
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N,N-Dimethylation of nitrobenzenes with CO2 and water by electrocatalysis†
Xiaofu
Sun
 ab,
Qinggong
Zhu
a,
Jiayin
Hu
ab,
Xinchen
Kang
ab,
Jun
Ma
a,
Huizhen
Liu
ab and
Buxing
Han
*ab
ab,
Qinggong
Zhu
a,
Jiayin
Hu
ab,
Xinchen
Kang
ab,
Jun
Ma
a,
Huizhen
Liu
ab and
Buxing
Han
*ab
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Colloid and Interface and Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: hanbx@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 7th June 2017
Abstract
We have proposed a strategy for the synthesis of N,N-dimethylanilines from nitrobenzene and its derivatives, CO2, and water via an electrochemical reaction under ambient conditions. H+ generated from H2O was used as the hydrogen source. Pd/Co–N/carbon, in which the Pd nanoparticles were supported on Co–N/carbon, was designed and used as the electrocatalyst. It was found that the electrocatalyst was very efficient for the reaction in MeCN solution with 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([Bmim]Tf2N) as the supporting electrolyte and 1-amino-methylphosphonic acid (AMPA) as the thermal co-catalyst. A series of control experiments showed that Pd/Co–N/carbon and AMPA cooperated very well in accelerating the reaction. This synthetic route has some obvious advantages, such as using CO2 and water as the reactants, ambient reaction conditions, and high yields of the desired products. This opens up a way to synthesize chemicals by the combination of an electrocatalyst and a thermal catalyst with organic compounds, CO2, and water as the reactants.
Introduction
Carbon dioxide (CO2) is an abundant, inexpensive, harmless and renewable C1 resource.1,2 CO2 chemistry has emerged as one of the most significant branches of chemistry. Owing to its thermodynamic stability and kinetic inertness, a well designed activation of CO2 and a thermodynamic driving force are required for efficient conversion.3–5 The transformation of CO2 into different chemicals has been reported, such as the direct hydrogenation of CO2 to organic acids and alcohols.6,7 In parallel, the direct introduction of CO2 into organic substrates is also an attractive goal in modern chemistry,8–11 and the exploration of new routes is very interesting.N-Methylation and N,N-dimethylation reactions are very important in the chemical industry, as they can obtain a series of useful intermediates for the synthesis of many valuable products, such as dyes, pesticides and perfumes.12–20 Traditionally, they can be synthesized through the methylation of amines with methanol or formaldehyde.19,20 Using CO2 as the C1 resource is a very promising route for the synthesis of N,N-dimethylanilines. In general, H2 or PhSiH3 has been used as the reducing agent for the N-methylation reaction of anilines.12–18 Anilines are usually synthesized by the hydrogenation of nitrobenzenes. Recently, the direct N-methylation of nitrobenzenes has also been studied using H2 as the reducing agent at high temperature and under high pressure (Fig. S1†).17 Obviously, searching for new reducing agents and performing the methylation reaction of nitrobenzenes under mild conditions are desirable.
The electrochemical method has been used to synthesize organic molecules in the past century.21–25 It possesses some obvious advantages, such as mild conditions, high functional group tolerance, and innate scalability and sustainability.21 Water is an ideal hydrogen source. H+ can be obtained via the oxygen evolution reaction (OER) at the anode electrode and transferred to the cathode electrode.26 It is well known that the efficiency and selectivity of electrochemical reactions depend strongly on the properties of the electrodes and electrolytes, and their coupling.22 Therefore, designing a suitable electrocatalyst with abundant active sites and high electrical conductivity is the key to promoting electrocatalytic reactions.
The use of CO2 and water as the reactants simultaneously in organic reactions is very attractive. Herein, we have developed a strategy for the synthesis of N,N-dimethylanilines by the N,N-dimethylation of nitrobenzene or its derivatives with CO2 using H2O as the hydrogen source. In the reactions, an electrochemical reaction and thermal reaction were combined with Pd/Co–N/carbon as the electrocatalyst and 1-amino-methylphosphonic acid (AMPA) as the thermal catalyst. It was demonstrated that Pd/Co–N/carbon and AMPA had excellent synergistic effects and the reactions proceeded efficiently under ambient conditions and high yields of the desired products could be reached. As far as we know, this is the first work on the synthesis of N,N-dimethylanilines using nitrobenzene (or its derivatives), CO2, and water as the reactants.
Results and discussion
The H-type reaction cell used was composed of a cathode, a platinum anode, and an Ag/Ag+ reference electrode (Fig. S2†), which was similar to that utilized in the previous works for the electroreduction of CO2.27–31 The cathode and anode compartments were separated by a Nafion 117 proton exchange membrane (PEM). H2SO4 aqueous solution was used as the anodic electrolyte. In the reaction, H+ from water could be transferred from the anode compartment to the cathode compartment through the PEM, which acted as the hydrogen source for the reaction. The detailed description of the apparatus is given in the ESI.†Pd is an efficient electrocatalyst for the reduction of CO2 to CO or formate.32,33 It is known that nanoporous carbon materials have high specific surface areas, high chemical and thermal stabilities, and good conductivities.34,35 Zeolitic imidazolate frameworks (ZIFs) have been reported to be outstanding carbon precursors.36,37 They can incorporate N atoms and metal species into the carbon lattice, which can enhance the electric conductivity and electron-donor tendency, and act as the hard template during the carbonization process.36 Meanwhile, the residual metals can also act as the co-catalyst. We have designed a new electrocatalyst, Pd/Co–N/carbon, using a ZIF as the carbon and nitrogen precursor, and the detailed preparation procedures are given in the ESI.† Very briefly, the Co–N/carbon support was first prepared by the carbonization of the Co–ZIF/graphene oxide (GO) at 800 °C under an Ar atmosphere. The Pdx/Co–N/carbon electrocatalysts were obtained by immobilizing Pd nanoparticles on the support, in which x denotes the average size of the Pd particles in nm. The size of the Pd nanoparticles could be controlled by the reduction temperature and the ratio of sodium citrate (stabilizing agent) to PdCl2. The size distributions of the Pd nanoparticles in different catalysts are shown in Fig. S3,† and were obtained from counting more than 200 particles in the transmission electron microscopy (TEM) images. The support had a specific surface area of 380 m2 g−1, which was determined by the N2 adsorption/desorption method. The elemental analysis of the Co–N/carbon support was conducted by inductively coupled plasma optical emission spectroscopy (ICP-OES). The results showed that the content of Co, C and N elements in the support is 37.69 wt%, 30.73 wt% and 1.05 wt%, respectively. The surface composition of the support detected by X-ray photoelectron spectroscopy (XPS) is given in Table S1.†
Fig. 1 shows high-resolution transmission electron microscopy (HR-TEM) images of Pd2.2/Co–N/carbon. It can be seen that the Pd nanoparticles were uniformly deposited on the Co–N/carbon support (Fig. 1A). Elemental distribution mappings also illustrate the co-existence of Pd and Co elements and the Pd nanoparticles were dispersed on the support homogeneously (Fig. 1B). The typical Pd (111) plane with the characteristic lattice spacing of 0.23 nm could be observed (Fig. 1C). XPS spectra (Fig. 1D) show the chemical nature of the catalyst, including the peaks belonging to Pd0 (Pd 3d: 340.7 and 335.4 eV), Pd2+ (Pd 3d: 343.0 and 336.9 eV), Co–N (Co 2p: 780.1 eV), Co–O (Co 2p: 795.4 eV), pyridinic N (N 1s: 399.5 eV) and graphitic N (N 1s: 402.2 eV).36,38 The results provided direct evidence that most of the Pd nanoparticles existed in the form of Pd0, and some electrochemical active sites for CO2 reduction such as the pyridinic N and Co–Nx moieties39,40 existed in the support. Furthermore, the actual Pd loadings in all of the catalysts were 18.3 ± 0.6 wt%, as measured by ICP-OES as listed in Table S2.† TEM and HR-TEM images of the other Pdx/Co–N/carbon catalysts with different Pd sizes are shown in Fig. S4.†
 | ||
| Fig. 1 Structural and elemental analysis of Pd2.2/Co–N/carbon. (A) TEM image of Pd2.2/Co–N/carbon. (B) Corresponding elemental mappings of Pd2.2/Co–N/carbon (scale bar: 50 nm). (C) HR-TEM image of Pd2.2/Co–N/carbon. The scale bar in the inset of (C) is 1 nm. (D) XPS spectra of Pd 3d, Co 2p and N 1s orbitals of Pd2.2/Co–N/carbon. | ||
To prepare the electrodes, the Pdx/Co–N/carbon catalysts were suspended in acetone with Nafion D-521 dispersion to form a homogeneous ink with the aid of ultrasound, which was spread onto carbon paper (CP) to obtain the working electrodes. Ionic liquids (ILs) are efficient supporting electrolytes for the reduction of CO2.27,41 The initial screening was performed for the methylation reaction of nitrobenzene, 1a, with CO2 and H+ from H2O over Pd2.2/Co–N/carbon, using MeCN containing 0.5 M 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([Bmim]Tf2N) as the electrolyte, in the presence of AMPA. As expected, the reduction potential was crucial for the conversion of 1a (Table 1, entries 1–6). The most effective potential was −2.3 V vs. Ag/Ag+, and afforded the full conversion of 1a with a 92% yield of N,N-dimethylaniline 1d at 30 °C. Lowering the reaction temperature to 20 °C resulted in a yield of only 65% after 10 h (Table S3†). Increasing the temperature to 40 °C, 50 °C and 60 °C resulted in 81%, 72% and 58% yields of 1d (Table S3†) due mainly to the lower solubility of CO2 in the electrolyte. Therefore, 30 °C is an optimal temperature. Table 1 (entries 5 and 7–10) and Fig. S5† indicated that reactant 1a could be converted completely after 10 h. Subsequently, the conversion of 1a was also studied using MeCN containing other ILs as the supporting electrolytes, but their performances were not as good as that of [Bmim]Tf2N (Table 1, entries 5 and 11–16). Furthermore, the solutions of [Bmim]Tf2N with AMPA in DMSO, DMF, MeNO2 and 1,4-dioxane were also used as the electrolytes, and lower yields of 1d were obtained (Table S4†). The results indicated that [Bmim]Tf2N in MeCN solution was the best electrolyte.
|
|
||||
|---|---|---|---|---|
| Entry | Electrolyteb | E /V | t/h | Yieldd/% |
| a Reaction conditions: nitrobenzene (1.0 mmol), AMPA (0.06 mmol), CO2 (0.1 MPa) and 30 °C. b Electrolyte (30 mL) is CO2-saturated MeCN, containing 0.5 M IL. c All potentials are reported with respect to Ag/Ag+. d Yields determined by 1H NMR spectroscopy. | ||||
| 1 | [Bmim]Tf2N | −1.9 | 10 | 13 |
| 2 | [Bmim]Tf2N | −2.0 | 10 | 36 |
| 3 | [Bmim]Tf2N | −2.1 | 10 | 68 |
| 4 | [Bmim]Tf2N | −2.2 | 10 | 89 |
| 5 | [Bmim]Tf2N | −2.3 | 10 | 92 |
| 6 | [Bmim]Tf2N | −2.4 | 10 | 92 |
| 7 | [Bmim]Tf2N | −2.3 | 1 | 7 |
| 8 | [Bmim]Tf2N | −2.3 | 5 | 56 |
| 9 | [Bmim]Tf2N | −2.3 | 8 | 85 |
| 10 | [Bmim]Tf2N | −2.3 | 12 | 92 |
| 11 | [Bmim]PF6 | −2.3 | 10 | 81 |
| 12 | [Bmim]BF4 | −2.3 | 10 | 82 |
| 13 | [Bmim]TfO | −2.3 | 10 | 85 |
| 14 | [Bmim]ClO4 | −2.3 | 10 | 23 |
| 15 | [Bmim]NO3 | −2.3 | 10 | 15 |
| 16 | [Bmim]H2PO4 | −2.3 | 10 | 30 |

Fig. S6† summarizes the yields of 1d from the electrochemical conversion of 1a over the Pdx/Co–N/carbon catalysts with different Pd particle sizes at different potentials. The electronic properties of the nanoparticles can be tuned by controlling the sizes.42,43 Generally, smaller nanoparticles have lower d-band centers, which in turn result in a decrease in their adsorption energies.44 With respect to the reduction of CO2, lowering the d-band center reduces the binding energy of the hypothetical intermediates,45 thereby enhancing the rate of CO2 reduction and the conversion of nitrobenzene.
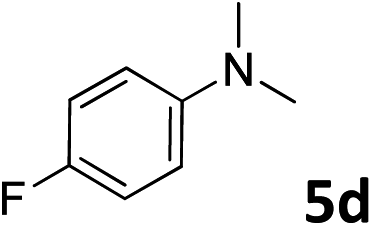
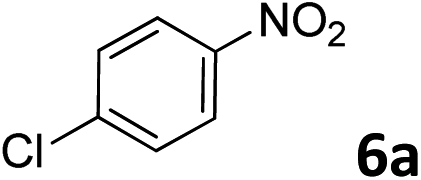
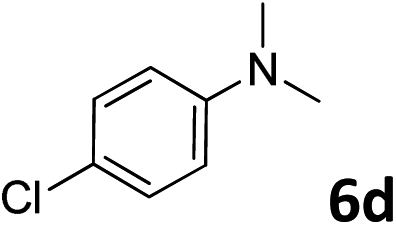
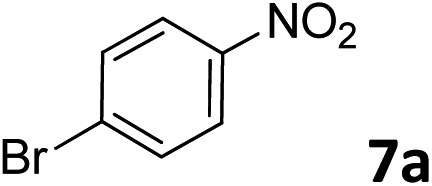
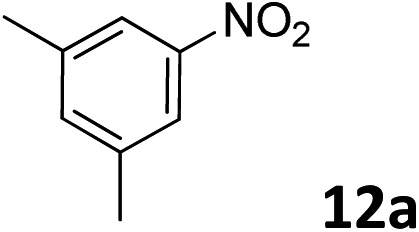
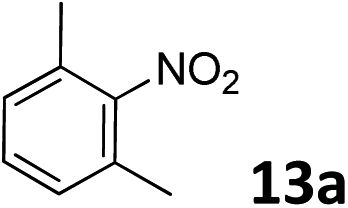
The methylation of various substituted nitrobenzenes over Pd2.2/Co–N/carbon was investigated (Table 2). The electron-donating methyl, methoxy and methylthio groups in the 4-position of the aromatic ring of 1a reduced the reactivity giving yields of 75% (2d), 74% (3d) and 71% (4d), respectively (Table 2, entries 1–3). For halogen-substituted nitrobenzenes, the desired products were obtained in 82–90% yields (Table 2, entries 4–8). When 4-nitrobiphenyl 10a and 2-nitrobiphenyl 11a were used, the corresponding products were generated in 78% (10d) and 76% (11d) yields, respectively (Table 2, entries 9 and 10). Di-substituted nitrobenzenes such as 5-nitro-m-xylene (12a), 2,6-dimethylnitrobenzene (13a) and 3-methyl-4-nitroanisole (14a) also showed high reactivity, which gave the desired products in 75–82% yields (Table 2, entries 11–13). To our delight, the N,N-dimethylation of benzonitrile and its derivatives could also be carried out with moderate yields (Table S5†).
| Entry | Substrates | Products | Yieldb/% |
|---|---|---|---|
| a Reaction conditions: substituted nitrobenzene (1.0 mmol), AMPA (0.06 mmol), CO2 (0.1 MPa), electrolyte (30 mL, CO2-saturated MeCN containing 0.5 M [Bmim]Tf2N), −2.3 V vs. Ag/Ag+, 30 °C and 10 h. b Yields determined by 1H NMR spectroscopy. | |||
| 1 |

|
|
75 |
| 2 |

|
|
74 |
| 3 |

|
|
71 |
| 4 |
|
|
90 |
| 5 |
|
|
87 |
| 6 |
|
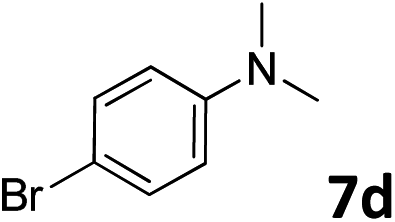
|
82 |
| 7 |
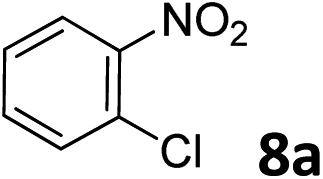
|

|
82 |
| 8 |

|

|
86 |
| 9 |

|
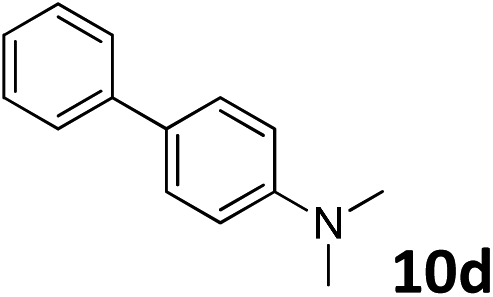
|
78 |
| 10 |

|
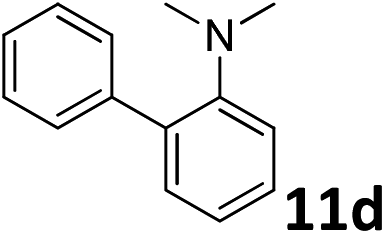
|
76 |
| 11 |
|

|
78 |
| 12 |
|

|
82 |
| 13 |

|

|
75 |
Some control experiments were conducted in order to obtain some evidence to study the mechanism. Fig. S7† shows that a very small amount of AMPA could promote the reaction to produce N,N-dimethylaniline efficiently. However, only aniline was formed in the absence of AMPA (Fig. S8†), and aniline could not be further converted even with extending the reaction time (Table S6†). These results indicate that AMPA is necessary and acts as a co-catalyst. To further understand the role of AMPA, NMR analysis was performed on aniline and its mixture with AMPA (Fig. S9†). It was found that the 1H signal of N–H in aniline shifted downfield from 4.88 to 5.11 ppm due to mixing with AMPA. The shift of the 1H signal of N–H in aniline was also affected by the amount of AMPA and the result was consistent with the reaction result that the yield of the product increased with an increase in the amount of AMPA from 0 to 0.06 mmol (Fig. S7†). From these results, it can be concluded that AMPA as a Brønsted base could activate the proton on aniline to promote the formation of N,N-dimethylaniline. In this way, the C atom on the intermediates obtained from the CO2 electroreduction could promote an easier electrophilic attack to the N atom on aniline, i.e. AMPA acted as the co-catalyst to promote the reaction of aniline with the intermediate from the CO2 electroreduction to form N-phenylformamide or N-methyl-N-phenylformamide. As a result, the next reaction in which N-methyl-N-phenylformamide reacted with H+ from water to synthesize N,N-dimethylaniline could proceed successfully. More details will be discussed in the mechanism part below.
On the basis of the experimental results and the related knowledge in the literature, we propose a speculative mechanism for the N,N-dimethylation of nitrobenzene, which is shown schematically in Fig. 2. Two parallel processes, CO2 and nitrobenzene hydrogenation, occur in the cathode compartment in the first step. For CO2 activation and reduction, a [Bmim-CO2]+ complex can form quickly via the hydrogen bonding interaction between CO2 and the IL cation,46 which can reduce the reaction barrier for the electron transfer to CO2.27 [Bmim-CO2]+ can be adsorbed on the electrode surface and CO2 is reduced to CO2˙−, which forms COads after receiving the second electron. The Pd nanoparticles facilitate these processes.32 After accepting an electron and proton, COads can be adsorbed on the support, and the pyridinic N as well as the Co–Nx moieties can further drive COads to generate CHOads.31,39,40 The next step is the insertion of CHOads into ArNH2Pd* to form adsorbed N-phenylformamide in the presence of the Brønsted base, AMPA. ArNH2Pd* can be obtained through the pathway of nitrobenzene → nitrosobenzene → N-phenylhydroxylamine → aniline on the electrode surface. It is usually observed in the electrochemical reduction of nitrobenzene to aniline.47 The adsorbed N-phenylformamide can be quickly hydrogenated into N-methylaniline. Following this, N-methylaniline can be adsorbed onto the electrode surface and reacted with CHOads to form the adsorbed N-methyl-N-phenylformamide. Finally, the protonation of the adsorbed N-methyl-N-phenylformamide leads to the formation of N,N-dimethylaniline.
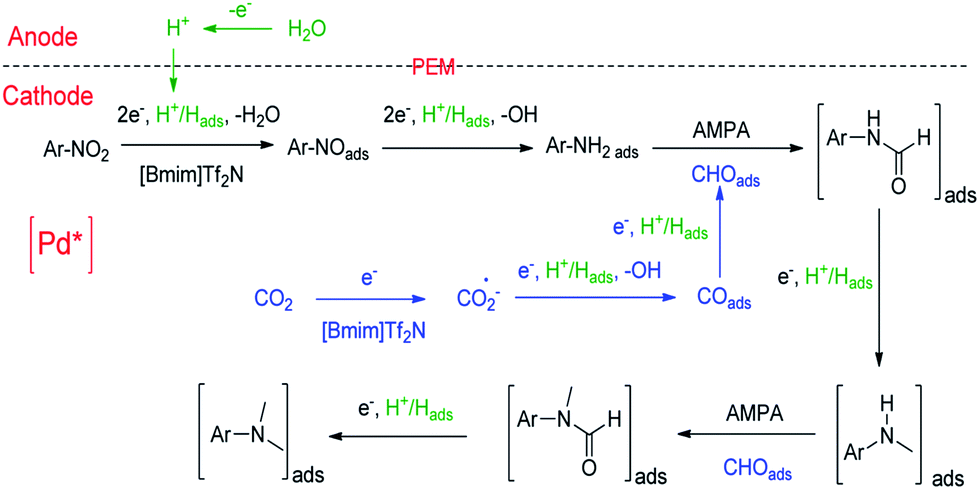 | ||
| Fig. 2 Possible pathway for the electrochemical reaction of nitrobenzene with CO2 and water to form N,N-dimethylaniline over Pd/Co–N/carbon. | ||
To further verify the reaction mechanism proposed, we conducted the experiments using some intermediates in Fig. 2 as the starting reactants, including aniline, N-phenylformamide, N-methylaniline and N-methyl-N-phenylformamide (Fig. S10†). 92% N,N-dimethylaniline, 5% N-methylaniline, a small amount of aniline, N-phenylformamide and N-methyl-N-phenylformamide can be detected in reaction (I). In reaction (II), the products contained 93% N,N-dimethylaniline, about 3% N-methylaniline as well as a small amount of N-phenylformamide and N-methyl-N-phenylformamide. The results were similar to those in reaction (I), suggesting that nitrobenzene was first converted to aniline completely in a short time. In reactions (III) and (V), when N-phenylformamide or N-methyl-N-phenylformamide was used as the starting reactant, the desired products N-methylaniline or N,N-dimethylaniline could be obtained. At the same time, most N-methylaniline could be transformed to N,N-dimethylaniline with the aid of AMPA in reaction (IV). In addition, a small amount of N-phenylformamide or N-methyl-N-phenylformamide as the reaction intermediates could also be detected in reactions (III) and (IV). All of this evidence supports the proposed reaction mechanism.
Conclusions
In conclusion, N,N-dimethylanilines can be synthesized by the methylation of nitrobenzene (or substituted nitrobenzenes) with CO2 and water. A Pd/Co–N/carbon electrode and an electrolyte composed of MeCN, [Bmim]Tf2N, and AMPA is a very efficient electrolysis system under ambient conditions. The N,N-dimethylation of a range of substrates, including nitrobenzene, substituted nitrobenzene and benzonitrile, can proceed smoothly with satisfactory yields. In the reaction, Pd/Co–N/carbon catalyzes the reduction of nitrobenzene to aniline quickly, which is further converted to N,N-dimethylaniline, catalyzed cooperatively by the electrocatalyst Pd/Co–N/carbon and the thermal catalyst AMPA. We believe that this route has potential application for producing N,N-dimethylanilines due to some obvious advantages. In addition, the combination of electrocatalysts and thermal catalysts can realize other reactions that cannot be conducted with only electrocatalysts or thermal catalysts.Experimental section
Preparation of Co–ZIF/GO
In this work, GO was prepared by the modified Hummers’ method according to ref. 48. 1.2 mmol of Co(NO3)2·6H2O and 1 mmol of 2-methylimidazole were dissolved in 12 mL and 20 mL of methanol, respectively. A Co(NO3)2 solution was added to the 2-methylimidazole solution to obtain a clear purple solution under continuous stirring. 10 mL of a GO dispersion (10 mg of GO in 10 mL of methanol/water, v/v, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was then immediately added to the above purple solution. After stirring for 3 h, the precipitate was collected by centrifugation and washing with methanol, followed by drying at 50 °C for 10 h. Co–ZIF/GO was obtained.
:1) was then immediately added to the above purple solution. After stirring for 3 h, the precipitate was collected by centrifugation and washing with methanol, followed by drying at 50 °C for 10 h. Co–ZIF/GO was obtained.
Preparation of Co–N/carbon support
The resulting Co–ZIF/GO precursor was heated in the tube furnace at 10 °C min−1 in an argon (Ar) medium to 800 °C and kept at this temperature for 3 h. The composites were then immersed in 2 M HCl aqueous solution for 24 h. The precipitate was collected by centrifugation and washing with water, followed by drying at 80 °C for 24 h. The Co–N/carbon support was obtained, of which the elemental composition is given in Table S1.†Preparation of Pdx/Co–N/carbon catalysts
The Pd/Co–N/carbon catalysts were synthesized with sodium borohydride as a reductive agent, sodium citrate as a stabilizing agent and Co–N/carbon as a support. The sizes of the Pd nanoparticles in the catalysts were controlled by varying the ratio of sodium citrate to PdCl2 as well as the reduction temperature.32 The average particle size was determined by counting more than 200 particles from the TEM images. Herein, the preparation of Pd2.2/Co–N/carbon is described in detail. 0.5 mmol of PdCl2 in 0.1 M HCl solution and 4 mmol of sodium citrate were dissolved in 200 mL of water under continuous stirring for 30 min. 200 mg of the as-prepared Co–N/carbon support was then added to the mixture with sonication for 1 h. 50 mL of 0.1 M sodium borohydride aqueous solution was then added to the above suspension dropwise at 0 °C under vigorous stirring to reduce the Pd2+. After stirring for 8 h, the black precipitate was collected by centrifugation and washing with water 3 times, and Pd2.2/Co–N/carbon was obtained after freeze drying.The other Pd/Co–N/carbon catalysts were prepared by controlling the reduction temperature and adjusting the ratio of sodium citrate (stabilizing agent) to PdCl2. For Pd3.6/Co–N/carbon and Pd4.5/Co–N/carbon, the temperatures for reducing Pd2+ were 25 °C and 60 °C, respectively, and the ratio of sodium citrate to PdCl2 was the same as that for preparing Pd2.2/Co–N/carbon. For Pd6.4/Co–N/carbon and Pd7.9/Co–N/carbon, the ratios of sodium citrate to PdCl2 were 2 and 0, respectively, and the reduction temperature was 25 °C. Pd10.2/Co–N/carbon was obtained by bubbling Pd3.6/Co–N/carbon with H2 for 12 h in 0.1 M HCl aqueous solution at 25 °C.
Materials characterization
XPS analysis was performed on the Thermo Scientific ESCALab 250Xi using 200 W monochromatic Al Kα radiation. The 500 μm X-ray spot was used. The base pressure in the analysis chamber was about 3 × 10−10 mbar. Typically, the hydrocarbon C 1s line at 284.8 eV from the adventitious carbon was used for energy referencing. The actual loading of Pd in the catalysts was determined by ICP-OES (Vista-MPX). The N2 adsorption/desorption isotherms of the Co–N/carbon nanosheets were determined using a Quadrasorb SI-MP system. The microstructures of the catalysts were studied using JEOL-2100F HR-TEM operated at 200 kV.Preparation of the electrode and electrochemical reaction
To prepare an electrode, the corresponding Pdx/Co–N/carbon catalyst was suspended in acetone with Nafion D-521 dispersion to form a homogeneous ink with the aid of ultrasound, which was spread onto carbon paper (CP: 1 × 1 cm−2) to obtain the working electrodes. The loading of the catalyst was 2.0 ± 0.1 mg cm−2. Before the experiment, all of the auxiliary electrodes were sonicated in acetone for 3 min and then washed with water and ethanol, followed by drying under a N2 atmosphere.An electrochemical workstation (CHI 6081E, Shanghai CH Instruments Co., China) was used in all of the experiments. The electrolysis experiments were conducted in a typical H-type cell that was similar to that used in previous works,28,29 and is schematically shown in Fig. S2.† It consisted of a cathode (working electrode), an anode (platinum gauze auxiliary electrode), and an Ag/Ag+ reference electrode. The cathode and anode compartments were separated by a Nafion 117 proton exchange membrane. A 0.5 M H2SO4 aqueous solution was used as the anodic electrolyte. H+ can be transferred from the anode compartment to the cathode compartment through the Nafion 117 proton exchange membrane, which is the hydrogen source. Under continuous stirring, CO2 was bubbled through the catholyte (2 mL min−1) for 30 min before electrolysis. The electrolysis experiments were then carried out with CO2 bubbling (2 mL min−1). In a typical reaction procedure, 1.0 mmol of nitrobenzene and 0.06 mmol of AMPA were added to 30 mL of the electrolyte (CO2-saturated MeCN containing 0.5 M [Bmim]Tf2N). The reaction was performed at 30 °C for the desired time under magnetic stirring. After the reaction, MeCN in the system was removed by rotary evaporation and the IL was extracted using ether. The isolated product was obtained and purified by column chromatography using petroleum ether/ethyl acetate (150:1).
Product analysis
The gaseous product of the electrochemical experiments was collected and analyzed by gas chromatography (GC, HP 4890D), which was equipped with FID and TCD detectors, using helium as the internal standard. For the methylation of nitrobenzene, the liquid product was analyzed using 1H NMR (Bruker Avance III 400 HD spectrometer) in chloroform-d, with TMS as an internal standard.Acknowledgements
The authors thank the National Natural Science Foundation of China (21533011, 21403253, 21673248), National Key Research and Development Program of China (2017YFA0403100), and the Chinese Academy of Sciences (QYZDY-SSW-SLH013).Notes and references
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296 CrossRef CAS PubMed.
- M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620 CrossRef CAS PubMed.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259 CrossRef CAS.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631 RSC.
- M. Cui, Q. Qian, Z. He, Z. Zhang, J. Ma, T. Wu, G. Yang and B. Han, Chem. Sci., 2016, 7, 5200 RSC.
- Q. Qian, J. Zhang, M. Cui and B. Han, Nat. Commun., 2016, 7, 11481 CrossRef PubMed.
- Z. Z. Yang, L. N. He, J. Gao, A. H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602 CAS.
- J. Hu, J. Ma, Q. Zhu, Z. Zhang, C. Wu and B. Han, Angew. Chem., Int. Ed., 2015, 54, 5399 CrossRef CAS PubMed.
- M. Cui, Q. Qian, Z. He, J. Ma, X. Kang, J. Hu, Z. Liu and B. Han, Chem.–Eur. J., 2015, 21, 15924 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- K. Beydoun, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 9554 CrossRef CAS PubMed.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568 CrossRef CAS PubMed.
- Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 12156 CrossRef CAS PubMed.
- I. Sorribes, J. R. Cabrero-Antonino, C. Vicent, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 13580 CrossRef CAS PubMed.
- E. Blondiaux, J. Pouessel and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 12186 CrossRef CAS PubMed.
- X. Cui, X. Dai, Y. Zhang, Y. Deng and F. Shi, Chem. Sci., 2014, 5, 649 RSC.
- O. Jacquet, X. Frogneux, C. Das Neves Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127 RSC.
- P. S. Niphadkar, P. N. Joshi, H. R. Gurav, S. S. Deshpande and V. V. Bokade, Catal. Lett., 2009, 133, 175 CrossRef CAS.
- F. M. Bautista, J. M. Campelo, A. Garcia, D. Luna, J. M. Marinas, A. A. Romero and M. R. Urbano, J. Catal., 1997, 172, 103 CrossRef CAS.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302 CrossRef CAS PubMed.
- J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265 CrossRef CAS PubMed.
- T. Morofuji, A. Shimizu and J.-i. Yoshida, J. Am. Chem. Soc., 2015, 137, 9816 CrossRef CAS PubMed.
- M. Rafiee, K. C. Miles and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 14751 CrossRef CAS PubMed.
- H. C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2010, 132, 2839 CrossRef CAS PubMed.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643 CrossRef CAS PubMed.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, Z. Zhang and B. Han, Angew. Chem., Int. Ed., 2016, 55, 6771 CrossRef CAS PubMed.
- Q. Zhu, J. Ma, X. Kang, X. Sun, H. Liu, J. Hu, Z. Liu and B. Han, Angew. Chem., Int. Ed., 2016, 55, 9012 CrossRef CAS PubMed.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845 CrossRef CAS PubMed.
- J. L. DiMeglio and J. Rosenthal, J. Am. Chem. Soc., 2013, 135, 8798 CrossRef CAS PubMed.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288 CrossRef CAS PubMed.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701 CrossRef CAS PubMed.
- X. Kang, X. Sun and B. Han, Adv. Mater., 2016, 28, 1011 CrossRef CAS PubMed.
- X. Sun, X. Kang, Q. Zhu, J. Ma, G. Yang, Z. Liu and B. Han, Chem. Sci., 2016, 7, 2883 RSC.
- J. Wei, Y. Hu, Y. Liang, B. Kong, J. Zhang, J. Song, Q. Bao, G. P. Simon, S. P. Jiang and H. Wang, Adv. Funct. Mater., 2015, 25, 5768 CrossRef CAS.
- Q. Yang, Q. Xu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 3685 CrossRef CAS PubMed.
- R. Nie, J. Shi, W. Du and Z. Hou, Appl. Catal., A, 2014, 473, 1 CrossRef CAS.
- A. S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H.-S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 10758 CrossRef CAS PubMed.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. P. Schouten, G. Mul and M. T. M. Koper, Nat. Commun., 2015, 6, 8177 CrossRef PubMed.
- G. P. S. Lau, M. Schreier, D. Vasilyev, R. Scopelliti, M. Grätzel and P. J. Dyson, J. Am. Chem. Soc., 2016, 138, 7820 CrossRef CAS PubMed.
- H. Mistry, R. Reske, Z. Zeng, Z.-J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, J. Am. Chem. Soc., 2014, 136, 16473 CrossRef CAS PubMed.
- M. Hou, Q. Mei and B. Han, J. Colloid Interface Sci., 2015, 449, 488 CrossRef CAS PubMed.
- W. P. Zhou, A. Lewera, R. Larsen, R. I. Masel, P. S. Bagus and A. Wieckowski, J. Phys. Chem. B, 2006, 110, 13393 CrossRef CAS PubMed.
- A. Chen and C. Ostrom, Chem. Rev., 2015, 115, 11999 CrossRef CAS PubMed.
- C. Cadena, J. L. Anthony, J. K. Shah, T. I. Morrow, J. F. Brennecke and E. J. Maginn, J. Am. Chem. Soc., 2004, 126, 5300 CrossRef CAS PubMed.
- C. Lagrost, L. Preda, E. Volanschi and P. Hapiot, J. Electroanal. Chem., 2005, 585, 1 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc01058c |
| This journal is © The Royal Society of Chemistry 2017 |