DOI:
10.1039/C7SC00891K
(Edge Article)
Chem. Sci., 2017,
8, 7476-7482
Manganese complex-catalyzed oxidation and oxidative kinetic resolution of secondary alcohols by hydrogen peroxide†
Received
25th February 2017
, Accepted 6th September 2017
First published on 7th September 2017
Abstract
The highly efficient catalytic oxidation and oxidative kinetic resolution (OKR) of secondary alcohols has been achieved using a synthetic manganese catalyst with low loading and hydrogen peroxide as an environmentally benign oxidant in the presence of a small amount of sulfuric acid as an additive. The product yields were high (up to 93%) for alcohol oxidation and the enantioselectivity was excellent (>90% ee) for the OKR of secondary alcohols. Mechanistic studies revealed that alcohol oxidation occurs via hydrogen atom (H-atom) abstraction from an α-CH bond of the alcohol substrate and a two-electron process by an electrophilic Mn–oxo species. Density functional theory calculations revealed the difference in reaction energy barriers for H-atom abstraction from the α-CH bonds of R- and S-enantiomers by a chiral high-valent manganese–oxo complex, supporting the experimental result from the OKR of secondary alcohols.
Introduction
The selective oxidation of organic substrates using earth-abundant transition metal catalysts (e.g. manganese and iron) and environmentally benign oxidants (e.g. molecular oxygen and hydrogen peroxide) is fundamentally important in enzymatic/biomimetic reactions and immensely useful in organic synthesis.1,2 Therefore, tremendous efforts have been devoted to elucidating the biomimetic oxidation reactions and developing highly efficient, selective (asymmetric) oxidation reactions using earth-abundant transition metal catalysts and environmentally benign oxidants under mild conditions.2–7 As a result, a great advance has been achieved recently in catalytic (asymmetric) epoxidation and hydroxylation reactions using synthetic iron and manganese catalysts and aqueous H2O2 as an environmentally benign oxidant in the presence of carboxylic acid as an additive.2–6 In these reactions, it has been proposed that high-valent metal–oxo intermediates are the active oxidants that affect the (asymmetric) epoxidation and hydroxylation reactions, and that the role of the carboxylic acid is to facilitate the heterolytic O–O bond cleavage of putative metal–hydroperoxo species to form high-valent metal–oxo intermediates.3–5 Very recently, we reported that a manganese complex bearing a tetradentate N4 ligand is an efficient catalyst in the asymmetric epoxidation of olefins by aqueous H2O2 in the presence of a small amount of H2SO4, affording high product yields with excellent stereo- and enantioselectivities.7 In the latter reaction, it was shown that carboxylic acid can be replaced by H2SO4 for activating H2O2 by manganese complexes, generating high-valent manganese–oxo species as active oxidants,7 although the role of H2SO4 remains elusive.
Another important research area in oxidation reactions is the oxidation of alcohols to aldehydes or ketones.8,9 Recently, nonporphyrinic manganese complexes have been employed as catalysts in the development of efficient catalytic systems for alcohol oxidation reactions, especially in those using H2O2 as an environmentally benign oxidant in the presence of carboxylic acid.9 Mechanistic studies have been performed to elucidate the alcohol oxidation reactions using synthetic metal–oxo complexes.10 In alcohol oxidation chemistry, the oxidative kinetic resolution (OKR) of racemic secondary alcohols has attracted much attention for developing efficient catalytic systems to obtain enantio-enriched alcohols,11–13 since chiral secondary alcohols are valuable synthetic intermediates in the pharmaceutical and fine chemical industries. In the OKR of racemic secondary alcohols, chiral metal complexes (e.g. the Mn(III) salen complex) with artificial oxidants (e.g. iodobenzene diacetate and sodium hypochlorite) have been used to produce chiral secondary alcohols.12d,13 However, to the best of our knowledge, the OKR of secondary alcohols has never been explored using synthetic mononuclear manganese catalysts and aqueous H2O2 as the terminal oxidant.
Herein, we report that manganese complexes bearing tetradentate N4 ligands, such as Mn(II)(P-MCP)(OTf)2 (1) and Mn(II)(Dbp-MCP)(OTf)2 (2) (Scheme 1A),7 are highly efficient catalysts for the oxidation of alcohols by aqueous 30% H2O2 in the presence of a catalytic amount of H2SO4 (Scheme 1B). Furthermore, to the best of our knowledge, the present study reports the first example of the use of a chiral manganese catalyst with low loading, aqueous H2O2 as the terminal oxidant, and a catalytic amount of H2SO4 as an additive in the OKR of racemic secondary alcohols (Scheme 1C). Density functional theory (DFT) calculations reveal that the energy barriers for the α-CH bond activation of chiral 1-phenylethanol (R- and S-enantiomers) by a high-valent manganese–oxo complex are significantly different, explaining the experimental observation of high enantioselectivity in the OKR of racemic secondary alcohols.
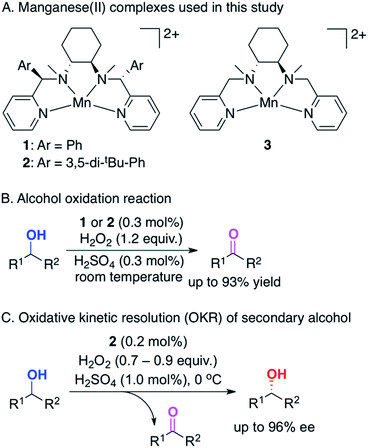 |
| | Scheme 1 (A) Schematic structures of manganese complexes bearing N4 ligands: MnII(P-MCP)(OTf)2 (1; P-MCP = (1R,2R)-N,N′-dimethyl-N,N′-bis-(phenyl-2-pyridinylmethyl)cyclohexane-1,2-diamine and OTf− = CF3SO3−), MnII(Dpb-MCP)(OTf)2 (2; Dbp-MCP = (1R,2R)-N,N′-di-methyl-N,N′-bis((R)-(3,5-di-tert-butylphenyl)-2-pyridinylmethyl)cyclohexane-1,2-diamine) and MnII(MCP)(OTf)2 (3; MCP = (1R,2R)-N,N′-dimethyl-N,N′-bis(2-pyridinylmethyl)cyclohexane-1,2-diamine). (B) Summary of the alcohol oxidation reaction. (C) Summary of the oxidative kinetic resolution (OKR) of secondary alcohols. | |
Results and discussion
Firstly, the reaction conditions for the catalytic oxidation of alcohols by manganese complexes and aqueous H2O2 in the presence of H2SO4 were optimized using 1-phenylethanol as a model substrate (see the Experimental section). Among the tested manganese catalysts (see Scheme 1A for the structures), Mn(II)(P-MCP)(OTf)2 (1) and Mn(II)(Dbp-MCP)(OTf)2 (2) exhibited high catalytic activity (Table 1, entries 1 and 2), whereas Mn(II)(MCP)(OTf)2 (3) was a poor catalyst (Table 1, entry 3). In the absence of the manganese catalyst, the oxidation of 1-phenylethanol to acetophenone was not observed (Table 1, entry 4). Since 1 can be prepared easily and cost-effectively, 1 was used as the catalyst to determine the optimal reaction conditions by varying the amount of catalyst (Table 1, entries 5 and 6), H2O2 (Table 1, entries 5 and 7) and H2SO4 (Table 1, entries 7–10). In addition, as reported in the olefin epoxidation reactions by nonporphyrinic Mn catalysts and H2O2,7 other Brønsted acids, such as HClO4, H3PO4, HCl and CF3SO3H, turned out to be poor additives (Table 1, entries 11–14).
Table 1 Optimization of reactions conditions for the oxidation of 1-phenylethanol by manganese complexes and H2O2a,b
|
|
| Entry |
Catalyst (mol%) |
Additive (mol%) |
H2O2 (equiv.) |
Yield (%) |
|
Reaction conditions: a CH3CN (0.50 mL) solution containing 30% H2O2 was added dropwise to a CH3CN (1.0 mL) solution containing 1-phenylethanol (0.50 mmol), the catalyst and the additive, using a syringe pump at 25 °C for 1 h.
Yields were determined by GC.
Yields were less than 5%.
Not detected.
|
| 1 |
1 (0.20) |
H2SO4 (1.0) |
1.5 |
90 |
| 2 |
2 (0.20) |
H2SO4 (1.0) |
1.5 |
91 |
| 3 |
3 (0.20) |
H2SO4 (1.0) |
1.5 |
Tracec |
| 4 |
— |
H2SO4 (1.0) |
1.5 |
NDd |
| 5 |
1 (0.30) |
H2SO4 (1.0) |
1.5 |
95 |
| 6 |
1 (0.40) |
H2SO4 (1.0) |
1.5 |
95 |
| 7 |
1 (0.30) |
H2SO4 (1.0) |
1.2 |
94 |
| 8 |
1 (0.30) |
H2SO4 (0.50) |
1.2 |
93 |
| 9 |
1 (0.30) |
H2SO4 (0.30) |
1.2 |
93 |
| 10 |
1 (0.30) |
H2SO4 (0.10) |
1.2 |
62 |
| 11 |
1 (0.30) |
HClO4 (1.0) |
1.5 |
13 |
| 12 |
1 (0.30) |
H3PO4 (1.0) |
1.5 |
18 |
| 13 |
1 (0.30) |
HCl (1.0) |
1.5 |
Tracec |
| 14 |
1 (0.30) |
CF3SO3H (1.0) |
1.5 |
Tracec |
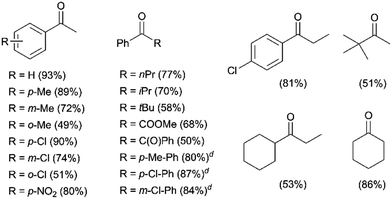
After optimizing the reaction conditions (Table 1, entry 9), we investigated the substrate scope for the oxidation of secondary alcohols by 1 and H2O2 in the presence of H2SO4 (Table 2). 1-Phenylethanol derivatives with electron-donating and -withdrawing substituents at the para-position of the phenyl group were oxidized to their corresponding ketones with good yields (e.g. >80%) (Table 2, left column). However, the product yields were moderate (e.g. ∼50%) for the oxidation of 1-phenylethanol derivatives with steric hindrance at the ortho-position of the phenyl group (Table 2, left column). Similarly, increasing the chain length and steric hindrance on the methyl side of the 1-phenylethanol derivatives, such as 1-phenylbutan-1-ol, 2-methyl-1-phenylpropan-1-ol and 2,2-dimethyl-1-phenylpropan-1-ol, decreased the product yields (Table 2, middle column). On the other hand, a series of diphenylmethanol derivatives were converted to the desired products with yields of >80% (Table 2, middle column). Unactivated aliphatic secondary alcohols were also oxidized to their corresponding ketones with moderate to high yields (Table 2, right column).
Table 2 Substrate scope for the oxidation of secondary alcoholsa,b,c
|
|
|
Reaction conditions: a CH3CN (0.50 mL) solution containing 30% H2O2 (1.2 equiv.) was added dropwise to a CH3CN (1.0 mL) solution containing the substrate (0.50 mmol), 1 (0.30 mol%) and H2SO4 (0.30 mol%), using a syringe pump at 25 °C for 1 h.
Yields were determined by GC.
Numbers in parentheses are product yields.
1.0 mol% H2SO4 was used.
|
|
We then investigated the OKR of secondary alcohols utilizing the manganese catalyst, H2O2 oxidant and H2SO4 additive system. Firstly, we optimized the reaction conditions by varying the amount of catalyst, oxidant and H2SO4, and the reaction temperature (see Fig. 1; also see Tables S1 and S2, ESI†). Obviously, the conversion of 1-phenylethanol would increase with an increasing amount of H2O2, as shown in Fig. 1. Due to the preference for oxidation of the S-enantiomer in the OKR of racemic secondary alcohols using the sulfuric acid-enabled manganese system, the ee value improved when increasing the number of equivalents of H2O2 from 0 to 0.80, while no significant change occurred when further increasing the number of equivalents of H2O2 up to 1.0. Therefore, the oxidant amount was chosen to be 0.80 equiv. for the OKR of 1-phenylethanol, as a result of the excellent ee with lower conversion. Besides, 2 was chosen as the catalyst since 2 afforded a higher enantiomeric excess (ee) value (90%) than 1 (65%) in the oxidation of 1-phenylethanol (Table 3, entry 1 and footnote c). Under the optimized catalytic conditions, we obtained high ee values (>90% ee) irrespective of the substituents on the phenyl group of the benzylic alcohols (i.e. no effect from steric hindrance or the electronic nature of the substrates) (Table 3, entries 2–6). Importantly, 1-phenylpropan-1-ol derivatives, which were reported to be poor substrates in most manganese salen systems,12d,13,14 also worked well in this sulfuric acid-enabled manganese system, with ee values of >90% (Table 3, entries 7–12). In addition, increasing the steric hindrance and chain length in the 1-phenylpropan-1-ol derivatives did not affect the ee values either (Table 3, entries 13–15).
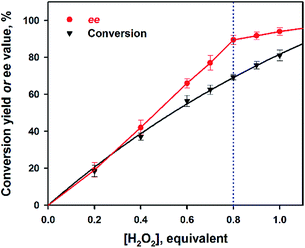 |
| | Fig. 1 Plots of the conversion yields (black triangles) of 1-phenylethanol and the ee values (red circles) of unreacted 1-phenylethanol against the number of equivalents of H2O2 obtained in the catalytic oxidation of 1-phenylethanol (0.50 mmol) by 2 (0.20 mol%) and H2O2 (0–1.0 equiv. based on the concentration of the substrate) in the presence of H2SO4 (1.0 mol%) in CH3CN at 0 °C for 1 h. | |
Table 3 Substrate scope for the oxidative kinetic resolution of secondary alcohols using the 2/H2O2/H2SO4 catalytic systema,b
In order to gain mechanistic insight into the manganese-catalyzed alcohol oxidation reactions, we firstly investigated the effect of para-substituents on the reactivity of the benzyl alcohol by carrying out competitive alcohol oxidation of the benzyl alcohol against para-substituted benzyl alcohols (see the Experimental section). A good linear correlation was obtained when the krel values were plotted against the Hammett parameters of the substituents (Fig. 2); the small but negative ρ value of −0.58 indicates that the active intermediate possesses electrophilic character, as reported in the oxidation of benzyl alcohol derivatives by synthetic metal–oxo complexes.10 Secondly, when the intermolecular competitive oxidation of 1-phenylethanol or its α-deuterated compound (1-deuterated 1-phenylethanol) was carried out together with 1-(p-chlorophenyl)ethanol as a mediator, a kinetic isotope effect (KIE) value of 1.8 was obtained (see the Experimental section for the detailed method), suggesting that hydrogen atom (H-atom) abstraction from an α–CH bond may be the rate-determining step, as observed in other manganese complex-catalyzed alcohol oxidation reactions.8b,c It is also notable that the KIE values determined in C–H bond activation reactions by synthetic metal–oxo complexes under stoichiometric conditions (e.g. KIE values of > 10)10,15 are much higher than those obtained in metal complex-catalyzed oxidation reactions under catalytic conditions (e.g. KIE values of < 4).8b,c It would be of interest to understand the reason for the difference in the KIE values obtained from the stoichiometric and catalytic reactions (e.g. the involvement of different metal–oxygen intermediates in the catalytic oxidation reactions). Thirdly, since cyclobutanol has often been used as a substrate probe to distinguish one-electron and two-electron processes in alcohol oxidation reactions,10a,c,16 we performed the oxidation of cyclobutanol and found that cyclobutanone was yielded exclusively and the ring-opened product, 4-hydroxylbutyraldehyde, was not detected (Scheme 2). Based on the mechanistic studies discussed above, we conclude that alcohol oxidation by an electrophilic manganese–oxo species is a two-electron process. This reactive manganese–oxo intermediate has been proposed previously in the asymmetric epoxidation of olefins by the manganese catalyst (2) and H2O2 in the presence of H2SO4.7
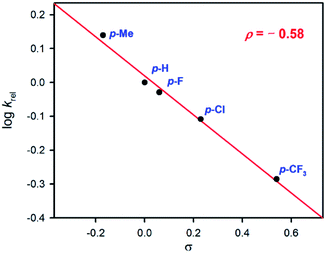 |
| | Fig. 2 Hammett plot of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) krel against the Hammett parameter (σ) for the catalytic oxidation of para-substituted benzyl alcohols by 1 (0.30 mol%) with H2O2 (0.40 equiv. based on the concentration of the substrate) as an oxidant in the presence of H2SO4 (0.30 mol%) in CH3CN at 25 °C. krel against the Hammett parameter (σ) for the catalytic oxidation of para-substituted benzyl alcohols by 1 (0.30 mol%) with H2O2 (0.40 equiv. based on the concentration of the substrate) as an oxidant in the presence of H2SO4 (0.30 mol%) in CH3CN at 25 °C. | |
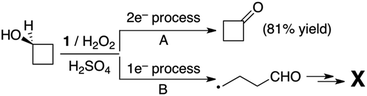 |
| | Scheme 2 Oxidation of cyclobutanol to cyclobutanone. | |
Density functional theory (DFT) computations were then carried out to explore the enantioselectivity in the α-CH bond activation of the chiral 1-phenylethanols (both R- and S-enantiomers) by a chiral [(P-MCP)Mn(V)(O)(SO4)]+ complex (I), which was proposed previously as the reactive intermediate in the reaction of 1 and H2O2 in the presence of H2SO4.7 The computational results reveal that H-atom abstraction from the α-CH bond of the S-enantiomer, with an energy barrier of 7.7/5.5 kcal mol−1 for the triplet/quintet spin state, is easier than that from the α-CH bond of the R-enantiomer, with an energy barrier of 10.6/6.8 kcal mol−1 for the triplet/quintet spin state (Table S3, ESI†). This reactivity trend was obtained by comparing the geometric character of these two transition states, in which TSS with rC–H = 1.203 Å has a smaller elongation of the C–H bond, while TSR with rC–H = 1.240 Å has a larger elongation of the C–H bond for the quintet ground state (Fig. S32, ESI†). In addition, the energy barrier difference for the two isomers might be due to the non-covalent anion–π interaction between the phenyl group of the substrate and the sulfuric acid anion ligand, which can stabilize the transition state, lowering the energy barrier. This kind of interaction only exists for TSS, with a distance of ca. 3.4 Å between the two groups. Thus, we may conclude that the S-enantiomer is an easier substrate than the R-enantiomer for oxidation by the high-valent Mn–oxo intermediate, regardless of the spin states; therefore the R-enantiomer remains in the reaction solution. Based on the Arrhenius equation, an energy barrier difference of 1.3 kcal mol−1 will give a rate constant ratio (k(R)/k(S)) of 0.112, which corresponds to a high ee value (∼80%), as obtained from the experiments (vide supra). From inspection of the spin densities (Table S4, Fig. S32 and S33, ESI†), we can see that for the quintet spin state, the sulfuric acid group has a spin density of ca. 0.5 in the 5I+ sub species and ca. 0.0 in the transition state. This indicates that the sulfuric acid group should be non-innocent to the reaction, and the non-innocence of the sulfuric acid group may make the reaction on the quintet spin state approachable. In addition, the low energy barrier indicates the high electrophilicity of the high-valent manganese–oxo species, which may originate from the existence of a low lying σ* orbital that can accept electronic density from the C–H bond (Fig. S33, ESI†).
Conclusions
In summary, we have reported the first example of sulfuric acid-enabled chemoselective oxidation of secondary alcohols by manganese catalysts and hydrogen peroxide.17 Secondary alcohols were oxidized to their corresponding ketones with good yields and an efficient OKR of racemic secondary alcohols was achieved with excellent enantioselectivities (>90% ee). Mechanistic studies revealed that the active manganese oxidant possesses electrophilic character, H-atom abstraction from an α-CH bond of the alcohol substrate is the rate-determining step, and alcohol oxidation occurs via a two-electron process. DFT calculations revealed that the difference in reaction energy barriers for H-atom abstraction from the α-CH bonds of the R- and S-enantiomers by a putative high-valent manganese–oxo intermediate is significant (i.e. 1.3 kcal mol−1), affording the enantioselectivity in the OKR of racemic secondary alcohols. Future studies will focus on the improvement of the catalytic activity as well as the enantioselectivity in the OKR of secondary alcohols, using synthetic nonheme iron and related manganese catalysts and environmentally benign oxidants such as molecular oxygen and hydrogen peroxide.
Experimental section
Materials
All chemicals were purchased from Aldrich, Alfa Aesar and TCI, which were of the best available purity and were used without further purification unless otherwise indicated. Solvents were dried according to published procedures and distilled under argon prior to use.18 PhCD(OH)CH3 was prepared from acetophenone according to the literature method.19 The ligands, MCP, P-MCP and Dbp-MCP, and their corresponding MnII complexes, MnII(MCP)(OTf)2, MnII(P-MCP)(OTf)2 and MnII(Dbp-MCP)(OTf)2, were prepared according to the reported methods.7,20
Instrumentation
1H and 13C NMR spectra were recorded on a Bruker AVANCE III 400 MHz spectrometer in CDCl3 at 25 °C. The reactions were monitored using thin layer chromatography (TLC). Commercial TLC plates (silica gel GF254) were developed and the spots were visualized under UV light at 254 or 365 nm. Silica gel column chromatography was performed with silica gel (particle size 200–300 mesh). Product analysis was performed on an Agilent Technologies 6890N gas chromatograph [GC; HP-5 column (length = 30 m and i.d. = 0.32 mm with 0.25 μm film thickness)] with a flame-ionization detector and a Thermo Finnigan (Austin, Texas, USA) FOCUS DSQ (dual stage quadrupole) mass spectrometer interfaced with a Finnigan FOCUS gas chromatograph (GC-MS). Gas chromatography (GC) analysis was performed on an Agilent Technologies 7890 with a flame-ionization detector and a CP-Chirasil-Dex CB column (length = 25 m and i.d. = 0.32 mm with 0.25 μm film thickness). High performance liquid chromatography (HPLC) analysis was performed on a Waters Breeze HPLC system (2487 Dual λ Absorbance Detector with a 1525 Binary HPLC Pump) equipped with a variable wavelength UV-220 detector. The Chiralpak IA column was purchased from Daicel Chemical Industries, Ltd.
Typical procedure for the oxidation of 1-phenylethanol
All reactions were performed under an Ar atmosphere using a dried solvent and standard Schlenk techniques. The catalyst (0.30 mol%), 1-phenylethanol (0.50 mmol) as a substrate and H2SO4 (0.30 mol%) were added into a Schlenk tube containing CH3CN (1.0 mL) at 25 °C. Subsequently, a solution of CH3CN (0.50 mL) containing 30% H2O2 (1.2 equiv.) was added dropwise using a syringe pump for 1 h. Then, the reaction solution was quenched with NaHCO3 and Na2S2O3, and n-decane was added into the solution as an internal standard. The yields were determined by GC.
Typical procedure for the OKR of 1-phenylethanol
All reactions were performed under an Ar atmosphere using a dried solvent and standard Schlenk techniques. The catalyst (0.20 mol%), racemic 1-phenylethanol (0.50 mmol) as a substrate and H2SO4 (1.0 mol%) were added into a Schlenk tube containing CH3CN (1.0 mL) at 0 °C. Subsequently, a solution of CH3CN (0.50 mL) containing 30% H2O2 (0.80 equiv.) was added dropwise using a syringe pump for 1 h. Then, the reaction solution was quenched with NaHCO3 and Na2S2O3, and n-decane was added into the solution as an internal standard. The yields were determined by GC (see Fig. 1 and Table 3; see also Tables S1 and S2, ESI†).
Determination of the relative rate constants (krel)
A solution of CH3CN (0.50 mL) containing 30% H2O2 (0.40 equiv.) was added dropwise into a CH3CN solution (1.0 mL) containing a mixture of 1-phenylethanol (0.50 mmol) and para-substituted 1-phenylethanol (0.50 mmol), the catalyst (1, 0.30 mol%) and H2SO4 (0.30 mol%) using a syringe pump at 25 °C for 1 h. Then, the reaction solution was quenched with NaHCO3 and Na2S2O3, and n-decane was added into the solution as an internal standard. The yields were determined by GC. The krel values were calculated using eqn (1),19a| | | krel = kR/kH = ln([R]f/[R]i)/ln([H]f/[H]i) | (1) |
where [R]i and [R]f are the initial and final concentrations of para-substituted 1-phenylethanol, respectively, and [H]i and [H]f are the initial and final concentrations of 1-phenylethanol, respectively.
Determination of the KIE value
A solution of CH3CN (0.50 mL) containing 30% H2O2 (0.40 equiv.) was added dropwise into a CH3CN solution (1.0 mL) containing a mixture of substrate (0.50 mmol; 1-phenylethanol or 1-deuterated 1-phenylethanol) and 1-(4-chlorophenyl)ethanol (0.50 mmol), 1 (0.30 mol%) and H2SO4 (0.30 mol%) using a syringe pump at 25 °C for 1 h. Then, the reaction solution was quenched with NaHCO3 and Na2S2O3, and n-decane was added into the solution as an internal standard. The yields were determined by GC. The KIE values were calculated using eqn (2)–(4),19a| | | kCl/kH = ln([Cl]f/[Cl]i)/ln([H]f/[H]i) | (2) |
| | | kCl/kD = ln([Cl]f/[Cl]i)/ln([D]f/[D]i) | (3) |
| | | KIE = (kCl/kD)/(kCl/kH) = kH/kD | (4) |
where [Cl]i and [Cl]f are the initial and final concentrations of 1-(4-chlorophenyl)ethanol, respectively, [H]i and [H]f are the initial and final concentrations of 1-phenylethanol, respectively, and [D]i and [D]f are the initial and final concentrations of 1-deuterated 1-phenylethanol, respectively.
Computational details
Density functional theory calculations were performed using Gaussian 09 software.21 The high-valent [(P-MCP)Mn5+(O2−)(SO42−)]+ species (I) was chosen as the reactive intermediate, and two enantiomers of the chiral 1-phenylethanol (S- and R-enantiomers) were used as the substrates. The spin-unrestricted B3LYP (UB3LYP) functional22,23 was employed with two basis sets: (1) the LACVP basis set for Mn and the 6-31G* basis set for the rest of the atoms, denoted as B1, were used to optimize the minima and transition states; (2) the LACV3P basis set for Mn and the 6-311+G** basis set for the rest of the atoms, denoted as B2, were used to obtain the single point energy corrections.24,25 The transition states and optimized minima were ascertained by vibrational frequency analysis with only one and zero imaginary frequencies, respectively. All calculations were performed in acetonitrile solvent using the self-consistent reaction field (SCRF) in the conductor-like polarizable continuum model (CPCM).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge financial support for this work from the National Natural Science Foundation of China (21473226 to W. S.), the Youth Innovation Promotion Association CAS (2015345 to C. M.), the Natural Science Foundation of Jiangsu Province (BK20161261 to C. M.) and the NRF of Korea through CRI (NRF-2012R1A3A2048842 to W. N.) and GRL (NRF-2010-00353 to W. N.).
Notes and references
-
(a) M. M. Abu-Omar, A. Loaiza and N. Hontzeas, Chem. Rev., 2005, 105, 2227 CrossRef CAS PubMed;
(b) C. Krebs, D. G. Fujimori, C. T. Walsh and J. M. Bollinger Jr, Acc. Chem. Res., 2007, 40, 484 CrossRef CAS PubMed;
(c) W. Nam, Acc. Chem. Res., 2007, 40, 522 CrossRef CAS PubMed;
(d) S. A. Cook and A. S. Borovik, Acc. Chem. Res., 2015, 48, 2407 CrossRef CAS PubMed;
(e) W. Nam, Acc. Chem. Res., 2015, 48, 2415 CrossRef CAS PubMed;
(f) M. Puri and L. Que Jr, Acc. Chem. Res., 2015, 48, 2443 CrossRef CAS PubMed;
(g) K. Ray, F. Heims, M. Schwalbe and W. Nam, Curr. Opin. Chem. Biol., 2015, 25, 159 CrossRef CAS PubMed;
(h) X. Engelmann, I. Monte-Pérez and K. Ray, Angew. Chem., Int. Ed., 2016, 55, 7632 CrossRef CAS PubMed;
(i) S. Sahu and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 11410 CrossRef CAS PubMed;
(j) D. A. Proshlyakov, J. McCracken and R. P. Hausinger, J. Biol. Inorg. Chem., 2017, 22, 367 CrossRef CAS PubMed;
(k) T. H. Yosca, A. P. Ledray, J. Ngo and M. T. Green, J. Biol. Inorg. Chem., 2017, 22, 209 CrossRef CAS PubMed;
(l) S. Kal and L. Que Jr, J. Biol. Inorg. Chem., 2017, 22, 339 CrossRef CAS PubMed.
-
(a) L. Que Jr and W. B. Tolman, Nature, 2008, 455, 333 CrossRef PubMed;
(b) K. Gopalaiah, Chem. Rev., 2013, 113, 3248 CrossRef CAS PubMed;
(c) P. Saisaha, J. W. de Boer and W. R. Browne, Chem. Soc. Rev., 2013, 42, 2059 RSC;
(d) F. G. Gelalcha, Adv. Synth. Catal., 2014, 356, 261 CrossRef CAS;
(e) Z. Chen and G. Yin, Chem. Soc. Rev., 2015, 44, 1083 RSC;
(f) T. J. Collins and A. D. Ryabov, Chem. Rev., 2017, 117, 9140 CrossRef CAS PubMed.
-
(a) Z. Codola, J. Lloret-Fillol and M. Costas, Prog. Inorg. Chem., 2014, 59, 447 CAS;
(b) O. Cussó, X. Ribas and M. Costas, Chem. Commun., 2015, 51, 14285 RSC;
(c) G. Olivo, O. Cussó and M. Costas, Chem.–Asian J., 2016, 11, 3148 CrossRef CAS PubMed;
(d) M. Canta, M. Rodríguez and M. Costas, Top. Curr. Chem., 2016, 372, 27 CrossRef CAS PubMed;
(e) G. Olivo, O. Cussó, M. Borrell and M. Costas, J. Biol. Inorg. Chem., 2017, 22, 425 CrossRef CAS PubMed;
(f) M. Milan, M. Bietti and M. Costas, ACS Cent. Sci., 2017, 3, 196 CrossRef CAS PubMed.
-
(a) W. N. Oloo and L. Que Jr, Acc. Chem. Res., 2015, 48, 2612 CrossRef CAS PubMed;
(b) W. N. Oloo, K. K. Meier, Y. Wang, S. Shaik, E. Münck and L. Que Jr, Nat. Commun., 2014, 5, 3046 Search PubMed;
(c) Y. Wang, D. Janardanan, D. Usharani, K. Han, L. Que Jr and S. Shaik, ACS Catal., 2013, 3, 1334 CrossRef CAS;
(d) Y. Feng, J. England and L. Que Jr, ACS Catal., 2011, 1, 1035 CrossRef CAS.
-
(a) E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418 CrossRef CAS;
(b) K. P. Bryliakov and E. P. Talsi, Coord. Chem. Rev., 2014, 276, 73 CrossRef CAS;
(c) R. V. Ottenbacher, E. P. Talsi and K. P. Bryliakov, Molecules, 2016, 21, 1454 CrossRef PubMed;
(d) R. V. Ottenbacher, E. P. Talsi and K. P. Bryliakov, Catal. Today, 2016, 278, 30 CrossRef CAS;
(e) A. M. Zima, O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2017, 7, 60 CrossRef CAS;
(f) K. P. Bryliakov, Chem. Rev., 2017, 117, 11406 CrossRef CAS PubMed.
-
(a) M. C. White, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 7194 CrossRef CAS PubMed;
(b) M. S. Chen and M. C. White, Science, 2010, 327, 566 CrossRef CAS PubMed;
(c) M. A. Bigi, S. A. Reed and M. C. White, Nat. Chem., 2011, 3, 216 CrossRef CAS PubMed;
(d) T. J. Osberger, D. C. Rogness, J. T. Kohrt, A. F. Stepan and M. C. White, Nature, 2016, 537, 214 CrossRef CAS PubMed.
- C. Miao, B. Wang, Y. Wang, C. Xia, Y.-M. Lee, W. Nam and W. Sun, J. Am. Chem. Soc., 2016, 138, 936 CrossRef CAS PubMed.
-
(a)
G. Tojo and M. Fernández, Oxidation of alcohols to aldehydes and ketones, Springer, New York, 2010 Search PubMed;
(b) J. Brinksma, M. T. Rispens, R. Hage and B. L. Feringa, Inorg. Chim. Acta, 2002, 337, 75 CrossRef CAS;
(c) K. Nehru, S. J. Kim, I. Y. Kim, M. S. Seo, Y. Kim, S.-J. Kim, J. Kim and W. Nam, Chem. Commun., 2007, 44, 4623 RSC;
(d) Z. Hu, H. Du, W.-L. Man, C.-F. Leung, H. Liang and T.-C. Lau, Chem. Commun., 2012, 48, 1102 RSC;
(e) X. Wu, X. Yang, Y.-M. Lee, W. Nam and L. Sun, Chem. Commun., 2015, 51, 4013 RSC;
(f) P. Tan, H.-K. Kwong and T.-C. Lau, Chem. Commun., 2015, 51, 12189 RSC.
-
(a) D. Shen, C. Miao, D. Xu, C. Xia and W. Sun, Org. Lett., 2015, 17, 54 CrossRef CAS PubMed;
(b) W. Dai, Y. Lv, L. Wang, S. Shang, B. Chen, G. Li and S. Gao, Chem. Commun., 2015, 51, 11268 RSC;
(c) P. Saisaha, J. J. Dong, T. G. Meinds, J. W. de Boer, R. Hage, F. Mecozzi, J. B. Kasper and W. R. Browne, ACS Catal., 2016, 6, 3486 CrossRef CAS.
-
(a) N. Y. Oh, Y. Suh, M. J. Park, M. S. Seo, J. Kim and W. Nam, Angew. Chem., Int. Ed., 2005, 44, 4235 CrossRef CAS PubMed;
(b) J. Yoon, S. A. Wilson, Y. K. Jang, M. S. Seo, K. Nehru, B. Hedman, K. O. Hodgson, E. Bill, E. I. Solomon and W. Nam, Angew. Chem., Int. Ed., 2009, 48, 1257 CrossRef CAS PubMed;
(c) M. Ghosh, Y. L. K. Nikhil, B. B. Dhar and S. S. Gupta, Inorg. Chem., 2015, 54, 11792 CrossRef CAS PubMed;
(d) H. Kotani, S. Kaida, T. Ishizuka, M. Sakaguchi, T. Ogura, Y. Shiota, K. Yoshizawa and T. Kojima, Chem. Sci., 2015, 6, 945 RSC.
-
(a) E. Vedejs and M. Jure, Angew. Chem., Int. Ed., 2005, 44, 3974 CrossRef CAS PubMed;
(b)
E. M. Carreira and L. Kvaerno, Classics in Stereoselective Synthesis, Wiley-VCH, Weinheim, Germany, 2009 Search PubMed;
(c) A. Lumbroso, M. L. Cooke and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 1890 CrossRef CAS PubMed;
(d)
E. J. Corey and L. Kurti, Enantioselective Chemical Synthesis: Methods, Logic, and Practice, Academic Press, New York, 2014 Search PubMed.
-
(a) T. Kunisu, T. Oguma and T. Katsuki, J. Am. Chem. Soc., 2011, 133, 12937 CrossRef CAS PubMed;
(b) K. Murakami, Y. Sasano, M. Tomizawa, M. Shibuya, E. Kwon and Y. Iwabuchi, J. Am. Chem. Soc., 2014, 136, 17591 CrossRef CAS PubMed;
(c) H. Mizoguchi, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2014, 53, 3178 CrossRef CAS PubMed;
(d) L. Ren, L. Li, Y. Li, G. Zhang, B. Huang, Z. Imam, A. Zheng and Y. Sun, J. Porous Mater., 2016, 23, 19 CrossRef CAS.
-
(a) W. Sun, H. Wang, C. Xia, J. Li and P. Zhao, Angew. Chem., Int. Ed., 2003, 42, 1042 CrossRef CAS PubMed;
(b) Y. Zhang, B. Gao, Q. Zhou and J. Zhao, Catal. Lett., 2014, 144, 1797 CrossRef CAS;
(c) M. K. Brown, M. M. Blewett, J. R. Colombe and E. J. Corey, J. Am. Chem. Soc., 2010, 132, 11165 CrossRef CAS PubMed.
-
(a) R. I. Kureshy, I. Ahmad, K. Pathak, N. H. Khan, S. H. R. Abdi, J. K. Prathap and R. V. Jasra, Chirality, 2007, 19, 352 CrossRef CAS PubMed;
(b) Y. Zhang, Q. Zhou, W. Ma and J. Zhao, Catal. Commun., 2014, 45, 114 CrossRef CAS;
(c) Q. Cheng, F. Guo, C. Xia and W. Sun, Tetrahedron: Asymmetry, 2008, 19, 2359 CrossRef CAS.
- W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146 CrossRef CAS PubMed.
-
(a) J. Roček and A. E. Radkowsky, J. Am. Chem. Soc., 1973, 95, 7123 CrossRef;
(b) O. Pestovsky and A. Bakac, J. Am. Chem. Soc., 2004, 126, 13757 CrossRef CAS PubMed;
(c) R. Ray, S. Chandra, D. Maiti and G. K. Lahiri, Chem.–Eur. J., 2016, 22, 8814 CrossRef CAS PubMed.
- E. P. Talsi, D. G. Samsonenko and K. P. Bryliakov, ChemCatChem, 2017, 9, 2599 CrossRef CAS.
-
W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Pergamon Press, Oxford, UK, 6th edn, 2009 Search PubMed.
-
(a) W.-H. Fung, W.-Y. Yu and C.-M. Che, J. Org. Chem., 1998, 63, 2873 CrossRef CAS;
(b) D. Klomp, T. Maschmeyer, U. Hanefeld and J. A. Peters, Chem.–Eur. J., 2004, 10, 2088 CrossRef CAS PubMed.
-
(a) A. Murphy, G. Dubois and T. D. P. Stack, J. Am. Chem. Soc., 2003, 125, 5250 CrossRef CAS PubMed;
(b) A. Murphy, A. Pace and T. D. P. Stack, Org. Lett., 2004, 6, 3119 CrossRef CAS PubMed;
(c) A. Murphy and T. D. P. Stack, J. Mol. Catal. A: Chem., 2006, 251, 78 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 37, 785 CrossRef CAS.
- J. P. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef.
- R. A. Friesner, R. B. Murphy, M. D. Beachy, M. N. Ringlanda, M. T. Pollard, B. D. Dunietz and Y. X. Cao, J. Phys. Chem. A, 1999, 103, 1913 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables S1–S4 and additional data: NMR spectra of the products, GC and HPLC chromatograms in the OKR of secondary alcohols, key geometric information for DFT, etc. See DOI: 10.1039/c7sc00891k |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Chungu
Xia
a,
Yong
Wang
a,
Wonwoo
Nam
c,
Chungu
Xia
a,
Yong
Wang
a,
Wonwoo
Nam












