
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual cobalt–copper light-driven catalytic reduction of aldehydes and aromatic ketones in aqueous media†
Arnau
Call‡
 a,
Carla
Casadevall‡
a,
Ferran
Acuña-Parés
a,
Alicia
Casitas
a and
Julio
Lloret-Fillol
*ab
a,
Carla
Casadevall‡
a,
Ferran
Acuña-Parés
a,
Alicia
Casitas
a and
Julio
Lloret-Fillol
*ab
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Avinguda Països Catalans 16, 43007 Tarragona, Spain. E-mail: jlloret@iciq.es
bCatalan Institution for Research and Advanced Studies (ICREA), Passeig Lluïs Companys, 23, 08010, Barcelona, Spain
First published on 1st June 2017
Abstract
We present an efficient, general, fast, and robust light-driven methodology based on earth-abundant elements to reduce aryl ketones, and both aryl and aliphatic aldehydes (up to 1400 TON). The catalytic system consists of a robust and well-defined aminopyridyl cobalt complex active for photocatalytic water reduction and the [Cu(bathocuproine)(Xantphos)](PF6) photoredox catalyst. The dual cobalt–copper system uses visible light as the driving-force and H2O and an electron donor (Et3N or iPr2EtN) as the hydride source. The catalytic system operates in aqueous mixtures (80–60% water) with high selectivity towards the reduction of organic substrates (>2000) vs. water reduction, and tolerates O2. High selectivity towards the hydrogenation of aryl ketones is observed in the presence of terminal olefins, aliphatic ketones, and alkynes. Remarkably, the catalytic system also shows unique selectivity for the reduction of acetophenone in the presence of aliphatic aldehydes. The catalytic system provides a simple and convenient method to obtain α,β-deuterated alcohols. Both the observed reactivity and the DFT modelling support a common cobalt hydride intermediate. The DFT modelled energy profile for the [Co–H] nucleophilic attack to acetophenone and water rationalises the competence of [CoII–H] to reduce acetophenone in the presence of water. Mechanistic studies suggest alternative mechanisms depending on the redox potential of the substrate. These results show the potential of the water reduction catalyst [Co(OTf)(Py2Tstacn)](OTf) (1), (Py2Tstacn = 1,4-di(picolyl)-7-(p-toluenesulfonyl)-1,4,7-triazacyclononane, OTf = trifluoromethanesulfonate anion) to develop light-driven selective organic transformations and fine solar chemicals.
Introduction
Catalysts developed for the reduction of H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 and CO22 in the context of artificial photosynthesis (AP) have the potential to provide greener light-driven methodologies for sustainable synthetic methods.3 The production of fine chemicals has less scaling and economic restriction than the synthesis of energy carriers. In this regard, using catalytic systems derived for AP to perform selective organic transformations is highly appealing.3c,d,4 Remarkable examples of catalytic systems based on semiconductors,5 enzymes,4a–c,i,6 and, more recently, homogeneous catalysts,4j,7 although with reduced scope or selectivity, have been shown to be active for specific transformations.
1 and CO22 in the context of artificial photosynthesis (AP) have the potential to provide greener light-driven methodologies for sustainable synthetic methods.3 The production of fine chemicals has less scaling and economic restriction than the synthesis of energy carriers. In this regard, using catalytic systems derived for AP to perform selective organic transformations is highly appealing.3c,d,4 Remarkable examples of catalytic systems based on semiconductors,5 enzymes,4a–c,i,6 and, more recently, homogeneous catalysts,4j,7 although with reduced scope or selectivity, have been shown to be active for specific transformations.
For instance, semiconductor materials, such as TiO2 or CdS, provide much lower redox potentials, promoting direct reduction reactions via one or two photoinduced electron transfer processes, but at the expense of using UV light, novel metals, and/or obtaining low to moderate selectivity (Scheme 1).5a–g The asymmetric reduction of acetophenones and α-ketoglutarate has been achieved by coupling (i) a sacrificial electron donor, (ii) a photosensitizer, (iii) a noble-metal-based catalyst, (iv) an electron carrier, and (v) an enzyme that carries out the enantioselective transformation.4a–c,i,6 Results of the latter case show that selective light-driven reduction of organic substrates, using water as a formal dihydrogen source, is a feasible transformation, albeit limited to a specific substrate.4a–f,i However, due to the complexity of these catalytic systems, optimization and mechanistic studies are difficult.
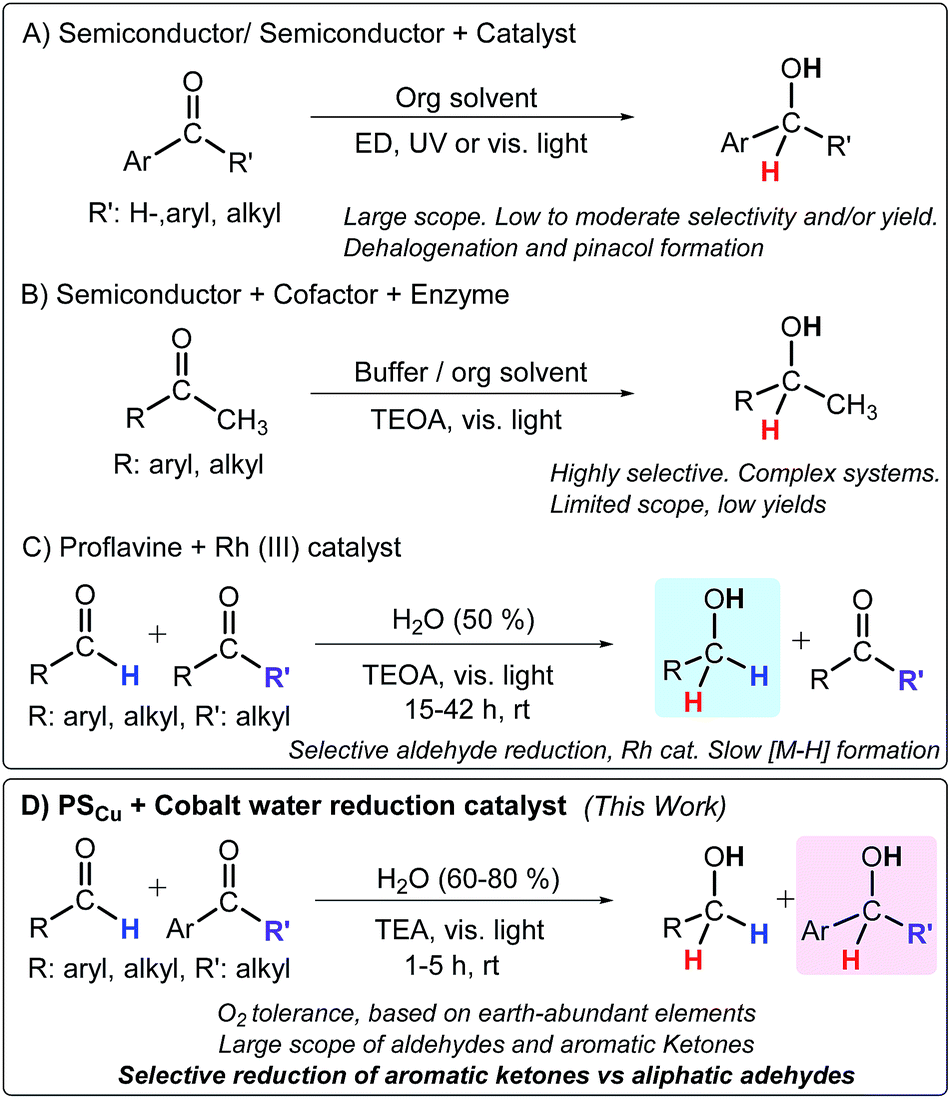 | ||
| Scheme 1 Methodologies for the light-driven reduction of carbonyl compounds. Abbreviations: ED: electron donor, TEOA: triethanolamine, TEA: triethylamine. Selected references: (A) ref. 5 and 7, (B) ref. 4a, e and 6, and (C) ref. 4j. (D) Developed methodology in this study. | ||
The combination of photoredox catalysts with well-defined molecular complexes is a powerful approach towards light-driven reduction of organic molecules.8 The introduction of a photoredox catalytic cycle potentially enables the use of water and an electron donor as reductive equivalents, while organometallic or coordination complexes can serve as selective hydrogenation catalysts with a broad scope. In this regard, coordination complexes based on Rh,4j Ru,7b and Ir7c have been explored. A remarkable example is the one reported by König and co-workers for the selective visible light photoreduction (λ = 455 nm) of aldehydes to alcohols in the presence of ketones. This catalytic system consists of proflavine (PF) as the photocatalyst and [Cp*RhIII(bpy)Cl]Cl as a well-known hydrogenation catalyst in the presence of triethanolamine (TEOA) as a sacrificial electron donor.4j Nevertheless, while aldehydes can be easily reduced using this system, ketones are not suitable substrates (Scheme 1).
Furthermore, remarkable efforts have been focused on developing hydrogenation catalysts based on earth-abundant elements.9 However, they are usually sensitive to O2. Therefore, catalysts based on earth-abundant elements that are resilient to O2, operate in H2O, and can reduce organic substrates using light as the energy source and water/an electron donor as a source of hydrides could have a beneficial impact in the synthesis of drugs, pesticides, and organic chemicals in general.10 In this vein, efficient catalysts for the reduction of water to hydrogen such as cobalt complexes based on glyoxime,11 diimine-glyoxime,1c and aminopyridine1b,d ligands are promising candidates as hydrogenation catalysts of organic substrates under similar reaction conditions.1f,h,12 Mechanistic investigations of these systems suggest that molecular [Co–H] species are key intermediates in H2 formation.13 Therefore, photochemically obtained [Co–H] intermediates based on these ligands could potentially be catalytic intermediates for the reduction of organic functionalities in H2O using amine/H2O as a hydride source (Scheme 1).
Herein, we present a methodology to reduce aromatic ketones and both aliphatic and aromatic aldehydes using dual cobalt–copper light-driven catalysis using H2O/(Et3N or iPr2EtN) as a hydride source (Scheme 2). The dual metal catalytic system is formed by an aminopyridine cobalt complex (1) and [Cu(bathocuproine)(Xantphos)](PF6)14 (PSCu) (bathocuproine = 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline and Xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene) as the photoredox catalyst (Scheme 2). Our reactivity studies suggest a common intermediate, most probably [Co–H], is responsible for the reduction of organic substrates and water. Using water/amine and light as reductive equivalents, we achieved a selective catalytic reduction of aromatic ketones in the presence of aliphatic ketones, aliphatic aldehydes, aliphatic alkenes, and alkynes. This unique selectivity is rationalised based on reactivity and isotopic labelling.
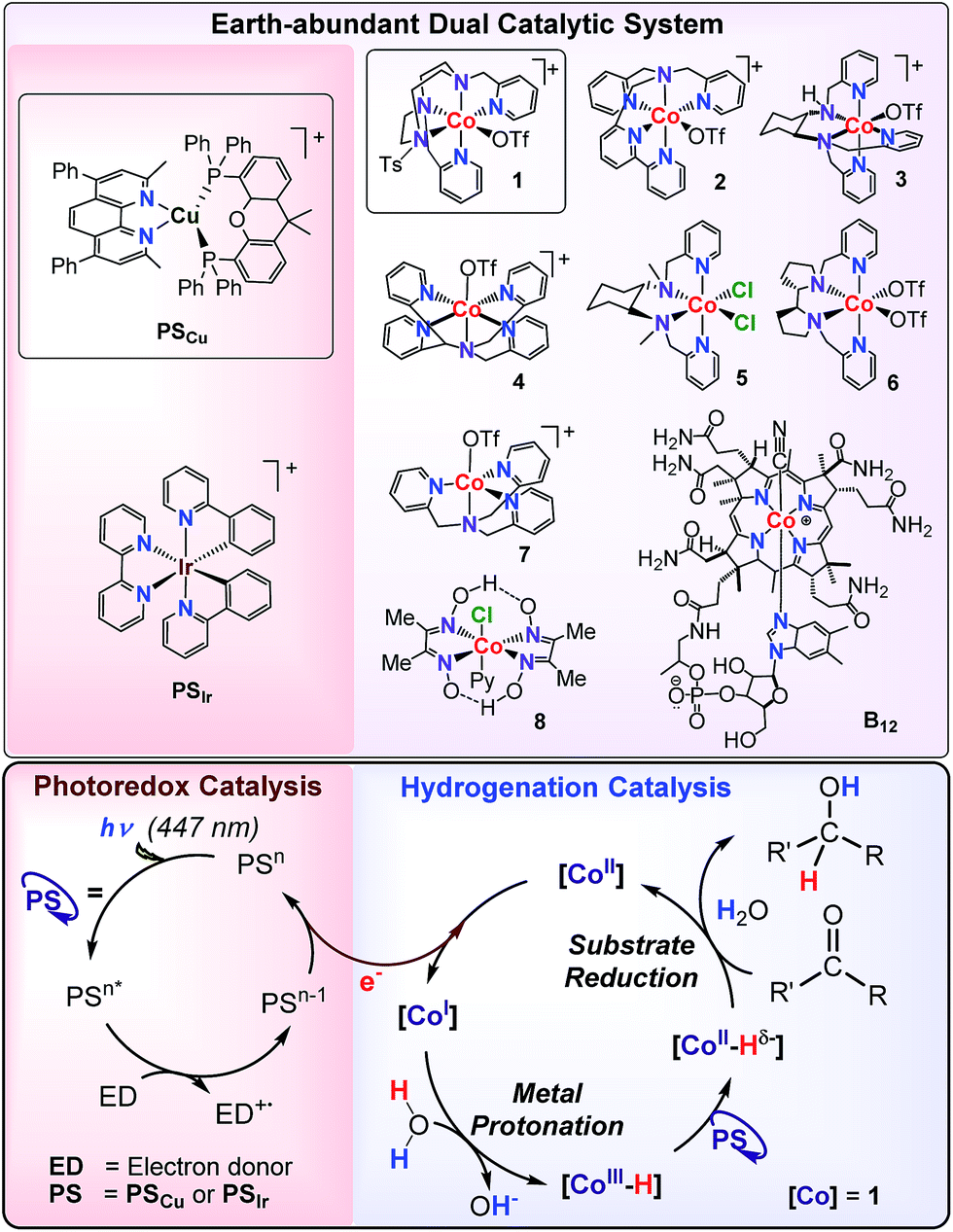 | ||
| Scheme 2 Earth-abundant dual catalytic system for the photoreduction of aromatic ketones and aldehydes. | ||
Results and discussion
For explorative purposes we examined the metal-catalyzed light-driven reduction of acetophenone (9a) as a model substrate under similar conditions to those recently reported by our group for the reduction of water to H2.15 Acetophenone (66 mM), [M(OTf)(Py2Tstacn)](OTf) (M = Co (1), Fe (1Fe), Ni (1Ni)), (Py2Tstacn = 1,4-di(picolyl)-7-(p-toluenesulfonyl)-1,4,7-triazacyclononane, OTf = trifluoromethanesulfonate anion) (1 mol%) as reduction catalysts, [Ir(bpy)(ppy)2](PF6) (PSIr, ppy = 2-phenylpyridine) (0.5 mol%) as the photoredox catalyst and Et3N (2.1 equiv.) as the electron donor were mixed in a solvent mixture H2O:CH3CN (3.5:1.5 mL) and irradiated for 5 h at 447 ± 20 nm using an in-house developed high throughput photoreactor, which allows for temperature and light intensity control (Fig. SI.1.1†). Cobalt complex 1 was the only complex yielding a significant amount of alcohol (23%), with quantitative recovery of the ketone 9a (ESI Table SI.1.1,† entry 1). The inactivity of the analogous iron (1Fe) and nickel (1Ni) complexes can be explained by the impossibility of generating stable M–H intermediates under photochemical conditions.15 Encouraged by this result, we screened catalyst/photosensitizer/electron-donor ratios and solvent mixtures, maximizing the alcohol yield up to 65% (initial rate = 0.065 mmol per h) (Table SI.1.1,† entry 5). Control experiments demonstrated that all components are required for the light-driven reduction of acetophenone (9a) (see Table SI.1.1,† entries 12–14).
Light–dark cycles for the reduction of 9a show that the formation of the corresponding reduced alcohol 10a stops in the dark and is restored upon irradiation with similar kinetics (Fig. 1). This indicates that the catalytic system is not degrading in the absence of light. The reaction is selective; the only organic products detected from 1H-NMR or GC monitoring are 9a and 10a (see Fig. SI.1.11†).
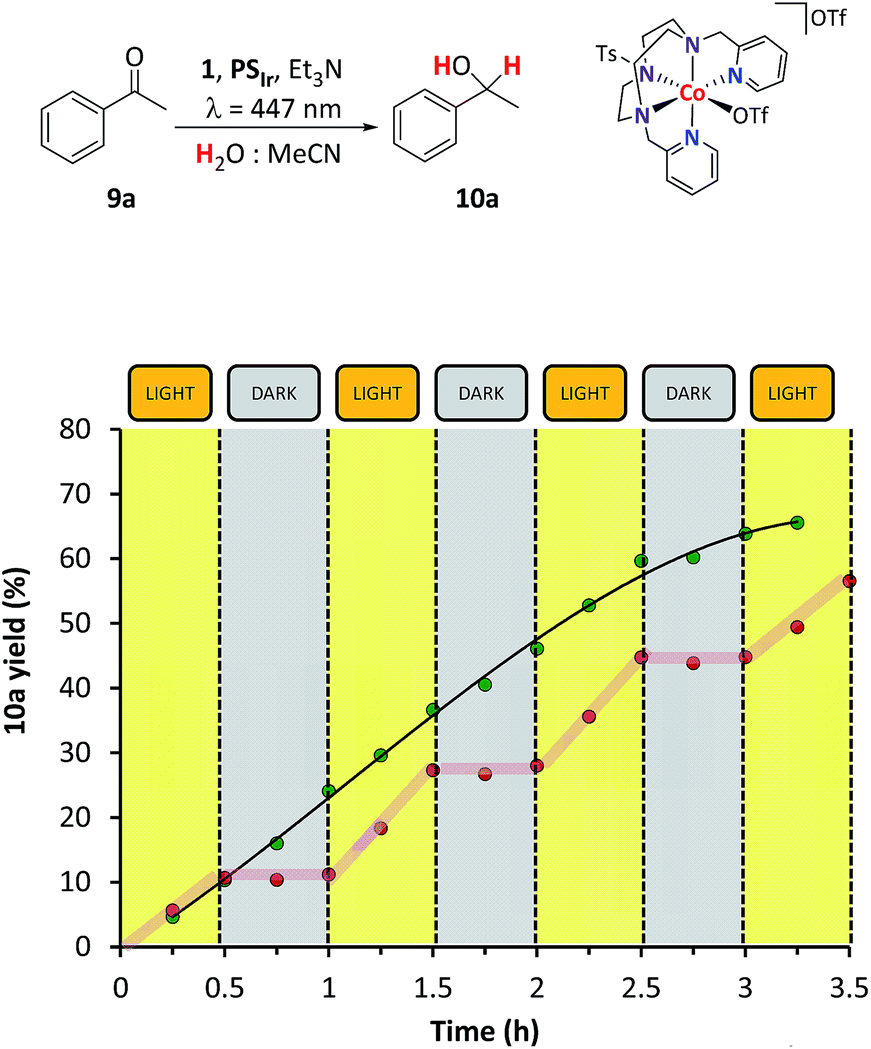 | ||
| Fig. 1 Comparison between light–dark cycles (red circles) and continuous irradiation (green circles) at 447 nm. The yields are determined by GC analysis relative to calibrated internal standard. Conditions: 1 (0.37 mM, 3 mol%), PSIr (0.25 mM, 2 mol%), substrate 9a (12.4 mM) in H2O:CH3CN:Et3N (8:2:0.2 mL) at 30 °C. | ||
An important amount of H2 was detected in the headspace of the reaction vials. Interestingly, the substrate inhibited the hydrogen evolution; the higher the substrate concentration the higher the inhibition. More importantly, the total amount of H2 + 10a formed was constant for all tested concentrations of 9a, which suggests competing pathways with a common intermediate (Fig. 2). Control experiments discard the idea that photogenerated H2 is the reducing agent of acetophenone. The reaction does not proceed in the dark under an H2 atmosphere and reaction rates under visible-light are essentially the same under H2 or N2 atmospheres, without induction time (see Fig. SI.1.12†). Finally, Hg poisoning experiments did not modify the reaction outcome (61% yield 10a), suggesting that the main catalytic activity is derived from a molecular system.16
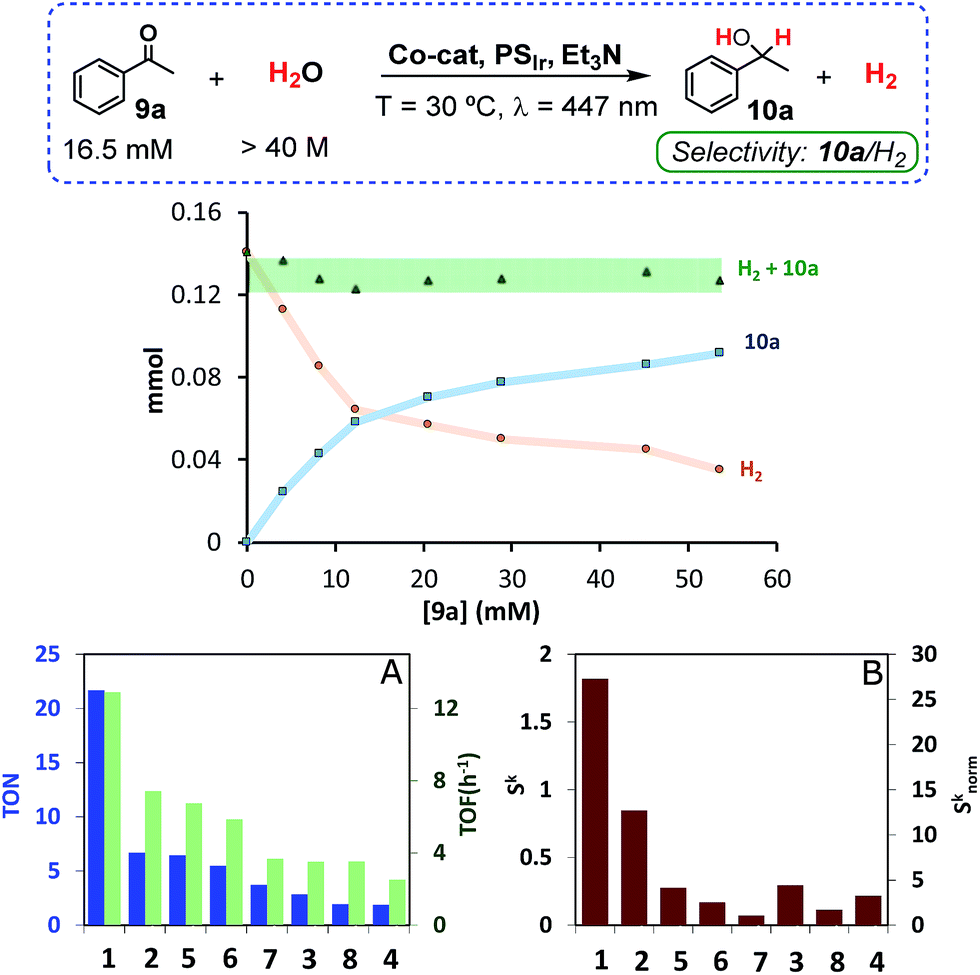 | ||
| Fig. 2 (Top) Alcohol (10a) and H2 formed at different concentrations of 9a under optimized photochemical conditions. (Bottom) Catalytic activity and selectivity for 9a reduction under optimized conditions, (A) TON = (n(10a)/n(Co-cat.)) and TOF = TON/t. (B) Selectivity ketone vs. H2 (Sk = (n(10a)/n(H2)), and normalized selectivity with respect to 7 (Sknorm). Conditions: Co-cat. (0.49 mM, 3 mol%) and PSIr (0.25 mM, 1.5 mol%), 9a (16.5 mM) in H2O:CH3CN:Et3N (8:2:0.2 mL) at 30 °C. | ||
Light-driven hydrogen evolution and ketone reduction activity of representative cobalt complexes: choosing the right cobalt catalyst
We hypothesised that the reaction mechanisms for the cobalt-catalysed light-driven catalytic reduction of both H2O and ketones share the same key cobalt–hydride intermediate. Therefore, a series of aminopyridine based cobalt complexes, known to be active for water reduction, were evaluated for the photocatalytic reduction of 9a. We studied cobalt complexes with pentadentate (2–4)17 and tetradentate (5–7)18 chelating nitrogen ligands as well as commercially available cobaloxime 819 and vitamin B12 (Scheme 2).Remarkably, all cobalt complexes (1–8) except vitamin B12 showed photocatalytic activity in the reduction of 9a (<0.5% yield of 10a) (Table SI.1.3†). The observed catalytic activity strongly depends on the nature of the cobalt complex employed. For instance, the most efficient complex, 1 (65% yield, initial rate = 0.065 mmol 10a per h) is about 2 fold more reactive than complex 2.
Next, we evaluated the selectivity of the reduction of 9avs. water. Without 9a, all complexes (except vitamin B12) produced large quantities of H2 under both typical conditions for H2 evolution and our optimized conditions for the reduction of ketones (Fig. SI.1.3 and 4†). However, in the presence of 9a, we observed notable differences in the selectivity of 9avs. H2O reduction among the cobalt catalysts tested, which illustrate that selectivity can be tuned by the ligand employed (Fig. 2). For instance, cobalt complexes bearing aminopyridine tetradentate ligands offer high selectivity towards H2 evolution (ratio (H2/10a): 3.6–13.9), while pentadentate ligands display an excellent selectivity towards the reduction of 9a (ratio (10a/H2): 1.7 and 0.8 for 1 and 2, respectively). Based on these results a general trend can be derived for each set of complexes: the higher the ligands’ basicity, the more active the catalyst is for ketone reduction.
Among the studied complexes, 1 is the most active and selective catalyst towards ketone reduction. The observed selectivity is remarkable since [9a] (16.5 mM) is about 2500 fold lower than [H2O] (>40 M) and it is expected that M–H intermediates react very rapidly with water to form H2.
Dual cobalt–copper light-driven catalytic reduction
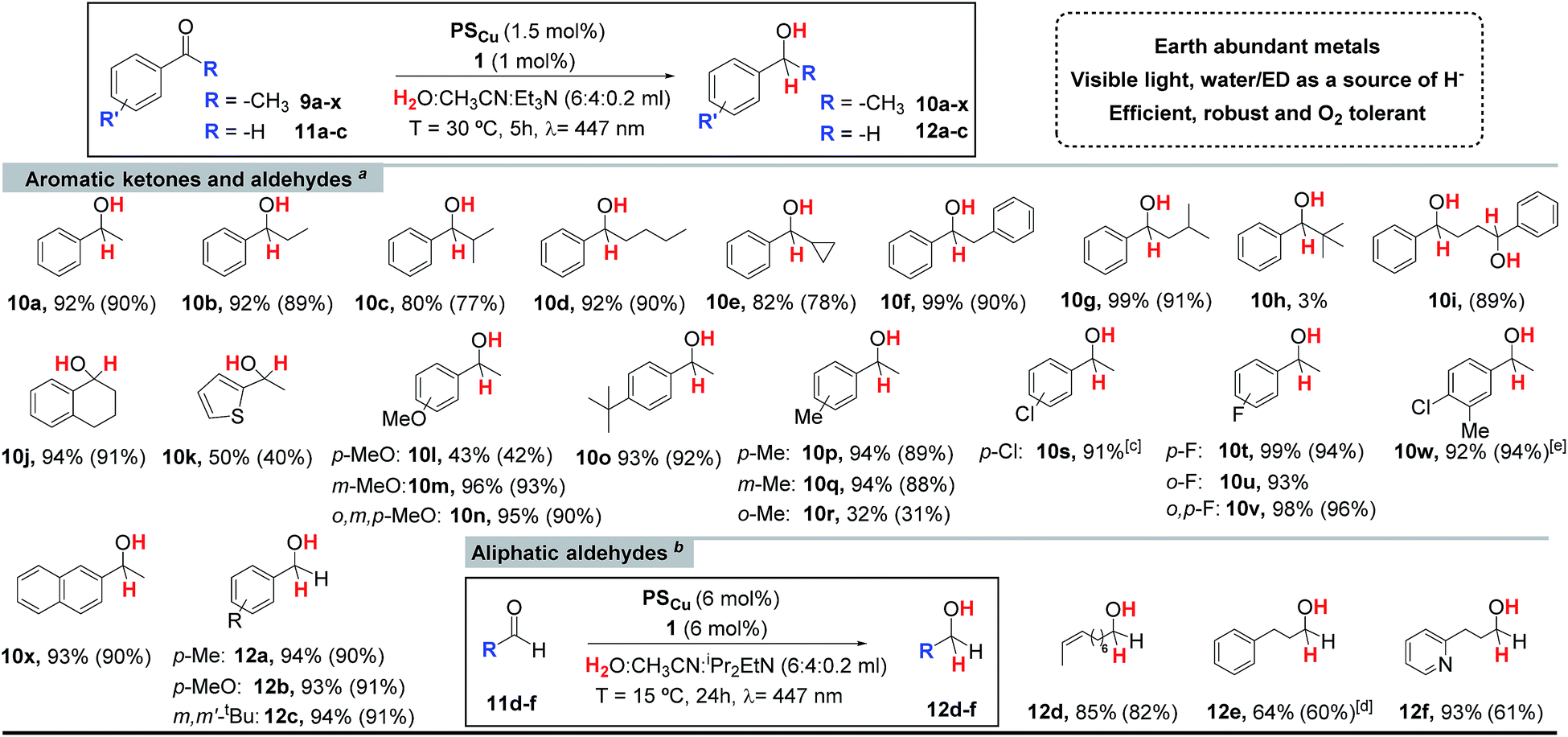
Encouraged by the excellent performance of 1 we carried out further optimization studies. The combination of complex 1 with the photoredox catalyst [Cu(bathocuproine)(Xantphos)](PF6) (PSCu), previously studied by Beller et al. for the reduction of H2O to H2,14 substantially improved the yield of alcohol 10a (92%) (Table SI.1.2,† entry 2). Control experiments show that the reaction does not proceed in the absence of complex 1. Under these conditions, the photocatalytic ketone reduction is compatible with O2 (Fig. SI.1.9†). Indeed, the reduction of 9a was quantitative when the reaction was carried out under air atmosphere and in non-degassed solvents in a crimped vial without headspace. We rationalize the result by the potential capacity of the catalytic system to reduce O2 to H2O and the remarkable stability of our catalytic system under air.15 Moreover, quantitative reduction of acetophenone can be obtained even at 0.25 mol% of cobalt catalyst (TON = 1400) (Table SI.1.6,† entry 14).A wide range of alkyl aryl ketones can be reduced to the corresponding alcohols by using 1 (1 mol%) and PSCu (1.5 mol%) in a H2O:CH3CN:Et3N (6:4:0.2 mL) solvent mixture under N2 with 5 h of irradiation at 447 nm. In general, isolated yields are high (Table 1), but are affected by the size of the alkyl group of the ketone. The electronic effects of the substrate influence the reaction as illustrated by the lower alcohol yields observed in the case of electron rich ketones such as 9k and 9l (40 and 42% respectively). The catalytic system developed is selective towards aromatic ketones. Aliphatic ketones, such as cyclohexanone (9z) or nonan-2-one (9aa) almost do not react (<5% yield). The developed methodology tolerates fluorine and chlorine substituted aromatic rings (9s–9x), which are reduced to the corresponding alcohols (10s–10x) in high yields (81–95%) (Table 1). However, brominated substrates such as 4-bromoacetophenone (9ab) and 2-bromo-4-methylacetophenone (9ac) quantitatively give the dehalogenated products 10a and 9p, respectively. The expected reactivity of aromatic ketones under reductive enough photochemical conditions, is the formation of ketyl radical species via single electron transfer, which finally dimerize to form pinacols.5c In this regard, efficient photoredox catalytic protocols for the reductive coupling of aldehydes and ketones have been developed by the groups of Sudo20 and Rueping.21 We noted that the formation of pinacols is suppressed for the tested ketones in the presence of the cobalt catalyst 1.
|
a Standard catalytic conditions: 1 (1 mol%), PSCu (1.5 mol%), Subs. (16.5 mM) in H2O:CH3CN:Et3N (6:4:0.2 mL) irradiated (447 nm) for 5 h at 30 °C under N2.
b Optimized catalytic conditions: 1 (6 mol%), PSCu (6 mol%), Subs. (4.4 mM) in H2O:CH3CN:iPr2EtN (6:4:0.2 mL) irradiated (447 nm) for 24 h at 15 °C under N2.
c Formation of 10a was detected (5% yield).
d Optimized conditions using 8.7 mM of substrate. Yields after workup (average of triplicates) determined by GC analysis relative to calibrated internal standard. Isolated yields between parentheses (average of 16 reactions).
|
|---|
|
The catalytic dual PSCu/1 system can be applied for the reduction of the aromatic aldehydes 11a–11c, which are converted to the corresponding alcohols with excellent yields (Table 1). On the other hand, the aliphatic aldehydes 11d–f were reduced with lower yields and required further optimization (see Tables SI.1.8 and 9†). We found that using a bulkier electron donor (diisopropylethylamine (DIPEA)), increasing catalyst loading (up to 6%), decreasing the substrate concentration (down to 8.7 mM), and performing the reaction at lower temperature (15 °C) improved the yield of the targeted alcohol 12e up to 64% (Table SI.1.8,† entry 24). In the case of aliphatic aldehydes, no pinacol products were detected either in the presence or absence of the cobalt catalyst. This is in agreement with the significantly lower redox potential of aliphatic aldehydes in comparison to those of aromatic ones or aromatic ketones.22 The redox potential of the aliphatic aldehydes 11d–f is lower than −2 V vs. SCE under catalytic conditions (H2O:CH3CN:Et3N, 6:4:0.2)23 (<−2.2 V vs. SCE22 in CH3CN), while the redox potential for ketones and aromatic aldehydes is much higher (Ered for 9l and 12a are −1.74, and −1.55 V vs. SCE, respectively). This serves as evidence that for the most electron rich substrates the formation of the ketyl radical is not viable, while for the most electron poor substrates it should be considered, since the redox potential of PSCu is about −1.53 V vs. SCE (Fig. SI.1.14†). This aspect will be further addressed in the mechanistic discussion.
Chemoselectivity
Stoichiometric competition experiments showed that the photocatalytic system is able to reduce acetophenone (9a) selectively in the presence of 1-phenylpropan-2-one (9ad), cyclohexanone (9ae) or 2-acetyl-1-methylpyrrole (9af) (Scheme 3). We also carried out competition experiments between 9a and aliphatic aldehydes (11d, 11e and 11f). Extraordinarily, the aromatic ketone was quantitatively reduced with an excellent unexpected selectivity in all three cases (Scheme 3 and Fig. 3). GC monitoring of the reactions showed that 9a was consumed and converted to 10a, whereas the aliphatic aldehyde remained virtually intact (Fig. 3 and SI.2.129–146†).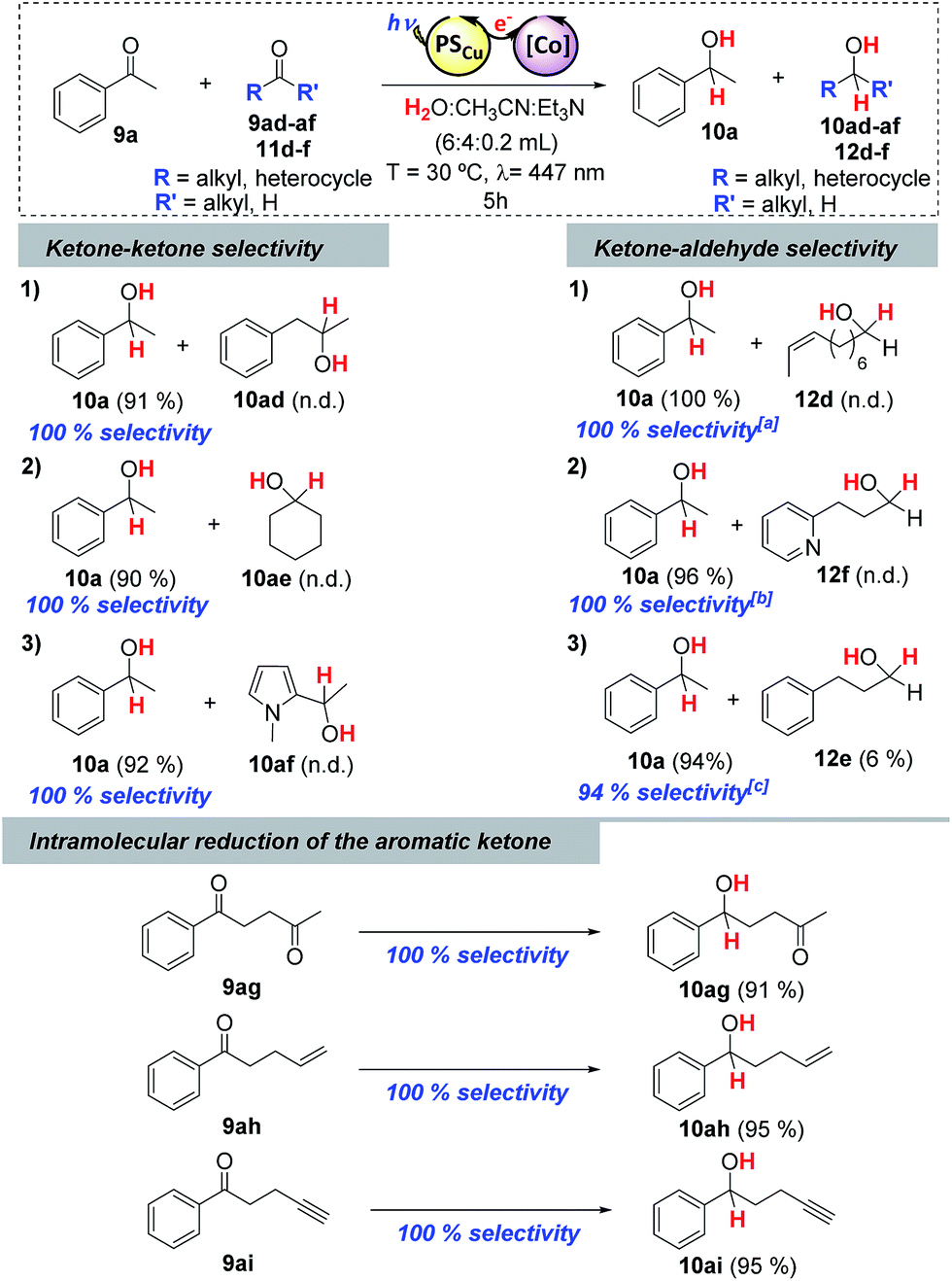 | ||
| Scheme 3 Competitive photoreductions. Conditions: 1 (1 mol%), PSCu (1.5 mol%), substrate A + B (16.5 mM, 1:1), in H2O:CH3CN:Et3N (6:4:0.2 mL) irradiated (447 nm) for 5 h at 30 °C under N2. [a]35 [b]50 and [c]30 min reaction, respectively. The isolated products were identified and characterized using NMR Spectroscopy. | ||
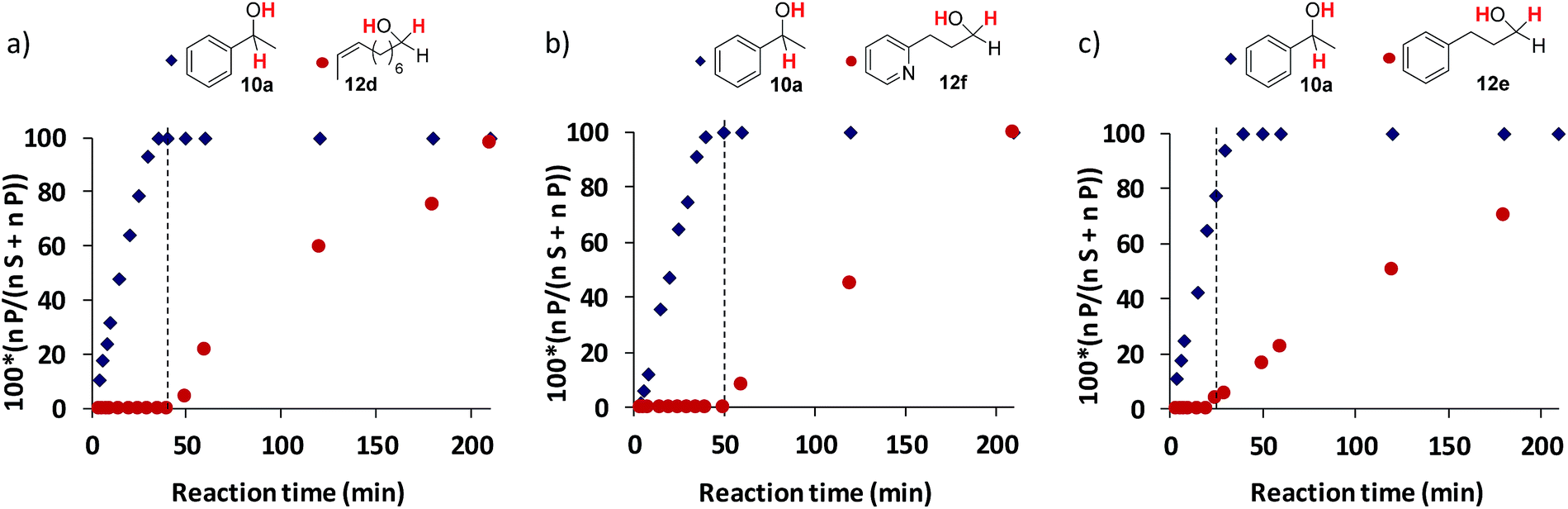 | ||
| Fig. 3 Conditions: 1 (1 mol%), PSCu (1.5 mol%), total substrate concentration (16.5 mM) in a H2O:CH3CN:iPr2EtN (3:2:0.1 mL) mixture, irradiated at 447 nm for 3.5 h at 30 °C under N2. The plotted data are the ratio between the amount of the reduced product formed and the sum of the amount of the reduced product formed and the unconverted starting material. The black dotted line indicates where substrates 11d (a), 11f (b), and 11e (c) start reacting. | ||
In addition, this preference for aromatic ketones is extended to aliphatic ketones, aliphatic alkenes, and aliphatic alkynes as showed by the reduction of 1-phenyl-1,4-pentanedione (9ag), 1-phenyl-1,4-penten-1-one (9ah), and 1-phenyl-4-pentyn-1-one (9ai) as model substrates to their corresponding aromatic alcohols (Scheme 3).
The selective reduction of aromatic ketones versus highly reactive aliphatic aldehydes is not straightforward. Current methods rely on protection–deprotection steps or on the trapping of the aldehyde using stoichiometric amounts of lanthanide salts.24 The latter methodology is known as the Luche reaction, and the most common conditions are the use of 1 equiv. of CeCl3 and 1.5 equiv. of NaBH4 in EtOH:H2O mixtures at low temperature.24a,24b,24f On this basis, we compared the selectivity of our methodology with both direct reduction with NaBH4 and the Luche reaction, for the reduction of substrates 11e and 11d in the presence of 9a (Scheme 4). As expected, NaBH4 did not yield satisfactory selectivity, however, neither did the Luche reaction, even when using fewer equivalents of NaBH4 (see Scheme SI.1.6† for further details). In contrast, the dual cobalt–copper light-driven catalytic reduction affords excellent selectivity (Scheme 4).
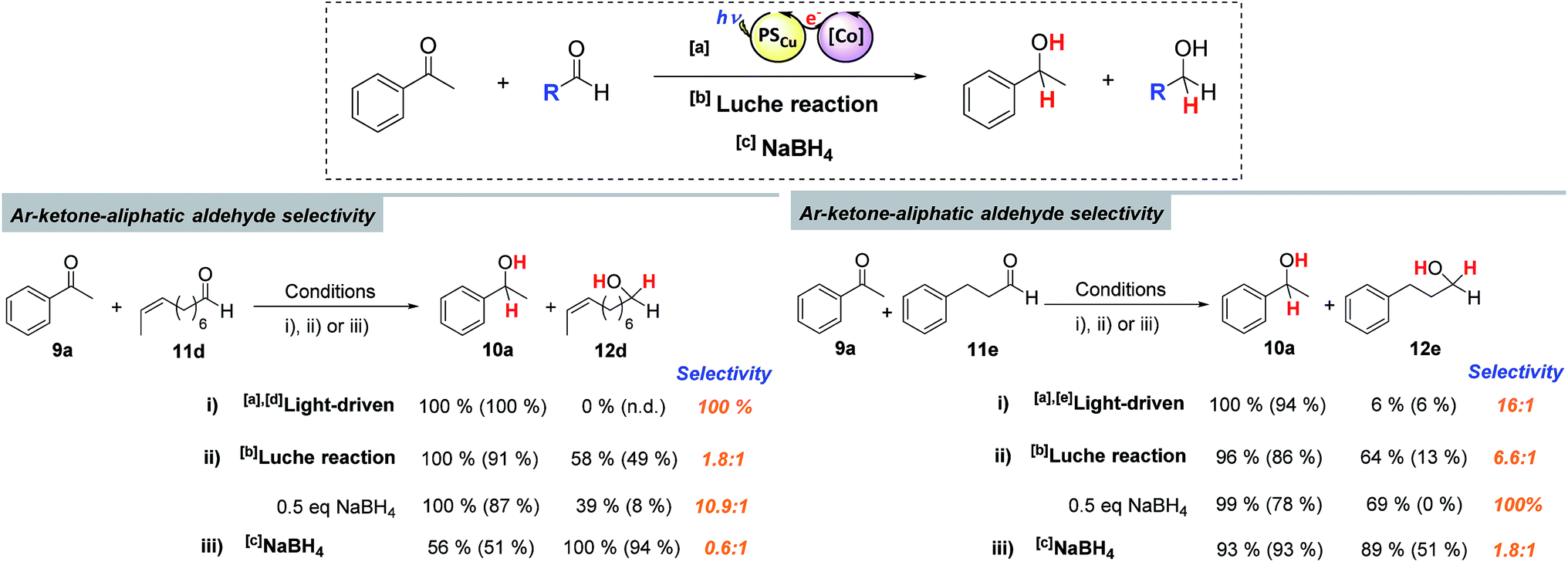 | ||
| Scheme 4 Competition experiments. [a]Light-driven conditions: 1 (1 mol%), PSCu (1.5 mol%), substrate A + B (16.5 mM, 1:1), in H2O:CH3CN:Et3N (6:4:0.2 mL) irradiated (447 nm) for 5 h at 30 °C under N2. [b]Luche reaction conditions: CeCl3·7H2O (1 equiv. molar), NaBH4 (1.5 equiv. molar), substrate A + B (16.5 mM, 1:1), in EtOH:H2O (4:6 mL) for 15 min at 0 °C under air. The same conditions but with NaBH4 (0.5 equiv. molar) are also shown. [c]NaBH4 (1 equiv. molar), substrate A + B (16.5 mM, 1:1), in MeOH (10 mL) for 15 min at rt. [d]Analysis after 35 and [e]30 minutes of irradiation. The percentages show the conversions of the substrate from which the product derives and percentages in brackets show the yield of the reduced product. | ||
Mechanistic investigations
We performed mechanistic studies based on isotopic labelling, radical clock experiments, and computational modelling to rationalize the unique selectivity of the developed dual Cu/Co photocatalytic system and to shed some light on the mechanism of action.Isotopic labelling studies
The photocatalytic reduction of several aromatic ketones (substrates 9a, 9b, and 9j) and the aliphatic aldehyde 11d with the dual PSCu/1 catalytic system in D2O and an excess of Et3N enables the incorporation of deuterium atoms in the resulting alcohol products with high yields (Scheme 5). All of the alcohol products analysed show nearly quantitative incorporation of deuterium atoms at the carbonyl and its α-position. Incorporation of deuterium at the α-position of the carbonyl is consistent with a keto–enol tautomerization due to the basic reaction conditions, as suggested by the blank experiments without cobalt. In the case of the aliphatic aldehyde 11d, the incorporation of a deuterium atom into the α-position of the carbonyl group was only about 40%. This is in contrast to the more than 90% incorporation of deuterium at the same position that was observed for the ketone derivatives.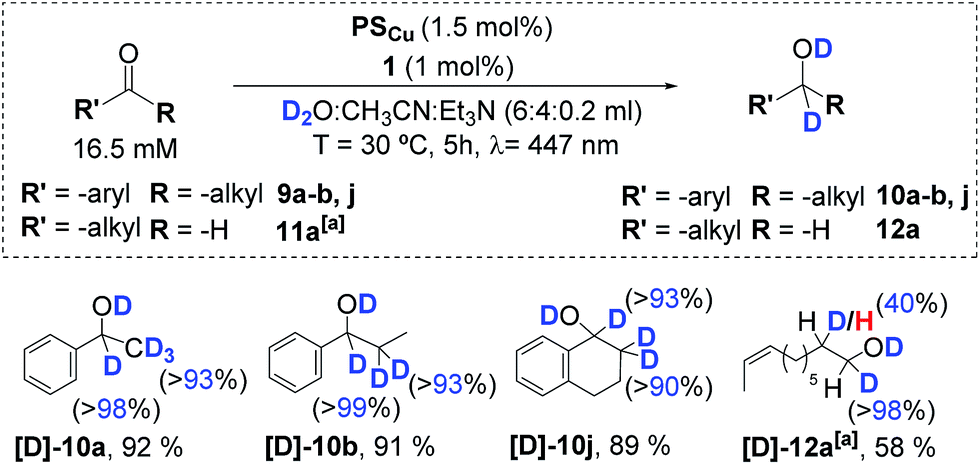 | ||
| Scheme 5 Deuterium labelling studies of aromatic ketones (9a–b, j), and aliphatic aldehydes (11a). [a]1 (6 mol%), PSCu (6 mol%), substrate (4.4 mM) in D2O:CH3CN:iPr2EtN (6:4:0.2 mL) irradiated (447 nm) for 24 h at −3 °C under N2. Isolated yields. Deuterium insertion analysed using NMR. | ||
Radical clock experiments
Ketones containing a 2-aryl-cyclopropyl moiety at the α-position are commonly used as diagnostic probes for reductions involving a single electron transfer (SET) mechanism. If a SET step is involved, it will trigger cyclopropane ring opening with a rate constant in the range from 105 to 107 s−1.25,26 Thus, we evaluated the cyclopropyl phenyl ketone 9ai under reaction conditions with and without the cobalt catalyst 1 (Scheme 6) with the aim of unravelling mechanistic information about the reduction step.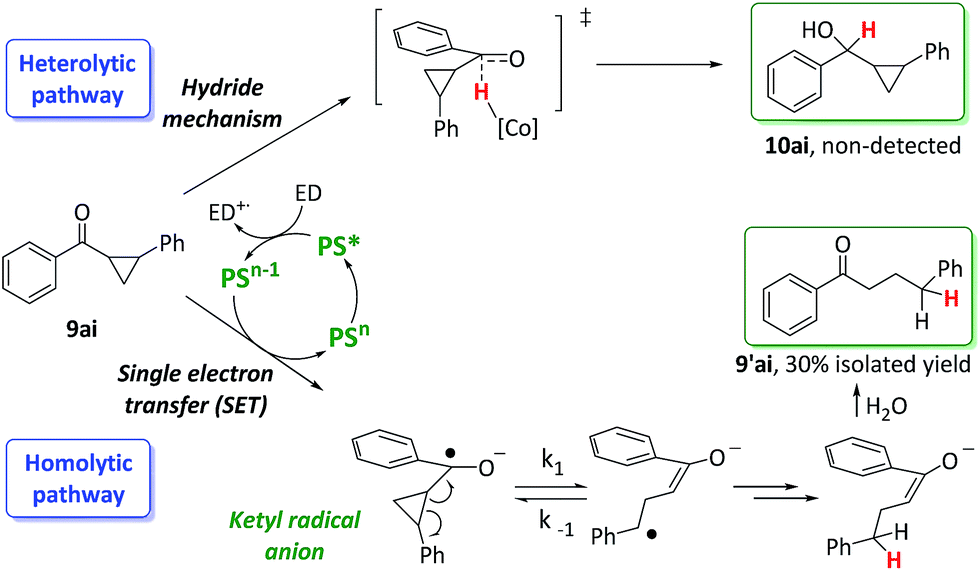 | ||
| Scheme 6 Considered reduction pathways for 9ai. | ||
In this regard, the reduction of phenyl cyclopropyl ketone 9ai forms only the ring-opening product 9′ai albeit in 30% yield. The same result was obtained in the absence of the cobalt catalyst 1. The reaction proceeds via a ring opening followed by a HAT from the [Co–H] intermediate to the benzylic radical (homolytic pathway) or by a reduction followed by a protonation. These results suggest that a SET from the photoredox catalyst to the ketone yields the corresponding ketyl radical anion. Indeed, this process is thermodynamically accessible (ΔG = 1.6 kcal mol−1) since the redox potential of 9ai is −1.6 V vs. SCE and the E1/2 of PSCu is only about 70 mV lower. This result indicates that, for ketones and aldehydes with similar redox potential, the ketyl radical anion can be formed under catalytic conditions. However, these radical clock experiments do not allow for the discrimination between possible interceptions of the ketyl radical anions by [Co–H] species via HAT and a possible direct reduction of the carbonyl groups via a hydride transfer mechanism.
DFT modelling
In order to explain the remarkable selectivity for acetophenone (9a) versus water reduction, we computed the free energy barriers associated with the reduction of water and 9a by the cobalt(II) hydride species ([1II-H]).Computational studies were conducted with the Gaussian09 software package. Geometry optimizations were performed with the B3LYP functional and the 6-31G* 6d basis set for all atoms, including the effect of the solvent (SMD implicit solvent model) and dispersion interactions (Grimme-D2 correction). A cluster of three water molecules has been introduced into the model to account for hydrogen bonding and the micro-solvation sphere around the ketone and water substrates. Free energy values (G) were obtained by including thermal (Gcorr.), solvation (Gsolv.) and dispersion corrections (Edisp.) to the potential energy computed with the 6-311+G** 6d basis set on equilibrium geometries:
| G = E6-311+G** + Gcorr. + Gsolv. + Edisp. | (1) |
Gibbs energies have also been adjusted to the concentration of all of the species as well as to the pH of the reaction. Concentration effects were quantified using computation of the free energy change associated with the conversion from a standard state of 1 atm in the gas phase to the desired concentration (ΔGo/*). Thus, the final absolute free energies for each species were evaluated as:
| Gfinal = G + ΔGo/* | (2) |
See ESI 3† for the complete computational details and Fig. SI.3.2–3.5† for relevant geometrical data.
Computational modelling supports the idea that under catalytic conditions the [1II-H] species is easily accessible (Fig. SI.3.6†). As expected, the initial reduction of [1II-NCCH3] to form [1I-NCCH3] by the reduced PSCu− is an exergonic process (ΔG = −3.6 kcal mol−1). Acetonitrile decoordination-protonation of [1I-NCCH3] to form CoIII–H is, under catalytic conditions (pH = 12), endergonic by 9.2 kcal mol−1, which is in agreement with reported DFT calculations on related cobalt complexes.1d,13d,27 Then [1III-H] can be further photochemically reduced to [1II-H] species. The overall photochemical formation of [1II-H], starting from [1II-NCCH3] and the copper photoredox catalyst (PSCu) as the reducing agent, is thermodynamically feasible (ΔG = −12.0 kcal mol−1). It has already been reported that the heterolytic pathway for H2 formation at CoII–H complexes is the most favoured mechanism for cobalt complexes bearing aminopyridine ligands.1d,11,28 In our case, the protonation of the [1II-H] species under catalytic conditions (pH = 12) has a low energy barrier of 12.2 kcal mol−1.
At this point, we have calculated two different reaction pathways for the reduction of 9a starting from [1II-H] (see Fig. SI.3.7–3.10† for complete free energy profiles). First, a hydride transfer from [1II-H] to the carbonyl group of the ketone was considered (Fig. 4). In this mechanism, the interaction between [1II-H], acetophenone, and water molecules has a remarkable free energy cost of 6.4 kcal mol−1, mainly due to concentration effects. The transition state (ΔG‡ = 11.1 kcal mol−1) is early since the metal-hydride distance is similar to [1II-H] (d(Co–H) = 1.660 Å, d(H–C) = 2.065 Å), and the metal center has a Hirshfeld charge of −0.18 and a relevant spin density (Fig. 5A). Finally, the hydride moiety is transferred to the ketone and the resulting negatively charged product is stabilized by an aqueous solvation sphere surrounding the oxygen atom (ΔG = −8.1 kcal mol−1). The thermodynamically feasible protonation of the alkoxide leads to the final product. For comparative reasons, the hydride transfer mechanism from a CoIII–H moiety has also been considered, but a ΔG‡ > 30 kcal mol−1 is obtained due to its reduced hydride character (Fig. SI.3.11†).
 | ||
| Fig. 4 DFT-modelled free energy profiles for the reduction of acetophenone (9a) and water with complex 1via hydride transfer or homolytic SET-HAT mechanisms. Gibbs energies are in kcal mol−1. | ||
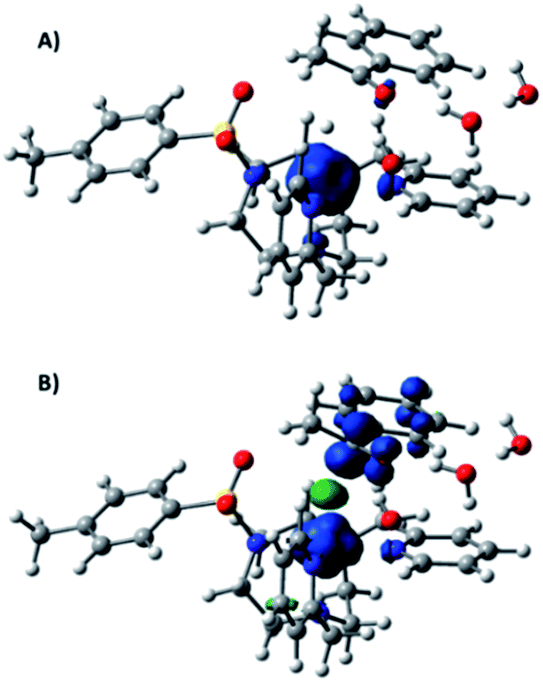 | ||
| Fig. 5 Spin density plots (isovalue = 0.006) of the transition states of (A) hydride transfer to acetophenone and (B) hydrogen atom transfer to a ketyl radical. | ||
Alternatively, we have explored a homolytic pathway for the ketone reduction. This mechanism starts with a single electron transfer (SET) from the reduced copper photoredox catalyst (E(exp) (PSCu) = −1.53 V vs. SCE) to 9a to give a ketyl radical species (9aketyl), which is then trapped by [1II-H]via a hydrogen atom transfer (HAT) mechanism (Fig. 4). In this regard, DFT calculations indicate that the reduction of 9a (Eo(theoretical) = −1.69 V, E(experimental) = −1.65 V vs. SCE) by PSCu− to form 9aketyl is slightly endergonic (ΔG = 3.8 kcal mol−1). Then, the subsequent HAT from CoII–H to the ketyl radical 9aketyl occurs through a transition state (ΔG‡ = 18.3 kcal mol−1, Fig. 4) that is higher in free energy than the direct hydride transfer of [1II-H] to 9a by 7.2 kcal mol−1.
In the late transition state structure, the transferred hydrogen (ρ(H) = −0.11) is closer to the C–O group (ρ(C–O) = 0.77) than in the previously discussed hydride pathway (d(Co–H) = 1.603 Å and d(H–C) = 1.873 Å). The ketone shows a substantial spin density (Fig. 5B) and is antiferromagnetically coupled to the cobalt, evolving the final alkoxide product and the formal one electron reduction of the cobalt center (ρ(Co) = 1.61). It has also been considered that the reduction reaction may start with a proton coupled electron transfer to generate the O-protonated ketyl radical (ΔG = 3.8 kcal mol−1 at pH = 12). However, the total energy barrier for the product formation is substantially higher in free energy (ΔΔG‡ = 8.4 kcal mol−1, see Fig. SI.3.10†).
The free energy profiles for ketone and water reduction are in agreement with the observed reactivity. They start with the [1II-H] species as a common intermediate for both reductions. Interestingly, the kinetic barrier of the hydride transfer mechanism for 9a (ΔG‡ = 11.1 kcal mol−1) is lower than that for the water reduction by 1.1 kcal mol−1 (Fig. 4 and SI.3.12†). This free energy difference is in agreement with the selectivity observed. Indeed, the large water content (>2500 fold) in comparison to acetophenone (about 15 mM) would explain the similar rates found for H2 evolution and 9a reduction (Fig. 2 and 4).
As a summary of the mechanistic studies, we propose that the hydride mechanism is prevalent, although we cannot discard the SET-HAT mechanism. It is known that under catalytic conditions PSCu is excited by light (*PSCu) and reductively quenched by the electron donor (ED) to give PSCu−,29 which is reductive enough (E = −1.53 V vs. SCE) to reduce complex 1 (E = −1.35 V vs. SCE) by one electron.15 This highly reactive low-valent intermediate, [1I], is protonated by water to form the putative [1III-H], which is easily reduced, giving the active [1II-H] species. Then, two different mechanistic scenarios are postulated for the reduction of ketones and aldehydes to alcohols. One possibility is a SET-HAT mechanism in which a single-electron transfer (SET) from PSCu− to the substrate generates a carbonyl radical anion intermediate, which is converted into the final product by a hydrogen atom transfer (HAT) from the [Co–H] species (homolytic pathway, Fig. 6, left). Alternatively, a direct nucleophilic attack of a putative [1II-H] intermediate (heterolytic pathway, Fig. 6, right) should be considered. Our DFT studies support the heterolytic mechanism. However, the observed pinacol formation in the absence of 1 as well as the observed ring-opening products in the radical clock experiments support a SET-HAT mechanism, which should therefore be considered as well, especially for those substrates that can be easily reduced. Further studies are needed to clarify the feasibility of SET-HAT under our catalytic conditions.
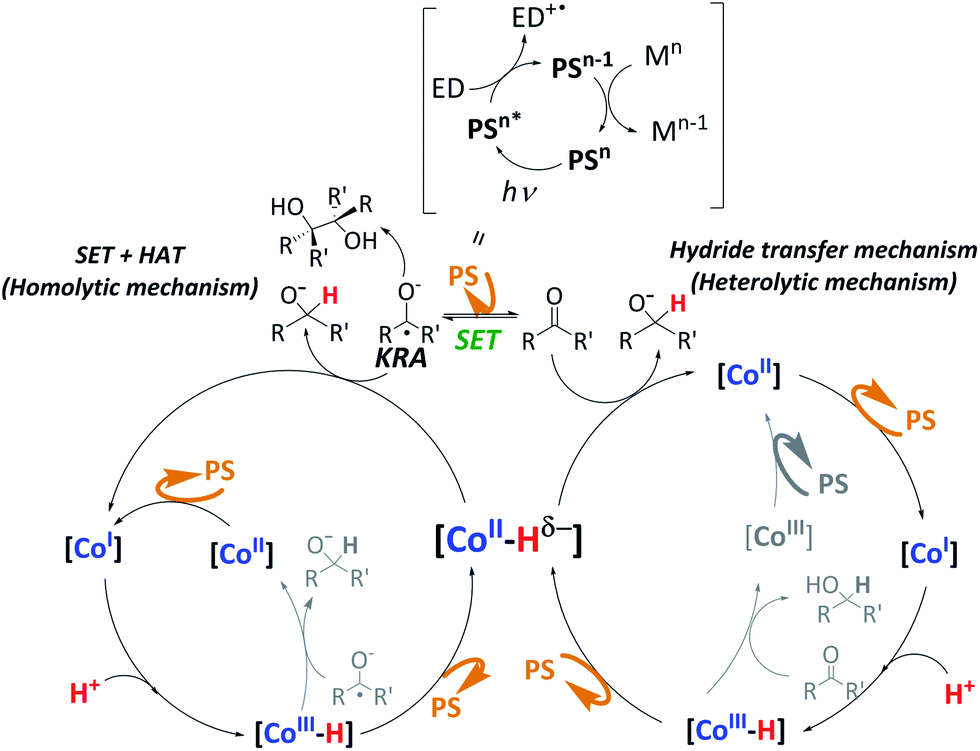 | ||
| Fig. 6 Possible mechanistic scenarios for the photoreduction of aromatic ketones and aldehydes. | ||
Conclusions
We report a new methodology based on a dual cobalt–copper catalytic (1 and PSCu) system that is able to reduce aromatic ketones and both aromatic and aliphatic aldehydes using H2O and an amine (Et3N or iPr2EtN) as the source of hydrides and visible light as the driving force. Remarkably, the system is highly selective towards the reduction of the organic substrates in the presence of water (>2000 fold). Moreover, replacement of H2O by D2O results in the formation of α,β-deuterated alcohols. Our results show that the selectivity towards the reduction of organic functionalities versus water is catalyst controlled, allowing for further improvements and developments. Moreover, the system presents a unique selectivity for the reduction of acetophenone versus aliphatic aldehydes. Indeed, this selectivity is unprecedented for metal catalysed transformations. The present system benefits from avoiding protecting–deprotecting steps and the use of stoichiometric amounts of lanthanides, which are required in other reduction methods of ketones and aldehydes.Our mechanistic studies and DFT modelling suggest that the well-defined cobalt hydride is a common intermediate in the reduction of both organic substrates and water. Reactivity experiments support a hydride transfer mechanism for substrates with low redox potentials (<−2 V), such as aliphatic aldehydes. Nevertheless, both homolytic and heterolytic pathways could coexist depending on the redox potential of the substrate.
We envision that other photocatalytic water reduction catalysts might also be active for the reduction of several organic functionalities. These results are in line with the development of selective organic reductions and synthesis of solar chemicals via artificial catalytic systems.
Conflict of interest
The authors declare no competing financial interests.Acknowledgements
We would like to thank the European Commission for the ERC-CG-2014-648304 (J. Ll.-F.) project. The Spanish Ministry of Science is acknowledged for a FPU fellowship to A. C. and C. C. and Juan de la Cierva contract to A. C. We also thank Catexel for a generous gift of tritosyl-1,4,7-triazacyclononane. The financial support from the ICIQ Foundation and the CELLEX Foundation through the CELLEX-ICIQ high throughput experimentation platform and the Starting Career Program is gratefully acknowledged. C. C. thanks the CELLEX Foundation for a pre-doctoral contract. We also thank the CERCA Programme (Generalitat de Catalunya) for financial support and MINECO (Severo Ochoa Excellence Accreditation 2014–2018; SEV-2013-0319). We are indebted to Dr M. Costas, Dr X. Ribas and Dr N. Rodríguez for their fruitful comments.Notes and references
- (a) T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS; (b) D. Z. Zee, T. Chantarojsiri, J. R. Long and C. J. Chang, Acc. Chem. Res., 2015, 48, 2027–2036 CrossRef CAS PubMed; (c) N. Kaeffer, M. Chavarot-Kerlidou and V. Artero, Acc. Chem. Res., 2015, 48, 1286–1295 CrossRef CAS PubMed; (d) N. Queyriaux, R. T. Jane, J. Massin, V. Artero and M. Chavarot-Kerlidou, Coord. Chem. Rev., 2015, 304–305, 3–19 CrossRef CAS PubMed; (e) M. Wang, L. Chen and L. Sun, Energy Environ. Sci., 2012, 5, 6763 RSC; (f) J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865–878 RSC; (g) Z. Han and R. Eisenberg, Acc. Chem. Res., 2014, 47, 2537–2544 CrossRef CAS PubMed; (h) V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC; (i) W. T. Eckenhoff and R. Eisenberg, Dalton Trans., 2012, 41, 13004–13021 RSC; (j) M. Rakowski DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62–72 RSC; (k) P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012 RSC.
- (a) A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis and J. G. Ferry, et al. , Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed; (b) Y. Izumi, Coord. Chem. Rev., 2013, 257, 171–186 CrossRef CAS; (c) J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC; (d) T. M. Monos, A. C. Sun, R. C. McAtee, J. J. Devery 3rd and C. R. Stephenson, J. Org. Chem., 2016, 81, 6988–6994 CrossRef CAS PubMed; (e) H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef CAS; (f) C. D. Windle and R. N. Perutz, Coord. Chem. Rev., 2012, 256, 2562–2570 CrossRef CAS; (g) J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC; (h) C. Costentin, M. Robert and J. M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- (a) T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed; (b) K. S. Joya, Y. F. Joya, K. Ocakoglu and R. van de Krol, Angew. Chem., 2013, 52, 10426–10437 CrossRef CAS PubMed; (c) J. Barber and P. D. Tran, J. R. Soc., Interface, 2013, 10, 20120984 CrossRef PubMed; (d) E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946–964 CrossRef CAS PubMed; (e) N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed; (f) S. Rau, D. Walther and J. G. Vos, Dalton Trans., 2007, 915–919, 10.1039/b615987g.
- (a) S. Choudhury, J. O. Baeg, N. J. Park and R. K. Yadav, Angew. Chem., Int. Ed., 2012, 51, 11624–11628 CrossRef CAS PubMed; (b) S. H. Lee, J. H. Kim and C. B. Park, Chem.–Eur. J., 2013, 19, 4392–4406 CrossRef CAS PubMed; (c) M. Mifsud, S. Gargiulo, S. Iborra, I. W. Arends, F. Hollmann and A. Corma, Nat. Commun., 2014, 5, 3145 Search PubMed; (d) J. H. Kim, D. H. Nam and C. B. Park, Curr. Opin. Biotechnol., 2014, 28, 1–9 CrossRef CAS PubMed; (e) J. Liu, J. Huang, H. Zhou and M. Antonietti, ACS Appl. Mater. Interfaces, 2014, 6, 8434–8440 CrossRef CAS PubMed; (f) A. Bachmeier, B. J. Murphy and F. A. Armstrong, J. Am. Chem. Soc., 2014, 136, 12876–12879 CrossRef CAS PubMed; (g) G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 3425–3437, 10.1039/b700395c; (h) R. E. Galian and J. Pérez-Prieto, Energy Environ. Sci., 2010, 3, 1488 RSC; (i) J. A. Macia-Agullo, A. Corma and H. Garcia, Chem.–Eur. J., 2015, 21, 10940–10959 CrossRef CAS PubMed; (j) T. Ghosh, T. Slanina and B. König, Chem. Sci., 2015, 6, 2027–2034 RSC.
- (a) C. Joyce-Pruden, J. K. Pross and Y. Li, J. Org. Chem., 1992, 57, 5087–5091 CrossRef CAS; (b) S. Yanagida, M. Yoshiya, T. Shiragami, C. Pac, H. Mori and H. Fujita, J. Phys. Chem., 1990, 94, 3104–3111 CrossRef CAS; (c) Photocatalytic Hydrogenation on Semiconductor Particles, ed. S. Kohtani, E. Yoshioka and H. Miyabe, InTech, 2012, chapters published Search PubMed; (d) S. Füldner, R. Mild, H. I. Siegmund, J. A. Schroeder, M. Gruber and B. König, Green Chem., 2010, 12, 400–406 RSC; (e) X. Ke, S. Sarina, J. Zhao, X. Zhang, J. Chang and H. Zhu, Chem. Commun., 2012, 48, 3509–3511 RSC; (f) S. Yanagida, Y. Ishimaru, Y. Miyake, T. Shiragami, C. Pac, K. Hashimoto and T. Sakata, J. Phys. Chem., 1989, 93, 2576–2582 CrossRef CAS; (g) S. Kohtani, T. Kurokawa, E. Yoshioka and H. Miyabe, Appl. Catal., A, 2016, 521, 68–74 CrossRef CAS; (h) X. Ke, X. Zhang, J. Zhao, S. Sarina, J. Barry and H. Zhu, Green Chem., 2013, 15, 236–244 RSC; (i) M. Zhang, W. D. Rouch and R. D. McCulla, Eur. J. Org. Chem., 2012, 2012, 6187–6196 CrossRef CAS.
- S. Choudhury, J.-O. Baeg, N.-J. Park and R. K. Yadav, Green Chem., 2014, 16, 4389 RSC.
- (a) J.-J. Zhong, Q. Liu, C.-J. Wu, Q.-Y. Meng, X.-W. Gao, Z.-J. Li and B. Chen, et al. , Chem. Commun., 2016, 52, 1800–1803 RSC; (b) X. Liu, D. Sun, R. Yuan, X. Fu and Z. Li, J. Catal., 2013, 304, 1–6 CrossRef CAS; (c) J. Li, J. Yang, F. Wen and C. Li, Chem. Commun., 2011, 47, 7080–7082 RSC; (d) H. Shimakoshi and Y. Hisaeda, ChemPlusChem, 2014, 79, 1250–1253 CrossRef CAS.
- X. Lang, J. Zhao and X. Chen, Chem. Soc. Rev., 2016, 45, 3026–3038 RSC.
- (a) T. Bleith and L. H. Gade, J. Am. Chem. Soc., 2016, 138, 4972–4983 CrossRef CAS PubMed; (b) T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2015, 137, 2456–2459 CrossRef CAS PubMed; (c) P. Jochmann and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 9831 CrossRef CAS PubMed; (d) R. Xu, S. Chakraborty, S. M. Bellows, H. Yuan, T. R. Cundari and W. D. Jones, ACS Catal., 2016, 6, 2127 CrossRef CAS; (e) N. Gorgas, B. Stöger, L. F. Veiros and K. Kirchner, ACS Catal., 2016, 6, 2664 CrossRef CAS PubMed; (f) S. Elangovan, B. Wendt, C. Topf, S. Bachmann, M. Scalone, A. Spannenberg and H. Jiao, et al. , Adv. Synth. Catal., 2016, 358, 820 CrossRef CAS; (g) C. Bornschein, S. Werkmeister, B. Wendt, H. Jiao, E. Alberico, W. Baumann and H. Junge, et al. , Nat. Commun., 2014, 5, 4111 CAS; (h) E. Alberico, P. Sponholz, C. Cordes, M. Nielsen, H. J. Drexler, W. Baumann and H. Junge, et al. , Angew. Chem., Int. Ed., 2013, 52, 14162 CrossRef CAS PubMed; (i) S. Chakraborty, G. Leitus and D. Milstein, Chem. Commun., 2016, 52, 1812 RSC; (j) B. Butschke, M. Feller, Y. Diskin-Posner and D. Milstein, Catal. Sci. Technol., 2016, 6, 4428–4437 RSC; (k) T. Zell, Y. Ben-David and D. Milstein, Catal. Sci. Technol., 2015, 5, 822 RSC; (l) T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979 CrossRef CAS PubMed; (m) R. H. Morris, Acc. Chem. Res., 2015, 48, 1494 CrossRef CAS PubMed; (n) S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann and R. Ludwig, et al. , J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed; (o) R. M. Bullock, Science, 2013, 342, 1054 CrossRef CAS PubMed; (p) H. Pellissier and H. Clavier, Chem. Rev., 2014, 114, 2775–2823 CrossRef CAS PubMed; (q) R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah and H. Huan, et al. , Science, 2013, 342, 1073–1076 CrossRef CAS PubMed; (r) F. Kallmeier, T. Irrgang, T. Dietel and R. Kempe, Angew. Chem., Int. Ed., 2016, 55, 11806–11809 CrossRef CAS PubMed; (s) B. A. F. Le Bailly and S. P. Thomas, RSC Adv., 2011, 1, 1435 RSC.
- (a) D. J. Ager, A. H. de Vries and J. G. de Vries, Chem. Soc. Rev., 2012, 41, 3340–3380 RSC; (b) J. G. de Vries and C. J. Elsevier, The Handbook of Homogeneous Hydrogenation, 2007 Search PubMed.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995 CrossRef CAS PubMed.
- (a) V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed; (b) W. T. Eckenhoff, W. R. McNamara, P. Du and R. Eisenberg, Biochim. Biophys. Acta, 2013, 1827, 958–973 CrossRef CAS PubMed.
- (a) W. K. Lo, C. E. Castillo, R. Gueret, J. Fortage, M. Rebarz, M. Sliwa and F. Thomas, et al. , Inorg. Chem., 2016, 55, 4564–4581 CrossRef CAS PubMed; (b) D. Moonshiram, C. Gimbert-Surinach, A. Guda, A. Picon, C. S. Lehmann, X. Zhang and G. Doumy, et al. , J. Am. Chem. Soc., 2016, 138, 10586–10596 CrossRef CAS PubMed; (c) G. Smolentsev, B. Cecconi, A. Guda, M. Chavarot-Kerlidou, J. A. van Bokhoven, M. Nachtegaal and V. Artero, Chem.–Eur. J., 2015, 21, 15158–15162 CrossRef CAS PubMed; (d) V. Artero and M. Fontecave, Coord. Chem. Rev., 2005, 249, 1518–1535 CrossRef CAS; (e) S. Losse, J. G. Vos and S. Rau, Coord. Chem. Rev., 2010, 254, 2492–2504 CrossRef CAS; (f) S. Varma, C. E. Castillo, T. Stoll, J. Fortage, A. G. Blackman, F. Molton and A. Deronzier, et al. , Phys. Chem. Chem. Phys., 2013, 15, 17544–17552 RSC; (g) A. Lewandowska-Andralojc, T. Baine, X. Zhao, J. T. Muckerman, E. Fujita and D. E. Polyansky, Inorg. Chem., 2015, 54, 4310–4321 CrossRef CAS PubMed.
- S. P. Luo, E. Mejia, A. Friedrich, A. Pazidis, H. Junge, A. E. Surkus, R. Jackstell, S. Denurra, S. Gladiali, S. Lochbrunner and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 419 CrossRef CAS PubMed.
- A. Call, Z. Codolà, F. Acuña-Parés and J. Lloret-Fillol, Chem.–Eur. J., 2014, 20, 6171–6183 CrossRef CAS PubMed.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338–2356 RSC.
- (a) A. Dossing, P. Engberg and R. Hazell, Inorg. Chim. Acta, 1998, 268, 159–162 CrossRef CAS; (b) W. M. Singh, T. Baine, S. Kudo, S. Tian, X. A. N. Ma, H. Zhou and N. J. DeYonker, et al. , Angew. Chem., Int. Ed., 2012, 51, 5941–5944 CrossRef CAS PubMed; (c) B. Shan, T. Baine, X. A. N. Ma, X. Zhao and R. H. Schmehl, Inorg. Chem., 2013, 52, 4853–4859 CrossRef CAS PubMed; (d) M. Vennampalli, G. Liang, L. Katta, C. E. Webster and X. Zhao, Inorg. Chem., 2014, 53, 10094–10100 CrossRef CAS PubMed; (e) A. Lewandowska-Andralojc, T. Baine, X. Zhao, J. T. Muckerman, E. Fujita and D. E. Polyansky, Inorg. Chem., 2015, 54, 4310–4321 CrossRef CAS PubMed; (f) X.-W. Song, H.-M. Wen, C.-B. Ma, H. Chen and C. Chen, New J. Chem., 2015, 39, 1734–1741 RSC; (g) J. Xie, Q. Zhou, C. Li, W. Wang, Y. Hou, B. Zhang and X. Wang, Chem. Commun., 2014, 50, 6520–6522 RSC; (h) W. K. C. Lo, C. E. Castillo, R. Gueret, J. Fortage, M. Rebarz, M. Sliwa and F. Thomas, et al. , Inorg. Chem., 2016, 55, 4564–4581 CrossRef CAS PubMed.
- (a) R. R. Fenton, F. S. Stephens, R. S. Vagg and P. A. Williams, Inorg. Chim. Acta, 1991, 182, 67–75 CrossRef CAS; (b) T. M. Kooistra, K. F. W. Hekking, Q. Knijnenburg, B. de Bruin, P. H. M. Budzelaar, R. de Gelder and J. M. M. Smits, et al. , Eur. J. Inorg. Chem., 2003, 648–655, DOI:10.1002/ejic.200390090; (c) A. L. Ward, L. Elbaz, J. B. Kerr and J. Arnold, Inorg. Chem., 2012, 51, 4694–4706 CrossRef CAS PubMed; (d) Z. Li, J.-D. Xiao and H.-L. Jiang, ACS Catal., 2016, 6, 5359–5365 CrossRef CAS.
- (a) P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 12576–12577 CrossRef CAS PubMed; (b) P. Du, J. Schneider, G. Luo, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2009, 48, 4952–4962 CrossRef CAS PubMed; (c) T. M. McCormick, B. D. Calitree, A. Orchard, N. D. Kraut, F. V. Bright, M. R. Detty and R. Eisenberg, J. Am. Chem. Soc., 2010, 132, 15480–15483 CrossRef CAS PubMed.
- S. Okamoto, K. Kojiyama, H. Tsujioka and A. Sudo, Chem. Commun., 2016, 52, 11339–11342 RSC.
- M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed. Engl., 2015, 54, 8828–8832 CrossRef CAS PubMed.
- D. Nicewicz, H. Roth and N. Romero, Synlett, 2015, 27, 714–723 CrossRef.
- The redox potential under photocatalytic conditions could not be determined due to the reduction of the solvent (H2O), which occurs at about −2 V vs. SCE.
- (a) A. L. Gemal and J. L. Luche, J. Am. Chem. Soc., 1981, 103, 5454–5459 CrossRef CAS; (b) J. L. Luche and A. L. Gemal, J. Am. Chem. Soc., 1979, 101, 5848–5849 CrossRef CAS; (c) A. F. Abdel-Magid, in Comprehensive Organic Synthesis, Elsevier Ltd., 2nd edn, 2014, vol. 8, pp. 1–84 Search PubMed; (d) F. J. Barrios, B. C. Springer and D. A. Colby, Org. Lett., 2013, 15, 3082–3085 CrossRef CAS PubMed; (e) G. Bastug, S. Dierick, F. Lebreux and I. E. Markó, Org. Lett., 2012, 14, 1306–1309 CrossRef CAS PubMed; (f) A. L. Gemal and J. L. Luche, J. Org. Chem., 1979, 44, 4187–4189 CrossRef CAS.
- (a) J. P. Stevenson, W. F. Jackson and J. M. Tanko, J. Am. Chem. Soc., 2002, 124, 4271–4281 CrossRef CAS PubMed; (b) Z. Lu, M. Shen and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 1162–1164 CrossRef CAS PubMed.
- (a) J. M. Tanko and R. E. Drumright, J. Am. Chem. Soc., 1992, 114, 1844–1854 CrossRef CAS; (b) D. Yang and D. D. Tanner, J. Org. Chem., 1986, 51, 2267–2270 CrossRef CAS; (c) J. M. Tanko and R. E. Drumright, J. Am. Chem. Soc., 1990, 112, 5362–5363 CrossRef CAS; (d) D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317–323 CrossRef CAS.
- C. Costentin, H. Dridi and J. M. Saveant, J. Am. Chem. Soc., 2014, 136, 13727–13734 CrossRef CAS PubMed.
- S. C. Marinescu, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15127–15131 CrossRef CAS PubMed.
- S. Fischer, D. Hollmann, S. Tschierlei, M. Karnahl, N. Rockstroh, E. Barsch and P. Schwarzbach, et al. , ACS Catal., 2014, 4, 1845–1849 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full materials and methods. See DOI: 10.1039/c7sc01276d |
| ‡ A. Call and C. Casadevall contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |