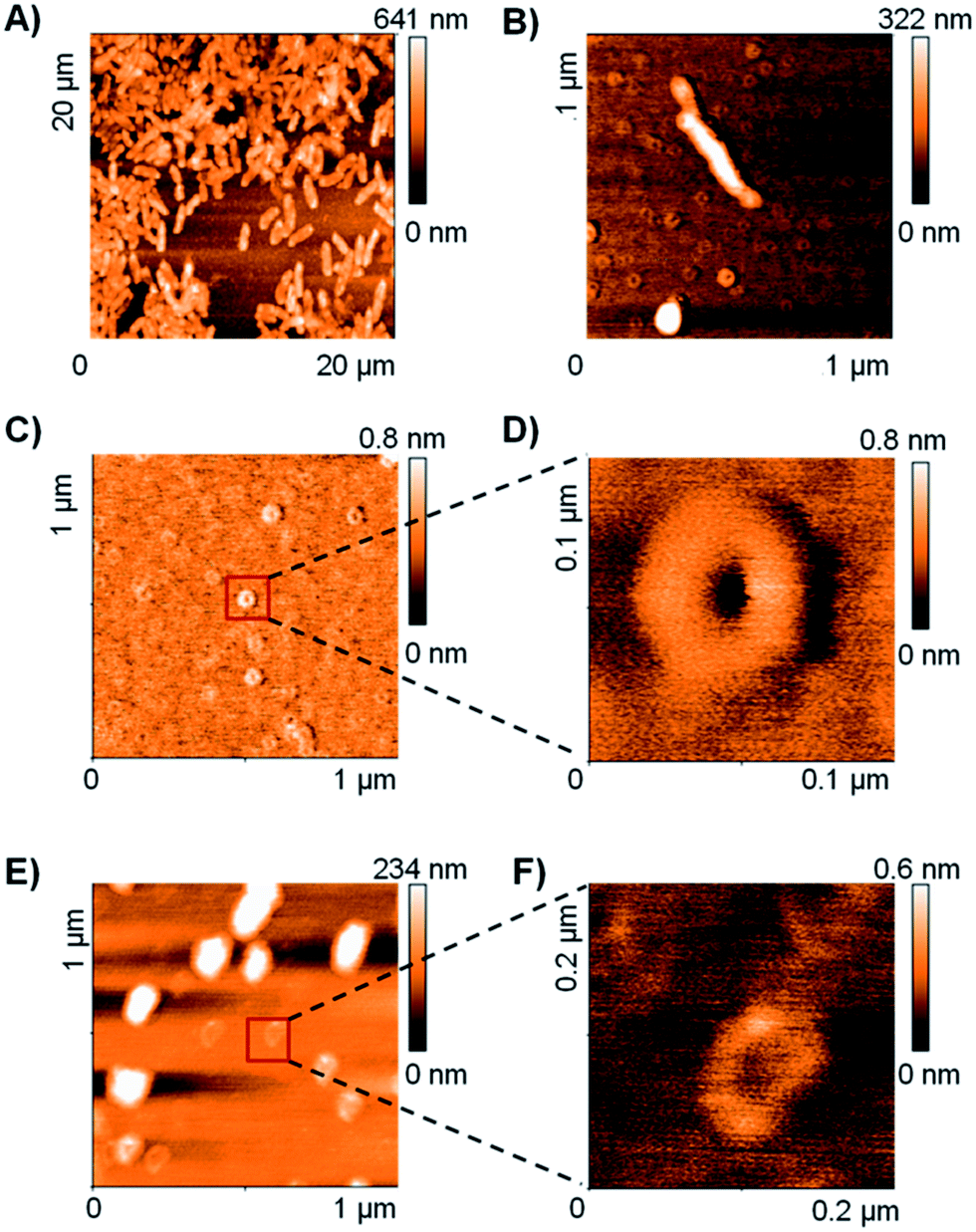 | ||
| Fig. 2 AFM image of B. subtilis cells on mica surface with or without treatment with CAMP RH11 at 4 × MIC (see methods for details). (A) Control cells without treatment; (B) cells after treatment with RH11, the AFM image shows doughnut-like structures around the bacteria. (C) Focus on doughnut-like structures observed with B. subtilis treated with RH11. (D) zoom image of (C). (E) AFM image of phosphatidylglycerol lipid vesicles treated with RH11. (F) zoom image of phosphatidylglycerol lipid vesicles with RH11. | ||
X-ray crystallography
In view of the above experimental evidences for membrane disruptive effects by RH11 we next determined its X-ray crystal structure to see if its amphiphilic character was comparable to that reported in the X-ray crystal structures of typical membrane disruptive CAMPs such as Gramicidin S23 and Tyrocidin A.26 Since our attempts to crystallize RH11 directly were unsuccessful, we prepared analogs of RH11 extended with an α-L-c-fucosylacetyl group at the N-terminus of the peptide in view of obtaining the CAMP structures as complexes with the fucose specific P. aeruginosa lectin LecB, which crystallizes readily.44–46,63 We prepared fucosylated derivatives for RH11, its enantiomer dRH11, as well as for the o-xylene and m-xylene analogs RH11o and RH11m, which were as active as RH11 itself, and for their respective enantiomers dRH11o and dRH11m. We screened crystallization conditions in all six cases either by co-crystallization of the ligands with LecB or by soaking preformed LecB crystals with the ligands.In the three cases of cyclic peptide–LecB complexes for which we obtained well-resolved data, which were all obtained by co-crystallization, the crystal symmetry resulted in four non-equivalent LecB monomers and the corresponding complexed fucosyl ligands in each LecB tetramer. Each of the four binding sites was occupied by a fucosylated ligand (Table S2†). Electron densities allowing structure determination of the cyclic peptide portion were observed for three out of four binding sites in the case of FdRH11o (the fucosylated enantiomer of RH11o, 5NF0, 1.27 Å resolution, Fig. 3a–e, S6a and b†), as well as for a single LecB binding site in the cases of FRH11o (fucosylated RH11o, 5NEY, 1.54 Å resolution, Fig. 3f and S6c†) and FdRH11m (fucosylated enantiomer of RH11m, 5NES, 1.60 Å resolution, Fig. 3 and S6d†). For all structures the peptide backbone as well as a large part of the amino acid side chains were well ordered, however side chains had very different B-factors in the different structures. The side chains of FdRH11o were completely ordered in all three conformers with very low B-factors, with almost every atom perfectly defined. By contrast FRH11o had higher side chain B-factors, and only its tryptophan side chains were ordered. In the case of FdRH11m the tryptophan side chains and the side chain of lys8 were ordered because it was immobilized by a 3.2 Å hydrogen bridge to the fucosyl group, however the other lysine side chains had high B-factors and were disordered.
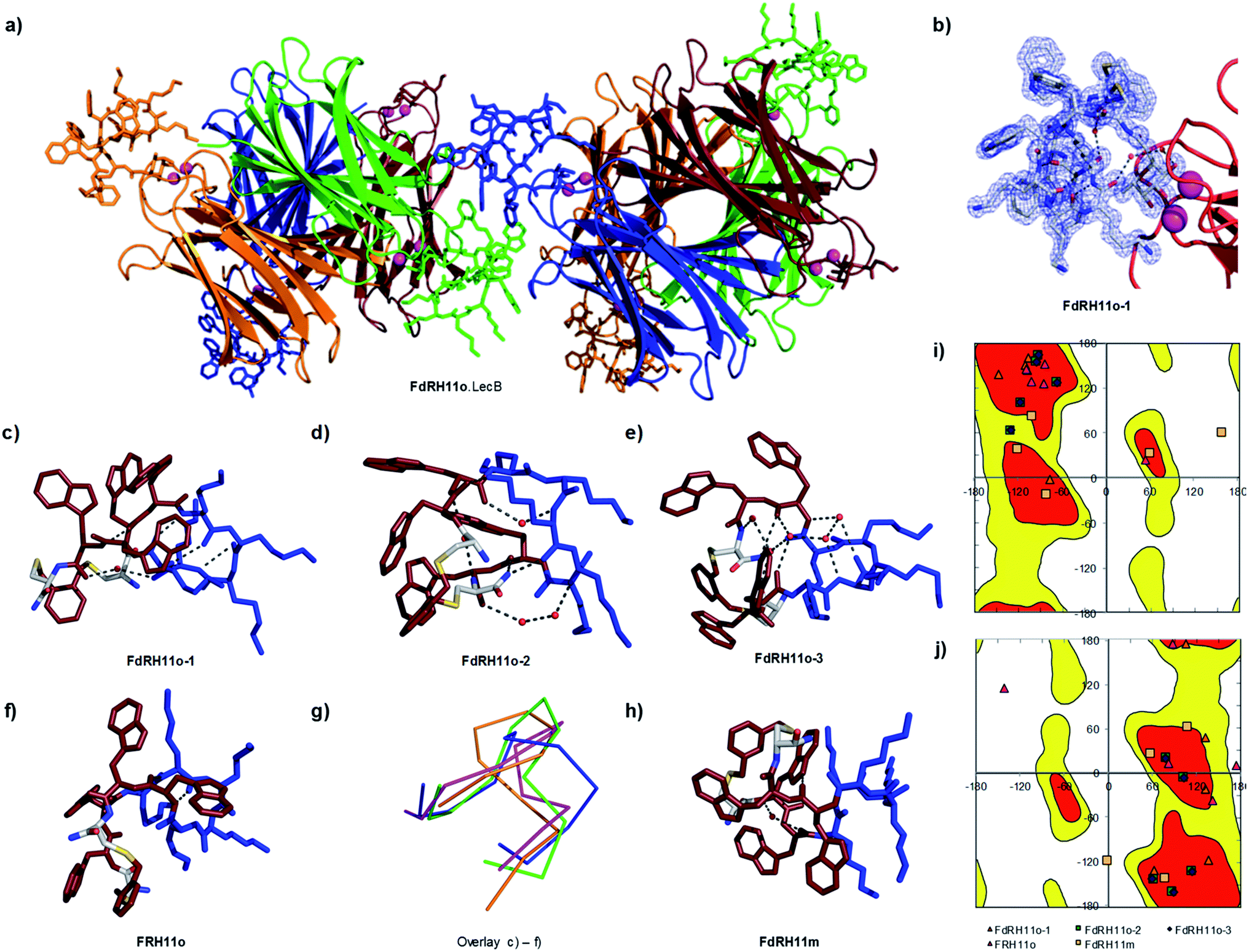 | ||
| Fig. 3 X-ray crystal structure of fucosylated analogs of RH11 in complex with lectin LecB. (a) Overview of the FdRH11o–LecB complex showing 2 symmetric copies of the LecB tetramer. The protein is shown as ribbons and the cyclic peptides in stick model, each symmetry non-equivalent LecB monomer and its bound ligand are shown in the same color. The orange (FdRH11o-1), green (FdRH11o-2), and blue (FdRH11o-3) monomers reveal a fully resolved bound ligand. (b) Detail of the FdRH11o-1 shown with blue dashed cloud density. (c) Cyclic peptide portion of FdRH11o-1 in stick model with cationic lysine side chains in blue and hydrophobic residues (Trp and the xylene bridge) in ruby, showing one water-bridged hydrogen bond and four backbone H-bonds forming an improper short β-sheet like structure. The chosen cut-off distance for hydrogen bridges is 3.2 Å. (d) Cyclic peptide portion of FdRH11o-2 in stick model with two backbone H-bonds and two water-bridged backbone H-bonds. (e) Cyclic peptide portion of FdRH11o-3 in stick model, with backbone H-bonds connected via four crystallographic water molecules, one backbone H-bond involving the C-terminus, and a backbone-to-lysine side chain H-bond. (f) Cyclic peptide portion of FRH11o in stick model as observed in its LecB complex, with missing side-chains modeled in, showing two backbone H-bonds. (g) Overlay of α-carbon backbone observed in the three occupied sites of the FdRH11o–LecB complex (orange, green and blue) and the single occupied site of FRH11o–LecB complex (magenta). (h) Cyclic peptide in stick model as observed in the FdRH11m–LecB complex, with missing side-chains modeled in, showing one water bridged backbone H-bond and one H-bond between an indole NH and a backbone carbonyl. (i) Ramachandran plot for all L-residue dihedrals in the cyclic peptides. (j) Ramachandran plot for all D-residue dihedrals in the cyclic peptides. In both plots the allowed regions are shown in red and yellow. See also Table S2 and Fig. S6.† | ||
Close analysis of each structure showed that cyclic peptides engaged in several backbone hydrogen bonds, several of them water-bridged (Fig. 3d–f and 2h). Dihedral angles resided in allowed regions of the Ramachandran plots except for two cases corresponding to poorly resolved portions of the peptide backbone with poor electron density indicating flexibility (Fig. 3i and j).
An overlay of the α-carbon backbone of the three structures of FdRH11o and the single structure of FRH11o provided direct structural evidence that this CAMP could adopt a variety of conformations, suggesting that these cyclic peptides were conformationally quite flexible, somewhat in contrast to the rather rigid structures observed with Gramicidin S in crystals23 or by NMR in membrane environment,64 or in the X-ray structure of the Tyrocidin A dimer (Fig. 3g).26 Nevertheless in all conformations observed in the X-ray structures our cyclic peptides adopted an amphiphilic structure grouping the hydrophobic xylene bridge and the tryptophan side chains on one side and the cationic lysine side chains on the other, in agreement with the original sequence design. Because there were almost no contacts between the cylic peptides and LecB apart from the fucose anchor we believe that these conformations are comparable to the conformation for the free peptides, and therefore support the membrane disruptive mechanism of action of RH11.
N-Methylation of backbone amides
The formation of intramolecular backbone hydrogen bonds including water-bridges observed in the crystal structures above suggested that our CAMPs were not preorganized for nanotube forming intermolecular backbone H-bonds despite of their alternating D,L-sequence inspired by Ghadiri's cyclic peptides. To test whether backbone H-bonds had any influence on the activity of RH11 we prepared three analogs RH18, RH19 and RH20, each featuring a different pair of N-methylated residues, using the 2-nitrobenzene sulfonamide method for N-methylation of the selected residues during SPPS.65 All three derivatives retained most of the antimicrobial activity of RH11 despite of each having two blocked N-methyl group unable to act as backbone H-bond donors, confirming that activity was independent of a precise pattern of backbone amide bonds (Table 1).Interestingly, N-methylation influenced hemolytic activity, with RH18 showing stronger hemolysis than RH11 and RH19/RH20 showing much weaker hemolysis than RH11. Although N-methylation is a standard backbone modification to tune the pharmacological properties of peptides,66 modulation of hemolytic properties by backbone N-methylation has not been reported previously for CAMPs and suggests a route for fine-tuning the properties of RH11.
Conclusion
In summary, investigating amphiphilic peptides with an alternating D,L-architecture cyclized by a xylene double thioether bridge led to the discovery of RH11 as a potent CAMP with activity against P. aeruginosa including MDR clinical isolates, good serum stability and moderate hemolysis. A sufficient ring size was essential in obtaining activity against P. aeruginosa. RH11 displayed membrane disruptive activity evidenced by DLS and TEM experiments. AFM images of bacteria exposed to RH11 showed that the interaction of RH11 with the membrane led to the formation of doughnut shaped structures tentatively attributed to supramolecular lipid–peptide aggregates. The amphiphilic nature of RH11 was confirmed by high resolution X-ray crystal structures of fucosylated analogs in complex with lectin LecB, which showed that these CAMPs can adopt a variety of conformations involving intramolecular and partly water-bridged backbone H-bonds, but are not preorganized for intermolecular aggregation. N-Methylation of backbone amides in RH11 led to derivatives with similar potency but reduced hemolysis, suggesting a route for further optimization.The structures of RH11 and analogs differ considerably from other CAMPs from the literature discussed in the introduction. In addition to the use of a xylene double thioether bridge, our choice of lysine as the only cationic residue was somewhat special because CAMPs usually contain combinations also involving arginine, ornithine and diaminobutyric acid. Our CAMPs also stand out by the unique combination of lysine with tryptophan as the only hydrophobic residue, leading to an unusually high fraction of aromatic groups (Fig. S7A†). High aromaticity is otherwise reported only in Arg/Trp cyclic peptides, while all other CAMPs also feature aliphatic hydrophobic residues (Leu, Pro, Val). Furthermore, with 120 heavy atoms and 6 positive charges, RH11 is among the largest and most highly charged CAMPs (Fig. S7B†). Although this property is shared with the P. aeruginosa specific CAMP POL7080 and its analogs as well as BacR, an analog of bactenecin optimized for P. aeruginosa,12 polymyxin B is significantly smaller and has only 5 positive charges. In addition to N-methylation, optimization of RH11 might therefore also consider non-aromatic hydrophobic groups as well as size and charge reduction to address not only MDR antimicrobial activity but also toxicity issues beyond hemolysis and eventually in vivo effects.
Experimental
General
All reagents were purchased from Sigma Aldrich and Fluka, TCI Europe, Advanced ChemTech and Iris Biotech, GL Biochem Ltd. and Dr. Gogg Chemie AG. Tental Gel S RAM (loading: 0.25 mmol g−1) were purchased from Rapp Polymere. Preparative RP-HPLC was performed on a Waters PrepLC facility apparatus using a Reprospher 100C-18-DE column, 5 μm, 100 × 30 mm column. Eluent A: 0.1% TFA in 100% milliQ-H2O; eluent B: 50% H2O, 50% MeCN; eluent C: 90% MeOH, 10% milliQ-H2O, 0.1% TFA; eluent D: 90% MeCN, 10% milliQ-H2O, 0.1% TFA. Detection with Waters 2489 UV Visible Detector at λ = 214 nm. MS spectra were provided by Mass Spectrometry and Protein Analysis of the Department of Chemistry and Biochemistry at the University of Bern. Analytical UPLC was performed on Dionex Ultimate 3000 using an Acclaim RSLC 120 C18-column, 2.2 μm, 3.0 × 10 mm eluent A: 0.1% TFA in 100% milliQ-H2O; eluent B: 50% milliQ-H2O, 50% MeCN, eluent; C: 20% MeOH 80% milliQ-H2O 0.1% TFA; eluent D: 90% MeCN, 10% milliQ-H2O, 0.1% TFA. Detection by UV at λ = 214 nm. LC-MS data were collected after coupling the analytical system described above with a LCQ Fleet Ion Trap mass spectrometer (Thermo Scientific, San Jose, CA, USA). LC-MS data recording and processing was done with Xcalibur (version 2.2, Thermo Scientific).Cyclic peptides
All linear peptides were synthesized by solid phase peptide synthesis (SPPS). The solid support was tentagel S RAM resin. Synthesis of peptides was done manually in polypropylene syringes. The resin was swelled in 7 mL DCM and the Fmoc protecting groups were removed with 7 mL of solution of 20% piperidine in DMF (2 × 20 min). For each coupling the amino acid (3 eq.) and PyBOP (3 eq.) was dissolved in NMP (7 mL) and added to the syringe. Then DIPEA (5 eq.) was added and the reaction was stirred for 1 hour. Than the resin was washed 3 times with NMP (7 mL), MeOH (7 mL) and DCM (7 mL). The Fmoc protecting groups were removed with 7 mL solution of 20% piperidine in DMF (2 × 20 min). The cleavage was carried out with TFA/TIS/H2O (94:5:1) for 4.5 h and the peptide was precipitated with methyl tert-butyl ether. The cyclization was performed in high dilution in H2O/ACN, in presence of potassium iodide and DIPEA. The cyclization was performed with α,α′-dichloro-p-xylene (3 eq.), or α,α′-dichloro-o-xylene (3 eq.) or α,α′-dichloro-m-xylene (3 eq.) as linkers for a double thioether ligation and were monitored by LC-MS and were quenched after complete disappearance of the starting material (see ESI†). The disulfide-linked peptide was catalyzed with aldrithiol-2 in high dilution in H2O with PH = 7.8. The peptides were dissolved in milli-Q H2O and purified by preparative HPLC to yield the TFA salts after lyophilization of the pooled pure fractions.
Fucosylated cyclic peptides
The linear sequence was assembled by SPPS as described above until removal of the last Fmoc group. Peracetylated α-L-fucosyl-acetic acid (5 eq.), HATU (4 eq.) and DIPEA (10 eq.) were dissolved in 4.5 mL of NMP and 1.5 mL of DCM and added to the syringe. The mixture was stirred overnight. The solvent was filtrated followed by washing with NMP (2 × 6 mL), MeOH (2 × 6 mL) and DCM (2 × 6 mL). Deacetylation when then performed on solid support by stirring the resin for 24 h in MeOH/H2O/NH3 (8:1:1). The resin was then filtered, washed, and the TFA cleavage of the peptide, cyclization and final HPLC purification were performed as above.
MS (ESI+) calculated for C88H119N21O11S2 [M + H]+: 1711.55, found: 1711.42.
MS (ESI+) calculated for C94H131N23O12S2 [M + H]+: 1838.98, found: 1838.58.
MS (ESI+) calculated for C88H119N21O11S2 [M + H]+: 1711.15, found: 1711.25.
MS (ESI+) calculated for C88H119N21O11S2 [M + H]+: 1711.15, found: 1711.33.
MS (ESI+) calculated for C88H119N21O11S2 [M + H]+: 1711.15, found: 1711.25.
MS (ESI+) calculated for C88H119N21O11S2 [M + H]+: 1711.15, found: 1711.67.
MS (ESI+) calculated for C88H119N21O11S2 [M + H]+: 1711.15, found: 1711.75.
MS (ESI+) calculated for C99H129N23O12S2 [M + H]+: 1897.36, found: 1897.33.
MS (ESI+) calculated for C93H117N21O11S2 [M + H]+: 1768.87, found: 1768.25.
MS (ESI+) calculated for C83H121N21O11S2 [M + H]+: 1652.90, found: 1652.42.
MS (ESI+) calculated for C90H123N21O11S2 [M + H]+: 1739.20, found: 1739.25.
Minimal inhibition concentration (MIC)
A colony of bacteria from glycerol stock was grown in Müller–Hinton (MH) medium overnight at 37 °C and 180 rpm shaking. The compounds were prepared as stock solutions of 8 mg mL−1 in MH medium, added to the first well of 96-well sterile, U-bottomed polypropylene microtiter plates and diluted serially by 1/2. Compounds containing solutions were sterile filtered (pore size 0.22 μm) prior to addition to the wells. The concentration of the bacteria was quantified by measuring absorbance at 600 nm and diluted to an OD600 of 0.022 in MH medium. The sample solutions (150 μL) were mixed with 4 μL diluted bacterial suspension with a final inoculation of about 1 × 105 CFU. For each test, two columns of the plate were kept for sterility control (MH medium only), growth control (MH medium with bacterial inoculum, no compound). Polymyxin B was used as a positive control. The plates were incubated at 37 °C for ∼18 hours. 15 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (1 mg mL−1 in sterilized milliQ deioinized water) were added to each well and the plates were incubated for 10 minutes at room temperature. The minimal inhibitory concentration (MIC) was defined as the lowest concentration of the monocyclic peptide that inhibits the visible growth of the tested bacteria (yellow) with the unaided eye.Minimal hemolytic concentration (MHC)
To determine the minimal hemolytic concentration (MHC) stock solutions of 8 mg mL−1 of the peptide in H2O were prepared and 50 μL were diluted serially by 1/2 in 50 μL PBS (pH 7.4) in 96-well plate (Cornstar or Nunc, polystyrene, untreated). Human red blood cells (hRBC) were obtained by centrifugation of 1.5 mL of whole blood, from the blood bank of Bern, at 3000 rpm for 15 minutes at 4 °C. Plasma was discarded and the pellet was re-suspended in a 15 mL falcon tube up to 5 mL of PBS. The washing was repeated three times and the remaining pellet was re-suspended in 10 mL of PBS at a final hRBC concentration of 5%. The hRBC suspension (50 μL) was added to each well and the plate was incubated at room temperature for 4 hours. Minimal hemolytic concentration (MHC) end points were determined by visual determination of the wells after the incubation period. Controls on each plate included a blank medium control (50 μL PBS + 50 μL of hRBC suspension) and a hemolytic activity control (mQ-deionized water 50 μL + 50 μL hRBC suspension).Serum stability assay
Human serum was diluted in DMEM (1:4, v/v). Selected peptide was diluted in TRIS buffer to a concentration of 685 μg mL−1. Aliquots of peptide solution (50 μL) were added to aliquots of serum (50 uL) in sterile Eppendorf tubes, to reach a peptide concentration of 200 μM during the assay. Samples were incubated at 37 °C under gentle stirring (350 rpm). Different samples (triplicates) were quenched at different time points (0/1/3/6/24 h) by precipitating serum proteins through the addition of (0.1 M) ZnSO4 × 7H2O/ACN (1:1) (0.1 M, 100 μL) and cooling down in ice bath. Protein precipitates were pelleted under centrifugation and the supernatants were sampled and evaporated to dryness in a centrifugal evaporator. Samples were re-suspended in a H2O/ACN (4:1, v/v) mixture and centrifuged again to remove residual protein precipitate. Supernatants were then sampled and analyzed by LC-MS. Experiment controls included a precipitation control for each peptide and serum blanks.
Kinetic assay
Kinetic assays were measured using a VARIAN, CARY Eclipse Fluorescence Spectrophotometer. P. aeruginosa PA01 was incubated overnight at 37 °C and diluted with 60 mL MH medium and incubated about 2 h to get an OD600 of 0.2–0.3. The bacteria were split in 2, 50 mL sterile falcon tubes and centrifuged for 20 min at 5000 rpm at 4 °C. Bacteria were washed with 45 mL sterile 0.9% NaCl and centrifuged at 5000 rpm at 4 °C. Bacteria were diluted with sterile 0.9% NaCl to OD600 = 0.1. The increase of fluorescence of 3 mL P. aeruginosa and 3 mL P. aeruginosa with 3 μL of SYTOX (0.5 mM in DMSO) was measured as references respectively at 37 °C. To 3 mL P. aeruginosa PA01 treated with 3 μL SYTOX either 15 μL of polymyxin B (1 mg mL−1) or peptides to get a concentration of 4 × MIC were added to the cuvette. The addition of the peptides was at time t = 0. The experiments were measured with the following parameters: 523 nm emission wavelength; 504 nm excitation wavelength; 5 nm excitation slit, 5 nm emission slit; temperature control at 37 °C and time control from 0 min to 120 min.Transmission electron microscopy
Exponential phase of P. aeruginosa PAO1 were washed with PBS and treated 10 × MIC of the corresponding compound in M63 minimal medium. Each time, 1 mL of the bacteria were centrifuged after 15, 30 and 60 min at 12000 rpm for 3 min and fixed overnight with 2.5% glutaraldehyde in 0.15 M HEPES with an osmolarity of 670 mOsm and adjusted to a pH of 7.35. The next day, PAO1 were washed with 0.15 M HEPES three times for 5 min, post-fixed with 1% OsO4 in 0.1 M Na-cacodylate-buffer at 4 °C for 1 h. Thereafter, bacteria cells were washed in 0.1 M maleate buffer three times for 5 min and dehydrated in 70, 80, and 96% ethanol for 15 min each at room temperature. Subsequently, they were immersed in 100% ethanol three times for 10 min, in acetone two times for 10 min, and finally in acetone–Epon (1:1) overnight at room temperature. The next day, bacteria cells were embedded in Epon and hardened at 60 °C for 5 days.
Sections were produced with an ultramicrotome UC6 (Leica Microsystems, Vienna, Austria), first semithin sections (1um) for light microscopy which were stained with a solution of 0.5% toluidine blue O and then ultrathin sections (70–80 nm) for electron microscopy. The sections, mounted on single slot copper grids, were stained with uranyl acetate and lead citrate with an ultrostainer (Leica Microsystems, Vienna, Austria).
Sections were then examined with a Tecnai Spirit transmission electron microscope equipped with two digital cameras (Olympus-SIS Veleta CCD Camera, FEI Eagle CCD Camera).
Dynamic light scattering
Apparent particle sizes of the compounds and LPS solutions and the mixtures were measured at 25 °C by dynamic light scattering (DLS) at 632.8 nm using a Malvern Zetasizer Nano ZS (Malvern, UK) and using original Ependorf disposable single sealed Cuvettes, 50–2000 μL RNase-/DNA-/protein-free. LPS (E. Coli, 0111:B4) aggregates and the compounds (polymyxin B, tobramycin and RH11) were diluted to 200 μg mL−1 in PBS. The mixtures were prepared with a ratio of 1:1 of the LPS and the compound of interest to reach a final concentration of 100 μg mL−1. As a control, LPS solution was diluted to 100 μg mL−1 in PBS. The experiments were repeated three times.
Atomic force microscopy
AFM imaging was performed under ambient conditions in air with a Nanosurf easyscan 2 (Nanosurf AG, Switzerland). All measurements were carried out in tapping mode employing PPP-NCHR-W cantilevers from Nanosensors (resonance frequency ∼ 280 kHz, tip radius ∼ 10 nm). The mica substrates (20 × 20 mm2) were attached to a steel baseplate with Scotch tape and freshly cleaved prior to each new experiment. Peptides at a concentration of 4 × MIC were incubated with bacterial cell (B. subtilis OD600 = 0.1) in NaCl 0.9% for 10 min. The mixed solution was deposited onto a freshly cleaved mica surface. The solution was removed by gently rinsing with Milli-Q water after 5 min of incubation time. After dried with Argon gas, the substrate was analyzed with atom force microscopy. Peptide without bacterial treatment and bacterial without peptides were also analyzed by AFM as control. For details see ESI Fig. S2 and S3.†X-ray crystallography
LecB lectin was expressed, purified and dialyzed as previously described.67 The crystals of the different RH11 fucosylated derivatives were carried out with the sitting drop method. In brief, lyophilized protein was dissolved in water (5 mg mL−1) in the presence of salts (1 mM CaCl2 and MgCl2). The compounds FdRH11o–LecB, FRH11o–LecB and FdRH11m–LecB were added to the protein at a 20:1 molar excess, taking in to account, that the biological unit of LecB is a homo-tetramer. Crystals were obtained within five days after mixing 1.5 μL of LecB ligand-complex with 1.5 μL of reservoir solution at 18 °C. The best diffracting crystals were found in the Index and Crystal screen of Hampton research. In Detail, for FdRH11o–LecB, the best condition was the condition F11 of the Index screen which contains 0.005 M cadmium chloride hydrate, while for FRH11o–LecB it was the Crystal screen F3 condition (0.5 M ammonium sulfate, 0.1 M sodium citrate tribasic dehydrate, 1.0 M lithium sulfate monohydrate, pH 5.6). Respectively for FdRH11m–LecB it was the condition Index screen F4 (0.005 M cobalt(II) chloride hexahydrate, 0.1 M HEPES, 12% polyethylene glycol 3350, pH 7.5). The structures were solved using the XDS,68 CCP4,69 the phenix70 program suite and the coot71 graphical program. Pictures were done with the help of pymol.72
Cheminformatics
The structures of all CAMPs analyzed were redrawn from the original literature using ChemDraw, and exported as SMILES. The combined SMILES file annotated with compound code and antimicrobial activity data was then imported into WebMolCS to visualize the spread of structural features in various 3D-chemical spaces.73 The key parameters identified by this analysis (net charge vs. size and aromatic carbon versus non-aromatic carbons) were then extracted from the corresponding descriptor analysis and replotted in Fig. S7A and B.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the Swiss National Science Foundation and the Swiss TransMed B5 Platform project.Notes and references
- K. M. G. O'Connell, J. T. Hodgkinson, H. F. Sore, M. Welch, G. P. C. Salmond and D. R. Spring, Angew. Chem., Int. Ed. Engl., 2013, 52, 10706–10733 CrossRef PubMed.
- M. Chatterjee, C. P. Anju, L. Biswas, V. Anil Kumar, C. Gopi Mohan and R. Biswas, Int. J. Med. Microbiol., 2016, 306, 48–58 CrossRef CAS PubMed.
- T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed. Engl., 2014, 53, 13020–13041 CrossRef CAS PubMed.
- A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC.
- E. Valeur, S. M. Guéret, H. Adihou, R. Gopalakrishnan, M. Lemurell, H. Waldmann, T. N. Grossmann and A. T. Plowright, Angew. Chem., Int. Ed. Engl., 2017, 56, 10294–10323 CrossRef CAS PubMed.
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- B. Yoo and K. Kirshenbaum, Curr. Opin. Chem. Biol., 2008, 12, 714–721 CrossRef CAS PubMed.
- S. A. Fowler and H. E. Blackwell, Org. Biomol. Chem., 2009, 7, 1508–1524 CAS.
- M. Amblard, J. A. Fehrentz, J. Martinez and G. Subra, Mol. Biotechnol., 2006, 33, 239–254 CrossRef CAS PubMed.
- Y. Y. Liu, Y. Wang, T. R. Walsh, L. X. Yi, R. Zhang, J. Spencer, Y. Doi, G. Tian, B. Dong, X. Huang, L. F. Yu, D. Gu, H. Ren, X. Chen, L. Lv, D. He, H. Zhou, Z. Liang, J. H. Liu and J. Shen, Lancet Infect. Dis., 2016, 16, 161–168 CrossRef CAS PubMed.
- P. Nordmann and L. Poirel, Clin. Microbiol. Infect., 2016, 22, 398–400 CrossRef CAS PubMed.
- M. Wu and R. E. Hancock, J. Biol. Chem., 1999, 274, 29–35 CrossRef CAS PubMed.
- N. Srinivas, P. Jetter, B. J. Ueberbacher, M. Werneburg, K. Zerbe, J. Steinmann, B. Van der Meijden, F. Bernardini, A. Lederer, R. L. A. Dias, P. E. Misson, H. Henze, J. Zumbrunn, F. O. Gombert, D. Obrecht, P. Hunziker, S. Schauer, U. Ziegler, A. Kach, L. Eberl, K. Riedel, S. J. DeMarco and J. A. Robinson, Science, 2010, 327, 1010–1013 CrossRef CAS PubMed.
- M. Vaara, J. Fox, G. Loidl, O. Siikanen, J. Apajalahti, F. Hansen, N. Frimodt-Moller, J. Nagai, M. Takano and T. Vaara, Antimicrob. Agents Chemother., 2008, 52, 3229–3236 CrossRef CAS PubMed.
- A. Gallardo-Godoy, C. Muldoon, B. Becker, A. G. Elliott, L. H. Lash, J. X. Huang, M. S. Butler, R. Pelingon, A. M. Kavanagh, S. Ramu, W. Phetsang, M. A. T. Blaskovich and M. A. Cooper, J. Med. Chem., 2016, 59, 1068–1077 CrossRef CAS PubMed.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed.
- L. T. Nguyen, E. F. Haney and H. J. Vogel, Trends Biotechnol., 2011, 29, 464–472 CrossRef CAS PubMed.
- P. D. Cotter, R. P. Ross and C. Hill, Nat. Rev. Microbiol., 2013, 11, 95–105 CrossRef CAS PubMed.
- J. L. Fox, Nat. Biotechnol., 2013, 31, 379–382 CrossRef CAS PubMed.
- B. Mojsoska and H. Jenssen, Pharmaceuticals, 2015, 8, 366–415 CrossRef CAS PubMed.
- C. Heinis and G. Winter, Curr. Opin. Chem. Biol., 2015, 26, 89–98 CrossRef CAS PubMed.
- T. Morioka, N. D. Loik, C. J. Hipolito, Y. Goto and H. Suga, Curr. Opin. Chem. Biol., 2015, 26, 34–41 CrossRef CAS PubMed.
- S. E. Hull, R. Karlsson, P. Main, M. M. Woolfson and E. J. Dodson, Nature, 1978, 275, 206–207 CrossRef CAS.
- G. N. Tishchenko, V. I. Andrianov, B. K. Vainstein, M. M. Woolfson and E. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 151–159 CrossRef CAS PubMed.
- A. L. Llamas-Saiz, G. M. Grotenbreg, M. Overhand and M. J. van Raaij, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 401–407 CAS.
- P. J. Loll, E. C. Upton, V. Nahoum, N. J. Economou and S. Cocklin, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 1199–1207 CrossRef CAS PubMed.
- L. H. Kondejewski, M. Jelokhani-Niaraki, S. W. Farmer, B. Lix, C. M. Kay, B. D. Sykes, R. E. Hancock and R. S. Hodges, J. Biol. Chem., 1999, 274, 13181–13192 CrossRef CAS PubMed.
- M. Jelokhani-Niaraki, L. H. Kondejewski, L. C. Wheaton and R. S. Hodges, J. Med. Chem., 2009, 52, 2090–2097 CrossRef CAS PubMed.
- V. S. Fluxa, N. Maillard, M. G. Page and J. L. Reymond, Chem. Commun., 2011, 47, 1434–1436 RSC.
- A. Wessolowski, M. Bienert and M. Dathe, J. Pept. Res., 2004, 64, 159–169 CrossRef CAS PubMed.
- D. I. Chan, E. J. Prenner and H. J. Vogel, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1184–1202 CrossRef CAS PubMed.
- D. Oh, J. Sun, A. Nasrolahi Shirazi, K. L. LaPlante, D. C. Rowley and K. Parang, Mol. Pharmaceutics, 2014, 11, 3528–3536 CrossRef CAS PubMed.
- S. Finger, A. Kerth, M. Dathe and A. Blume, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 2998–3006 CrossRef CAS PubMed.
- M. L. Huang, S. B. Y. Shin, M. A. Benson, V. J. Torres and K. Kirshenbaum, ChemMedChem, 2012, 7, 114–122 CrossRef CAS PubMed.
- M. L. Huang, M. A. Benson, S. B. Y. Shin, V. J. Torres and K. Kirshenbaum, Eur. J. Org. Chem., 2013, 2013, 3560–3566 CrossRef CAS.
- S. Fernandez-Lopez, H. S. Kim, E. C. Choi, M. Delgado, J. R. Granja, A. Khasanov, K. Kraehenbuehl, G. Long, D. A. Weinberger, K. M. Wilcoxen and M. R. Ghadiri, Nature, 2001, 412, 452–455 CrossRef CAS PubMed.
- D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed. Engl., 2001, 40, 988–1011 CrossRef CAS PubMed.
- V. Dartois, J. Sanchez-Quesada, E. Cabezas, E. Chi, C. Dubbelde, C. Dunn, J. Granja, C. Gritzen, D. Weinberger, M. R. Ghadiri and T. R. Parr, Antimicrob. Agents Chemother., 2005, 49, 3302–3310 CrossRef CAS PubMed.
- J. T. Fletcher, J. A. Finlay, M. E. Callow, J. A. Callow and M. R. Ghadiri, Chem.–Eur. J., 2007, 13, 4008–4013 CrossRef CAS PubMed.
- P. Timmerman, J. Beld, W. C. Puijk and R. H. Meloen, ChemBioChem, 2005, 6, 821–824 CrossRef CAS PubMed.
- P. Timmerman, W. C. Puijk and R. H. Meloen, J. Mol. Recognit., 2007, 20, 283–299 CrossRef CAS PubMed.
- M. Q. Wentink, T. M. Hackeng, S. P. Tabruyn, W. C. Puijk, K. Schwamborn, D. Altschuh, R. H. Meloen, T. Schuurman, A. W. Griffioen and P. Timmerman, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12532–12537 CrossRef CAS PubMed.
- Y. V. Schlippe, M. C. Hartman, K. Josephson and J. W. Szostak, J. Am. Chem. Soc., 2012, 134, 10469–10477 CrossRef PubMed.
- E. Mitchell, C. Houles, D. Sudakevitz, M. Wimmerova, C. Gautier, S. Perez, A. M. Wu, N. Gilboa-Garber and A. Imberty, Nat. Struct. Biol., 2002, 9, 918–921 CrossRef CAS PubMed.
- G. Michaud, R. Visini, M. Bergmann, G. Salerno, R. Bosco, E. Gillon, B. Richichi, C. Nativi, A. Imberty, A. Stocker, T. Darbre and J.-L. Reymond, Chem. Sci., 2016, 7, 166–182 RSC.
- P. Roethlisberger, A. Istrate, M. J. Marcaida Lopez, R. Visini, A. Stocker, J. L. Reymond and C. J. Leumann, Chem. Commun., 2016, 52, 4749–4752 RSC.
- M. Stach, T. N. Siriwardena, T. Kohler, C. van Delden, T. Darbre and J. L. Reymond, Angew. Chem., Int. Ed. Engl., 2014, 53, 12827–12831 CrossRef CAS PubMed.
- J. Pires, T. N. Siriwardena, M. Stach, R. Tinguely, S. Kasraian, F. Luzzaro, S. L. Leib, T. Darbre, J. L. Reymond and A. Endimiani, Antimicrob. Agents Chemother., 2015, 59, 7915–7918 CrossRef CAS PubMed.
- I. Di Bonaventura, X. Jin, R. Visini, D. Probst, S. Javor, B.-H. Gan, G. Michaud, A. Natalello, S. M. Doglia, T. Kohler, C. van Delden, A. Stocker, T. Darbre and J.-L. Reymond, Chem. Sci., 2017 10.1039/c7sc01314k.
- D. Mandal, A. Nasrolahi Shirazi and K. Parang, Angew. Chem., Int. Ed. Engl., 2011, 50, 9633–9637 CrossRef CAS PubMed.
- G. Lattig-Tunnemann, M. Prinz, D. Hoffmann, J. Behlke, C. Palm-Apergi, I. Morano, H. D. Herce and M. C. Cardoso, Nat. Commun., 2011, 2, 453 CrossRef PubMed.
- R. Wallbrecher, L. Depre, W. P. Verdurmen, P. H. Bovee-Geurts, R. H. van Duinkerken, M. J. Zekveld, P. Timmerman and R. Brock, Bioconjugate Chem., 2014, 25, 955–964 CrossRef CAS PubMed.
- G. A. Eggimann, E. Blattes, S. Buschor, R. Biswas, S. M. Kammer, T. Darbre and J. L. Reymond, Chem. Commun., 2014, 50, 7254–7257 RSC.
- H. Traboulsi, H. Larkin, M. A. Bonin, L. Volkov, C. L. Lavoie and E. Marsault, Bioconjugate Chem., 2015, 26, 405–411 CrossRef CAS PubMed.
- B. L. Roth, M. Poot, S. T. Yue and P. J. Millard, Appl. Environ. Microbiol., 1997, 63, 2421–2431 CAS.
- M. Koike, K. Iida and T. Matsuo, J. Bacteriol., 1969, 97, 448–452 CAS.
- J. E. Shaw, J. R. Alattia, J. E. Verity, G. G. Prive and C. M. Yip, J. Struct. Biol., 2006, 154, 42–58 CrossRef CAS PubMed.
- S. Lu, G. Walters, R. Parg and J. R. Dutcher, Soft Matter, 2014, 10, 1806–1815 RSC.
- A. Meincken, D. L. Holroyd and M. Rautenbach, Antimicrob. Agents Chemother., 2005, 49, 4085–4092 CrossRef PubMed.
- A. Li, P. Y. Lee, B. Ho, J. L. Ding and C. T. Lim, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 411–418 CrossRef CAS PubMed.
- U. Khoe, Y. L. Yang and S. G. Zhang, Langmuir, 2009, 25, 4111–4114 CrossRef CAS PubMed.
- V. A. H. Ramirez, A. Pailleret, S. Joiret, F. d'Orlye, M. Lazerges, H. Perrot, S. G. Granados, F. Bedioui and L. M. De Leon-Rodriguez, New J. Chem., 2014, 38, 3637–3643 RSC.
- E. M. Johansson, S. A. Crusz, E. Kolomiets, L. Buts, R. U. Kadam, M. Cacciarini, K. M. Bartels, S. P. Diggle, M. Camara, P. Williams, R. Loris, C. Nativi, F. Rosenau, K. E. Jaeger, T. Darbre and J. L. Reymond, Chem. Biol., 2008, 15, 1249–1257 CrossRef CAS PubMed.
- M. Berditsch, S. Afonin, A. Steineker, N. Orel, I. Jakovkin, C. Weber and A. S. Ulrich, Appl. Environ. Microbiol., 2015, 81, 3593–3603 CrossRef CAS PubMed.
- S. C. Miller and T. S. Scanlan, J. Am. Chem. Soc., 1998, 120, 2690–2691 CrossRef CAS.
- J. Chatterjee, F. Rechenmacher and H. Kessler, Angew. Chem., Int. Ed. Engl., 2013, 52, 254–269 CrossRef CAS PubMed.
- H. Funken, K. M. Bartels, S. Wilhelm, M. Brocker, M. Bott, M. Bains, R. E. Hancock, F. Rosenau and K. E. Jaeger, PLoS One, 2012, 7, e46857 CAS.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- Schrodinger, LLC, The Pymol Molecular Graphics System, version 1.8, 2015 Search PubMed.
- M. Awale, D. Probst and J. L. Reymond, J. Chem. Inf. Model., 2017, 57, 643–649 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Compounds analytical data, crystallographic table and data, details of AFM, and list of structures as SMILES. See DOI: 10.1039/c7sc01599b |
| ‡ The authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2017 |