
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterizing the structural ensemble of γ-secretase using a multiscale molecular dynamics approach†
Rodrigo
Aguayo-Ortiz
a,
Cecilia
Chávez-García
a,
John E.
Straub
b and
Laura
Dominguez
 *a
*a
aDepartamento de Fisicoquímica, Facultad de Química, Universidad Nacional Autónoma de México, Mexico City, 04510, Mexico. E-mail: lauradd@unam.mx
bDepartment of Chemistry, Boston University, Boston, Massachusetts 02215, USA
First published on 5th June 2017
Abstract
γ-Secretase is an intramembrane-cleaving aspartyl protease that plays an essential role in the processing of a variety of integral membrane proteins. Its role in the ultimate cleavage step in the processing of amyloid precursor protein to form amyloid-β (Aβ) peptide makes it an important therapeutic target in Alzheimer's disease research. Significant recent advances have been made in structural studies of this critical membrane protein complex. However, details of the mechanism of activation of the enzyme complex remain unclear. Using a multiscale computational modeling approach, combining multiple coarse-grained microsecond dynamic trajectories with all-atom models, the structure and two conformational states of the γ-secretase complex were evaluated. The transition between enzymatic state 1 and state 2 is shown to critically depend on the protonation states of the key catalytic residues Asp257 and Asp385 in the active site domain. The active site formation, related to our γ-secretase state 2, is observed to involve a concerted movement of four transmembrane helices from the catalytic subunit, resulting in the required localization of the catalytic residues. Global analysis of the structural ensemble of the enzyme complex was used to identify collective fluctuations important to the mechanism of substrate recognition and demonstrate that the corresponding fluctuations observed were uncorrelated with structural changes associated with enzyme activation. Overall, this computational study provides essential insight into the role of structure and dynamics in the activation and function of γ-secretase.
Introduction
γ-Secretase is a membrane-embedded aspartyl protease that cleaves different integral membrane proteins within the lipid bilayer including amyloid precursor protein (APP), Notch, N-cadherin and ErbB4.1 APP is initially cleaved by β-secretase to release the APP C-terminal fragment (APP-C99), which is subsequently processed by γ-secretase to yield a variety of amyloid-β (Aβ) peptides of different lengths. The pattern of cleavage of APP-C99 results in a primary isoform, Aβ40 consisting of 40 amino acids, in addition to minor isoforms including Aβ38 and Aβ42.2 Aberrant accumulation of Aβ42 over Aβ40 has been associated with the formation of amyloid-β plaques in the brain of Alzheimer's disease (AD) patients.2,3 As such, developing an understanding of the mechanism of cleavage of APP-C99 by γ-secretase is an important goal for the field.The mature γ-secretase consists of four components (Fig. 1A): presenilin 1 (PS1), presenilin enhancer 2 (PEN-2), anterior pharynx-defective 1A (APH-1A) and nicastrin (NCT).4–6PS1 is the catalytic component of γ-secretase. It contains nine transmembrane helices (TMs) organized into a horseshoe-shaped structure with two catalytic Asp residues located at TM6 (Asp257) and TM7 (Asp385) within the convex side of the protein surface.7–10 Autoproteolysis of the intracellular loop connecting TM6 and TM7 leads to the formation of the PS1 N-terminal (NTF, TMs 1–6) and C-terminal fragments (CTF, TMs 7–9) that interact with PEN-2 and APH-1A, respectively.9PEN-2 consists of three TMs, of which TM1 and TM2 form a re-entrant loop extending halfway through the membrane from the intracellular side.7,11 The association of PEN-2 with PS1 has been related to the autocatalytic maturation of PS1 and γ-secretase activity. APH-1A consists of seven TMs helices and a C-terminal juxtamembrane region.12,13 Several experimental studies suggest that this component is required for proper γ-secretase assembly.14,15NCT is a transmembrane glycoprotein with a large N-terminal extracellular domain (ECD) and a single TM helix located in its C-terminal region.16,17 The globular ECD is comprised of a large and a small lobe composed of α-helices and β-strands.12,18,19 It has been proposed that Glu333 and Tyr337, located at NCT's large lobe, interact with the substrate's extracellular domain and play a critical role in substrate recognition.17,20 These residues are buried in a hydrophilic pocket covered by a lid formed by the small lobe.12 It is thought that a rotation of NCT's large lobe around a central pivot may cause lid opening, exposing the substrate-binding site.17 However, mutagenesis studies involving Glu333 contradict this theory, suggesting that Glu333 is involved in the maturation assembly of the complex rather than in substrate recognition.21,22
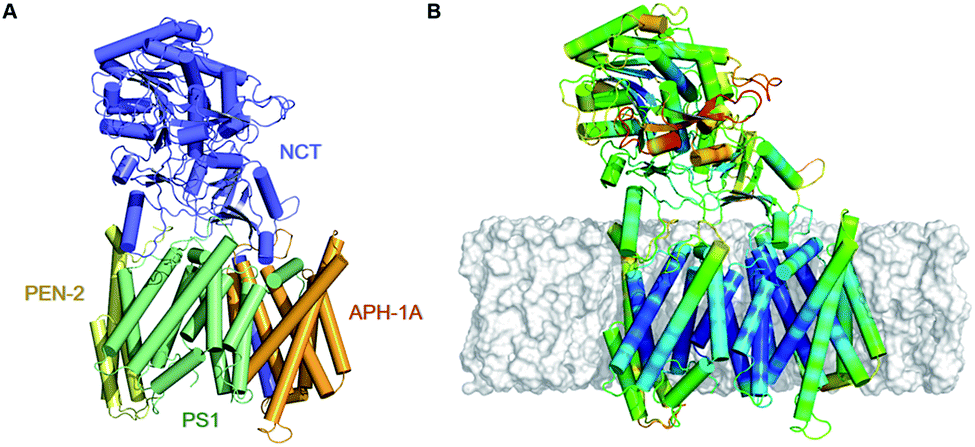 | ||
| Fig. 1 (A) Depiction of the all atom model of γ-secretase, derived from the 5FN2 PDB structure, colored by its subunits: PS1, NCT, PEN-2, and APH-1A in green, blue, yellow, and orange, respectively. (B) Fluctuation analysis of 5FN2-derived atomistic model of γ-secretase in POPC bilayer (gray) color-coded by the normalized per-residue root mean square fluctuation (RMSF) from more flexible (red) to less flexible (blue). The analysis was performed during the last 500 ns of the simulation. | ||
Novel high-resolution cryo-EM structures of human γ-secretase have been resolved.7,12,19 In the first proposed structure, Shi and coworkers7 reported the overall architecture of the complex at 4.5 Å resolution (PDB ID: 4UIS). Nevertheless, due to the limited resolution, partial assignment of the TMs side chains was only possible through sequence homology modeling of PS1 using a presenilin homologue (PSH) (PDB ID: 4HYG).8 Subsequently, Bai, et al.12 obtained an atomic-level γ-secretase structure with 3.4 Å resolution employing cryo-EM single particle analysis (PDB ID: 5A63). In this structure, the main-chain connectivity and side-chains were mostly displayed, facilitating identification of new interactions between the TMs, the presence of the juxtamembrane region of APH-1A, and a complete structure of the NCT ECD.
The structure of TM2 and the N-terminal region of TM6 in PS1 are typically not resolved due to the high flexibility of these domains. Importantly, four new γ-secretase structures exhibiting varying conformational states in this critical region have been resolved (PDB IDs: 5FN2, 5FN3, 5FN4 and 5FN5).19 The first was obtained in complex with the dipeptidic inhibitor N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT); however, the structure of the inhibitor was not assigned. The second and third γ-secretase structures were solved in complex with peptide fragments. Interestingly, the authors suggest that the peptide fragment found in 5FN3, located between PS1 TM2, TM3 and TM4, belongs to the N-terminal region of APP. Finally, the last structure did not appear to be complexed with any external agent, and as in the case of PDB ID: 5A63, it was not possible to resolve TM2.
A previous molecular dynamics (MD) study examined the dynamic properties and activation of a single human PS1 subunit embedded in a variety of membrane lipid compositions, with an initial PS1 conformation derived from homology modeling.23 However, in the absence of the PEN-2, APH-1A, and NCT subunits, the simulations exhibited large instabilities and flexibility in the TMs. More recently, Han and coworkers24 explored the initial substrate binding site of the transmembrane region of γ-secretase using a multiscale MD approach. In that work, the authors demonstrated the importance of using atomistic and coarse grained models to assess the behaviour of the TMs of forming the complex. Moreover, their results suggest that TM2/6/9 (including the PAL motif) constitute the initial APP-C99 binding site.
Despite these recent advances, many essential aspects of the structure and dynamics of the multicomponent γ-secretase complex remain undescribed. In this study, we employed a multiscale simulation approach that combines atomistic and coarse-grained models starting from a variety of cryo-EM structures to explore the dynamic structural ensemble of γ-secretase embedded in a POPC lipid bilayer. Conformational changes are analyzed using order parameters that characterize the essential dynamics of the enzyme complex, including the transition between active and inactive functional states. Our study provides critical insight into (1) the nature of large-scale conformational transitions in the γ-secretase complex, (2) the identification of two conformational states of PS1, constituting a mechanism for its activation, and (3) the characterization of NCT ECD mobility and its relationship to the observed PS1 states.
Methods
Model preparation
Two cryo-EM structures of γ-secretase (PDB IDs: 5FN2 and 5FN3) resolved at 4.2 and 4.1 Å resolution, respectively, were used as initial 3D coordinates to perform the multiscale MD simulations of the γ-secretase complexes. The missing side-chains of the structures were completed using the WHAT IF web server.25 The spatial arrangement of the γ-secretase models in the POPC lipid bilayer was assigned with the Orientation of Proteins in Membranes (OPM) web server.26Molecular dynamics simulations
We used a multiscale computational approach (Table 1) to characterize the structure and dynamics of the γ-secretase enzyme complex embedded in a POPC bilayer, combining all-atom and CG representations, using CHARMM36 and MARTINI force field models, respectively, for the protein, membrane, and solvent environments. We simulated two distinct γ-secretase models, each in three different protonated states: two models with one of the two catalytic aspartic acid residues protonated (Asp257 and Asp385) and a third model with both catalytic residues unprotonated.
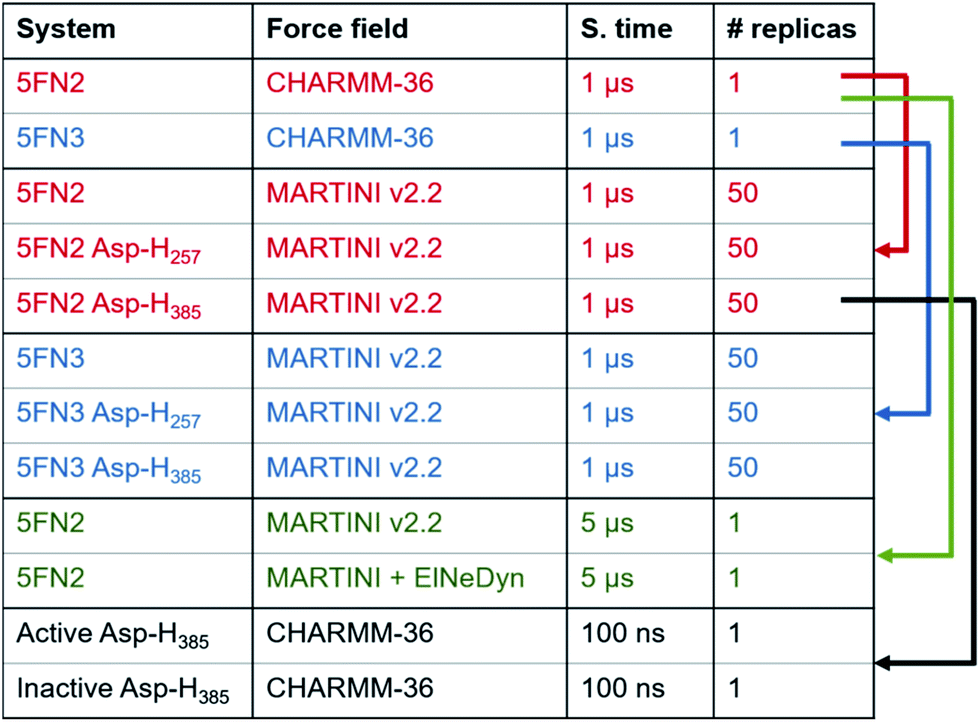
|
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 078 water particles, and 143 Cl− and 148 Na+ ions; the 5FN3 derived system contained the reconstructed protein complex, 326 POPC lipids, 46827 water particles, and 145 Cl− and 150 Na+ ions. The systems were energy minimized and equilibrated with NVT and NPT dynamics. Equilibration of each simulation was followed by a 1 μs production run with a time step of 2 ps. The temperature was set to 310 K using the Nose–Hoover coupling thermostat algorithm and the pressure was set to 1.0 bar using the semi-isotropic Parrinello–Rahman barostat algorithm. The Lennard-Jones potential was truncated using a shift function between 0.9 and 1.2 nm. Full electrostatic interactions were calculated between 0 and 1.2 nm, after which the electrostatic interactions were calculated using the Particle Mesh Ewald (PME) approach. Neighbor lists were updated every 20 steps and bonds involving hydrogens were constrained using the Linear Constraint Solver (LINCS) algorithm.29 The simulations were performed using GROMACS 5.0.6 (ref. 30) with the CHARMM36 force field31 and the TIP3P water model. The apolar contribution to the energy assigned to interaction of PS with adjacent components was calculated with the g_mmpbsa.py32 script. Finally, the MD simulations were analyzed using the inbuilt GROMACS tools and the MdAnalysis libraries33 for python and the images were generated using Bendix,34 VMD v1.9.2 (ref. 35) and PyMOL v0.9 (ref. 36).
078 water particles, and 143 Cl− and 148 Na+ ions; the 5FN3 derived system contained the reconstructed protein complex, 326 POPC lipids, 46827 water particles, and 145 Cl− and 150 Na+ ions. The systems were energy minimized and equilibrated with NVT and NPT dynamics. Equilibration of each simulation was followed by a 1 μs production run with a time step of 2 ps. The temperature was set to 310 K using the Nose–Hoover coupling thermostat algorithm and the pressure was set to 1.0 bar using the semi-isotropic Parrinello–Rahman barostat algorithm. The Lennard-Jones potential was truncated using a shift function between 0.9 and 1.2 nm. Full electrostatic interactions were calculated between 0 and 1.2 nm, after which the electrostatic interactions were calculated using the Particle Mesh Ewald (PME) approach. Neighbor lists were updated every 20 steps and bonds involving hydrogens were constrained using the Linear Constraint Solver (LINCS) algorithm.29 The simulations were performed using GROMACS 5.0.6 (ref. 30) with the CHARMM36 force field31 and the TIP3P water model. The apolar contribution to the energy assigned to interaction of PS with adjacent components was calculated with the g_mmpbsa.py32 script. Finally, the MD simulations were analyzed using the inbuilt GROMACS tools and the MdAnalysis libraries33 for python and the images were generated using Bendix,34 VMD v1.9.2 (ref. 35) and PyMOL v0.9 (ref. 36).
Additionally, in order to evaluate the stability of our simulations and validate our methodology and results, two longer simulations of 5 μs were performed for the unprotonated γ-secretase complex using our CG Martini model and a CG Martini with an Elastic Network in Dynamics (CG-ElNeDyn).40 The systems were prepared following the same methodology described above and results are presented in the ESI.†
The most representative structures of the state 1 and state 2 PS1 conformers from the 5FN2 derived CG simulations were selected using the GROMACS cluster tool with a root mean square deviation (RMSD) threshold of 2.0 Å. These structures were back-mapped from coarse-grained (MARTINI force field) models to all-atom (CHARMM36 force field) models using the script backward.py.41 The final systems were simulated for 100 ns of MD following the previously described all-atom simulation protocols.
Finally, three structures were selected from the 5FN2 derived CG simulations with protonated Asp385 to represent the compact, intermediate, and extended conformations of the γ-secretase state models. The density maps of the structures were generated with the molmap command of UCSF Chimera v1.11 (ref. 42) with a resolution of 8.0 Å.
Results
We employed multiscale molecular dynamics (MD) simulations, combining coarse-grained (CG) and all-atom models, to investigate different conformational states of the γ-secretase complex. The initial models were derived from the available experimental information (PDB IDs: 5FN2 and 5FN3). Our initial all-atom models were simulated for 1 μs in order to relax the structure into a POPC lipid bilayer environment and derive secondary and tertiary structural information required to build valid CG models. Using the constructed CG model of the full enzyme complex in a POPC lipid bilayer, 50 independent 1 μs trajectories were simulated in order to thoroughly sample the dynamics on a time scale sufficient to observe critical large-scale structural transitions. Subsequently, all-atom models of the γ-secretase complex were constructed based on conformational distributions derived from the extensive CG simulations. Finally, the all-atom models of the bilayer-embedded γ-secretase complex were simulated for 100 ns each, in order to assess the overall behavior of the complex at the atomic scale.In the following sections, we provide a detailed picture of (1) the relative roles of the enzyme subunits in stabilizing the structure of the PS1 catalytic subunit, (2) the activation mechanism of the enzyme resulting from protonation of the catalytic aspartic acid residues, and (3) the characterization of the principal motions of NCT ECD related to transitions between two conformational states of the enzyme complex. Overall, this study provides the first complete picture of the γ-secretase complex and the relative role of the enzyme subunits in the activation mechanism.
Protein dynamics and inter-subunit interactions of γ-secretase
γ-Secretase is a highly stable protein complex, the maturation and activation of which is related to the assembly of its four components.18 To develop a detailed atomistic description and evaluate the general dynamics of γ-secretase and its subunit interactions, we performed all-atom MD simulations of 5FN2 and 5FN3 derived models in a POPC lipid bilayer using the CHARMM36 force field (Fig. S1 and S2†). The initial structures display two important differences: (1) 5FN3 lacks the carboxyl terminal fragment of PS1 TM6 (264–288) while 5FN2 contains the carboxyl terminal fragment of PS1 TM6 and (2) 5FN3 presents a larger distance between the catalytic Asp residues (5.06 Å) compared to a shorter distance (3.89 Å) observed in 5FN2. Fig. 1B shows the all-atom fluctuation analysis of the 5FN2 structure model, depicting the high mobility of the NCT extracellular fragment and relatively low mobility of its TMs. PEN-2, TM5-7 of APH1, and TM2 of PS1 exhibited higher mobility due to their exposure to lipids and fewer protein–protein contacts. In contrast, the PS1 TMs in close contact with PEN-2 and APH-1A exhibited low mobility and stable protein–protein interactions during the all-atom simulations. These findings are consistent with previous experimental observations suggesting that PEN-2 and APH-1A play key roles in the catalytic subunit stabilization and activation.43 Our results also provide an explanation for the structural instabilities observed in an earlier simulation study of the isolated PS1 subunit.23Analysis of the apolar contributions to the inter-subunit binding free energies of the four γ-secretase components suggests that PS1-TM1, PS1-TM8 and PS1-TM9 are involved in critical interactions with APH-1A, contributing to the low observed energy values (Fig. S3A†). Similarly, we observed favorable interaction between PS1-TM4 and the first and third TMs of PEN-2. It is worth noting that we found significant interactions between the PS1 TM3–TM4 loop and the N-terminal fragment of PEN-2, which was involved in significant contacts with a major lobe helix of NCT. This network of interactions may play an essential role in the communication pathway between these subunits and the PS1 catalytic site. Fluctuation analysis reveals that the γ-secretase complex, including the catalytic subunit, remains structurally stable throughout the 1 μs all-atom simulations, consistent with previous experimental studies.7,12 An equivalent analysis of our 5FN3 model is shown in the ESI (Fig S1–S3†).
Presenilin structural ensemble
To complement the atomic-scale fluctuation analysis, we explored in detail the structure, flexibility, and orientation of the PS1 TMs in the POPC bilayer. Furthermore, we analyzed the dependence of the complex structure on the protonation state of the catalytic aspartic acid residues and the impact of protonation on the orientation of TMs defining the PS1 structure in the active and inactive states of the enzyme.The TM6 cytosolic fragment also showed high flexibility during the MD simulations. It has been proposed that alterations in the distance between cytosolic sides of TM6 and TM7 correlate with Aβ42 production, suggesting that the TM6 conformation is a critical regulator of PS1 catalytic activity.46 The large RMSF values and highly conserved helicity observed in TM2 and TM6 suggest the presence of collective motion modulating the relative orientation of these helices. The mobility of TM2 and TM6 has also been associated with the significant plasticity of the active site suggested by cryo-EM structural analysis.12 In Fig. 2 we present the computed tilt angle distribution of TMs, which agrees well with distributions derived from available γ-secretase structures. In particular, the wide tilt angle distribution of TM2 confirms the high mobility of this helix associated with the PS1 subunit plasticity.
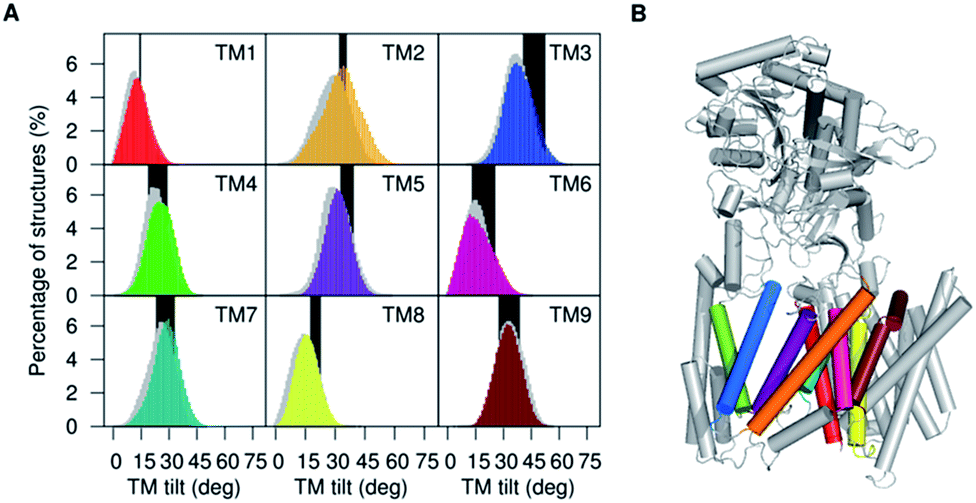 | ||
| Fig. 2 (A) Distribution of tilt angles of the PS1 TMs calculated for simulations of 5FN2 (color coded by TM helix number) and 5FN3 (in gray shade) and compared with TMs tilt angle ranges obtained from available experimental structures of γ-secretase (PDB IDs: 5A63, 4UIS, 5FN2, 5FN3, 5FN4 and 5FN5) (black bars). (B) Depiction of the 3D structure of γ-secretase (color coding PS1 TMs as (A)). | ||
PS1 populates two principal conformational states
PS1 is an aspartyl protease that contains two catalytic Asp residues located in the intramembrane regions of TM6 and TM7.8,47 Similar to other aspartyl proteases, the mechanism of catalysis is believed to involve formation of an initial intramolecular hydrogen-bond between the catalytic aspartic residues in coordination with a water molecule.48,49 In this acid–base mechanism, one Asp residue should be deprotonated (as aspartate, Asp−) to activate the water molecule, while the second Asp residue (as aspartic acid, Asp-H) donates a proton to the carbonyl group of the substrate.50 The initial hydrogen-bond formed between the aspartates is believed to be essential to the proton transfer step necessary to the overall enzymatic reaction.51,52 These observations suggest that the distance between the catalytic Asp residues should be less than 0.5 nm in order to form the crucial hydrogen-bond required for the formation of the PS1 active state.8 Previous structural studies of γ-secretase indicate that the relative proximity and orientation of the catalytic residues in the PS1 active site depend on the tilt angle orientation of different TMs of PS1.12,18 Inspired by this idea, large-scale conformational changes of γ-secretase were simulated for three coarse grained (CG) Martini models varying protonation states of the catalytic residues with either or both charged. Trajectories of 50 replicas were each simulated for 1 μs MD on each model. A pKa calculation with the PROPKA v3.1 (ref. 53) module of PDB2PQR server54 provides estimates of Asp385 (pKa = 9.91) and Asp257 (pKa = 5.12), suggesting these residues should be protonated and unprotonated, respectively, at pH = 7.0. As a consequence of the many replicas used and long simulation time, we expected to observe no dependence on the initial conditions (Fig. S4†). In order to validate our Martini CG model, we simulated 5 μs of dynamics for a CG Martini model and a CG Martini model including an Elastic-Network in Dynamics (ElNeDyn) (Fig. S5 and S6†).40 The CG-ElNeDyn simulation exhibited smaller RMSD values (∼0.2 nm) throughout the simulation. However, the RMSF showed that our CG-ElNeDyn system restricted motion of the HL1 loop, the CTF of TM6, and the loop between TM8 and TM9 (PAL motif). Importantly, the distance between the catalytic Asp residues in the CG-ElNeDyn model remained practically fixed. As mentioned above, experimental studies show that mobility of these regions is essential to conformational changes in the complex required to form the active and inactive states of the catalytic subunit. It is important to note that despite these differences, measurements of key distances between the TMs led to similar results in 5 μs simulations of a Martini CG model and CG-ElNeDyn model. Finally, the curvature and kinks of the PS1 TMs helices are consistent in all CG and AA simulation models (Fig. S7–S10†). Taken together these results validate our CG Martini models for the study of conformational changes of the γ-secretase enzyme complex.Order parameters used to characterize the PS1 ensemble include the TM tilt angles relative to the membrane normal (TTM) and the distance between the catalytic Asp (ddAsp). These order parameters are highly effective in differentiating the conformational state 1 (inactive state, long ddAsp and proper TTM angles) and state 2 (active state, short ddAsp and proper TTM angles) of γ-secretase. Short ddAsp conformations involve Asp conformations suitable to form the crucial hydrogen bond required for the catalytic reaction.
In order to explore the correlation between the TMs orientation and proximity of the catalytic residues, the set of 50 1 μs trajectories for the 5FN2 and 5FN3 derived CG models, varying Asp257 and Asp385 protonation states, were analyzed by projecting the simulated structural ensemble onto the ddAsp distance and TTM angles for TM2, TM6, TM7 and TM9. Fig. 3 displays the probability distributions for the ddAsp distance and the TTM angles for 5FN2 model of γ-secretase with unprotonated Asp and protonated Asp385 (similar results for 5FN3 and the two systems with the protonated Asp257 are available in Fig. S11†). There are notable changes in the relative probabilities of the conformational states of γ-secretase characterizing the structural ensemble of PS1: (1) the state 1, predominant in the unprotonated 5FN2 and 5FN3 derived CG models, is characterized by an inactive PS1 catalytic conformation with a ddAsp distance larger than 0.5 nm; (2) the state 2 observed in the protonated 5FN2 and 5FN3 derived CG models is characterized by a short ddAsp distance consistent with active site formation. The TTM angle distributions for the identified states indicate that TM6 and TM7 undergo an important TTM angle modification that is strongly correlated with modulation of the ddAsp distance. Moreover, the principal component analysis (PCA) of the PS1 catalytic subunit suggests that a concerted motion of all TMs is required for the state 1 to state 2 transition (Fig. S12†).
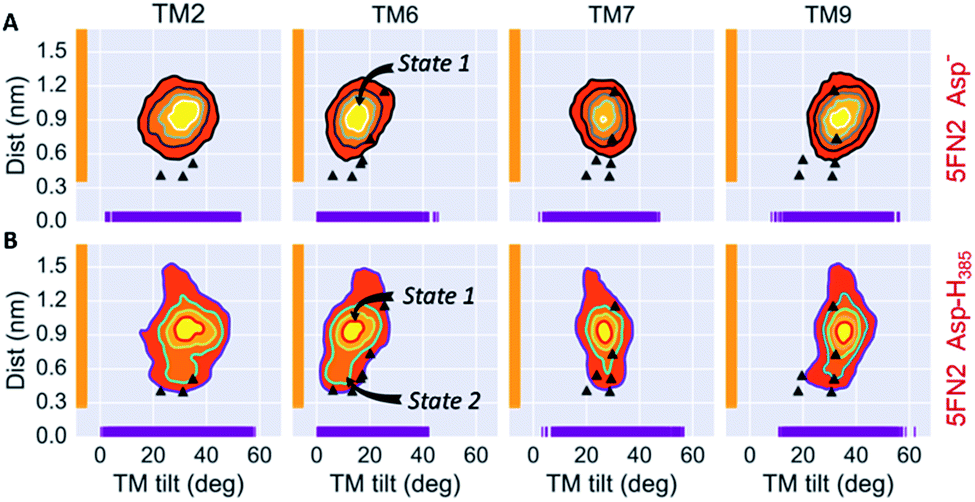 | ||
| Fig. 3 Simulated distributions of 5FN2 derived CG model in POPC bilayer projected onto (1) the distance between the catalytic residues (Asp257 and Asp385) and (2) the calculated TM2, TM6, TM7 and TM9 tilt angles in the (A) unprotonated and (B) Asp385 protonated states. The black triangles depict the values of ddAsp and TTM angles obtained from the experimental structures of γ-secretase (PDB IDs: 5A63, 4UIS, 5FN2, 5FN3, 5FN4 and 5FN5). | ||
Similarly, a slight change in the TTM angles of TM2 and TM9 is observed to be correlated with a conformational change in TTM angles of TM6 and TM7. Importantly, previous experimental studies have provided evidence that TM2 and TM9 constitute the initial substrate binding site in PS1, suggesting two different mechanisms of substrate entry into the active site: between TM6 and TM9 or between TM2 and TM6.43,45 Given the high flexibility and broad tilt angle distribution observed in our study, which is consistent with the conformational ensemble derived from cryo-EM studies, we specifically conjecture that TM2 acts as a gateway for substrate entry through TM2 and TM6.55
In order to obtain a detailed atomistic description of the identified conformational states of PS1 structures, we performed 100 ns all-atom MD simulations (Fig. 4) of the γ-secretase complex employing the CHARMM force field in a POPC lipid bilayer. The most probable state 1 and state 2 γ-secretase conformations were selected using the GROMACS cluster tool with a RMSD threshold of 2.0 Å from the CG structural ensemble. As observed in the CG studies, the state 2 conformation is showed to fluctuate between more and less active conformations (ddAsp distance does not exceed 0.75 nm). This state 2 (active/less-active) equilibrium is associated with Asp dihedral angle fluctuation with unaltered inclination of the key TMs. Equivalent fluctuations between state 1/state 2 (inactive/active) states were observed in the CG model simulations, indicating that the state 2 (active) conformation may require the substrate to be bound to the catalytic site in order to stabilize the active conformation. On the other hand, data derived from atomistic MD simulation of the PS1 state 1 (inactive) conformations display substantial structural fluctuations with a 1.0 nm ddAsp distance. This observation is consistent with the previously characterized inactive CG simulations. Furthermore, the analysis of water molecules at the active site showed the presence of intramolecular hydrogen-bonds between both catalytic aspartic residues and a coordinated water molecule within 3.0 Å distance and 150-degree angle (Fig. 4C). This hydrogen bond network was present 43.92% of the time of the last 50 ns of the state 2 all-atom simulation. Interestingly, this water coordination is a crucial step for PS1 activation and proteolytic processing of γ-secretase substrates.
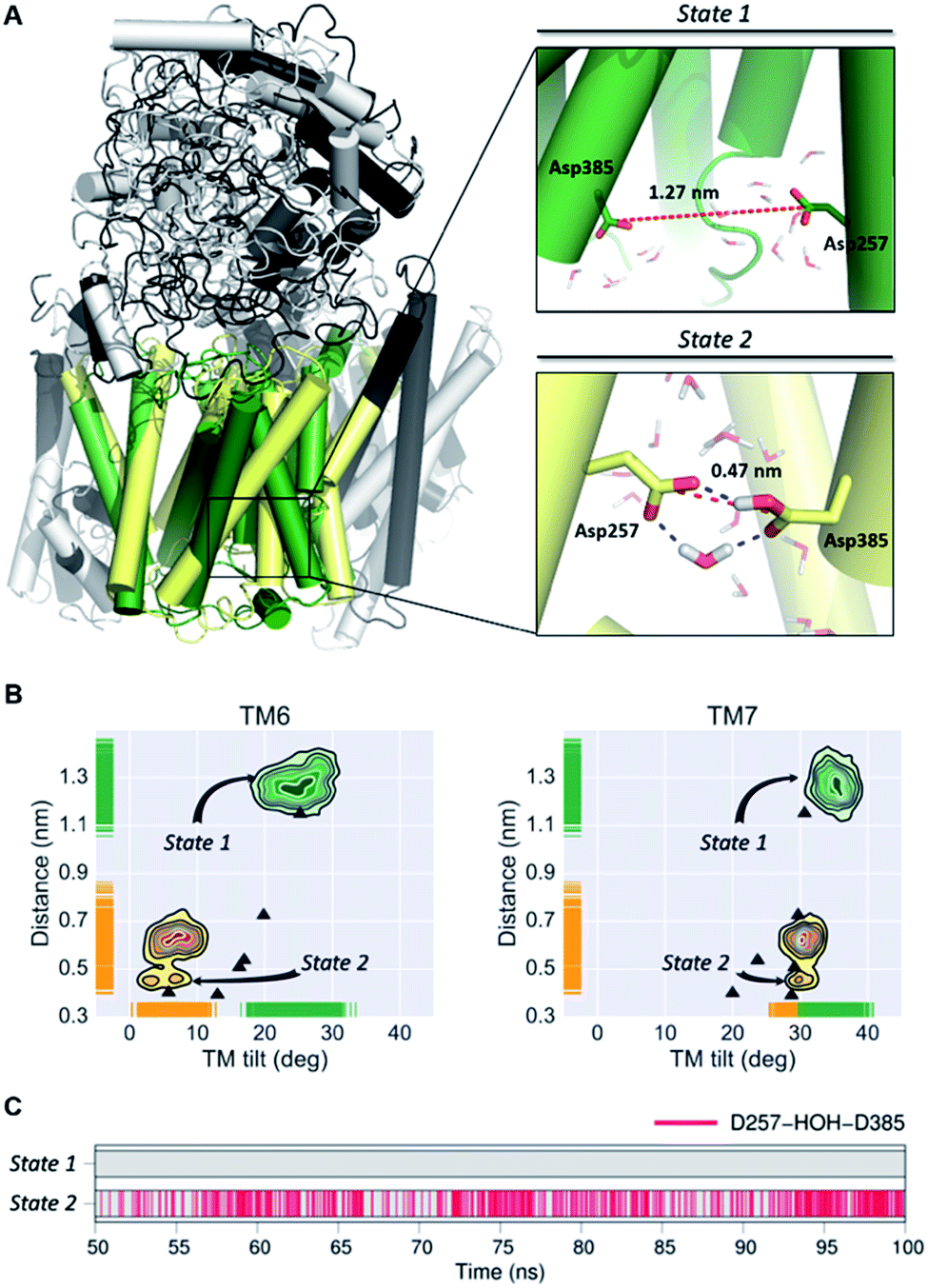 | ||
| Fig. 4 (A) Most representative structures of the state 1 (inactive) and state-2 (active) conformations of the PS1 subunit of γ-secretase obtained from all-atom MD simulations. The dotted red line represents the distance between the gamma carbons of Asp257 and Asp385. (B) Simulated distribution of both conformational state models projected onto the distance between the catalytic aspartic acid residues and the PS1 tilt angles of TM6 and TM7. The black triangles depict ddAsp distances and TTM angles obtained from the experimental structures of γ-secretase (PDB IDs: 5A63, 4UIS, 5FN2, 5FN3, 5FN4 and 5FN5). (C) Time evolution of hydrogen bonds between a coordinated water molecule and both catalytic aspartic residues through the last 50 ns of the state 1 and state 2 simulations. | ||
Nicastrin shows dramatic up-down movement and left-right rotation of the NCT extracellular domain
The large conformational modifications observed during cryo-EM studies of the complex have led to several hypotheses that seek to define the substrate recognition mechanism of γ-secretase.20,22,56 The observed structural changes in γ-secretase complex can be related to the large-scale motion of the NCT extracellular domain. Employing the 50 1 μs CG MD simulations of our 5FN2 and 5FN3 derived models, we performed PCA to identify the most significant collective motions of the complex, for both protonated and unprotonated states. The PCA analysis was performed with the GROMACS inbuilt tools (covar and anaeig).30The first two eigenvectors, involving the NCT ECD and accounting for the majority (23%) of overall γ-secretase motion, were selected to analyze the NCT movement. Analysis of conformations projected onto the first two PC eigenvectors revealed that the first eigenvector is related to NCT “up/down” movement while the second eigenvector corresponds to NCT-ECD “left/right” rotation. Fig. 5 depicts motion along the first and second principal components (PCs) using a porcupine representation. Interestingly, we found the same two principal component motions during the 5 μs simulation of our CG-ElNeDyn model with a slower dynamics and a smaller set of collective vectors (Fig. S6†).
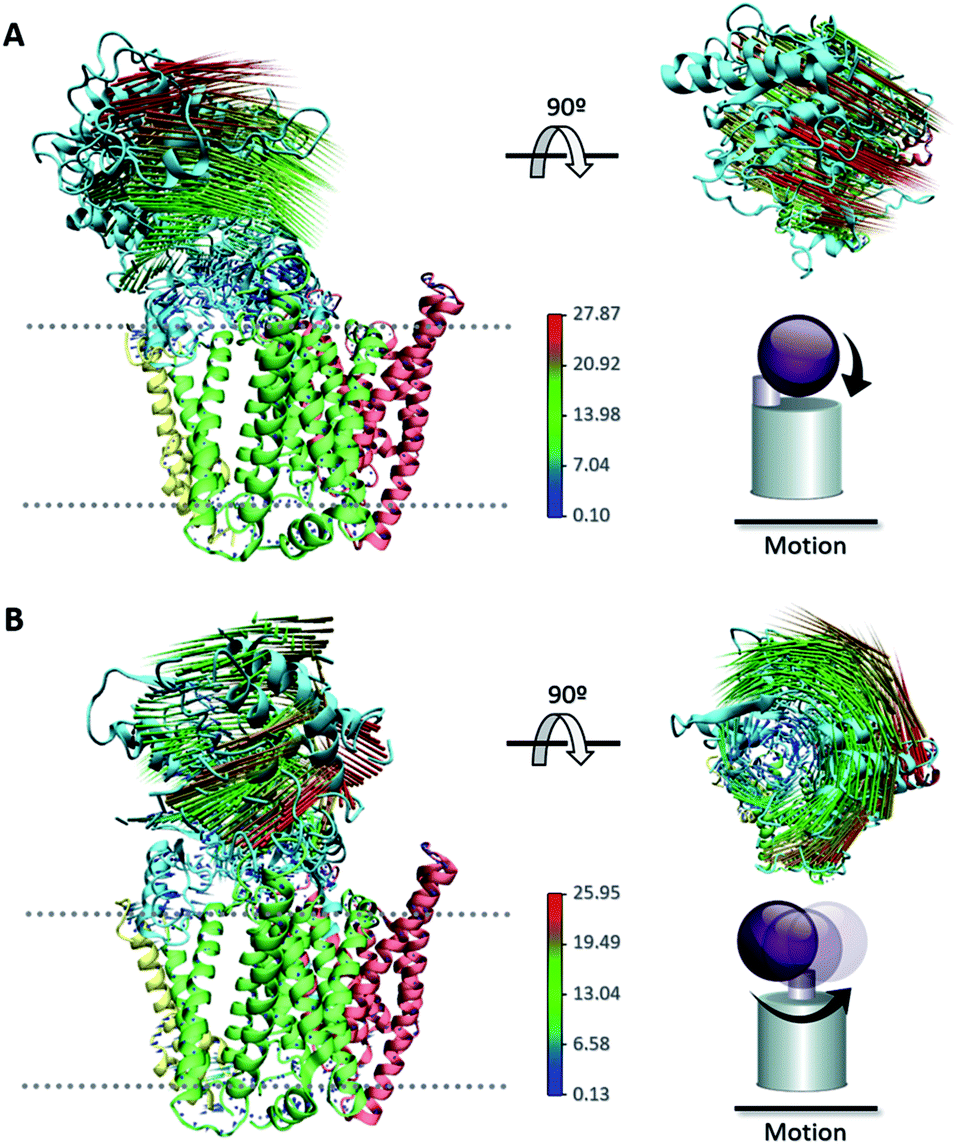 | ||
| Fig. 5 Porcupine representation of the (A) “up/down” movement and (B) “left/right” rotation of the NCT ECD obtained from PCA. | ||
In order to further analyze structural fluctuations of NCT, we constructed two order parameters based on the first and second PCs obtained from the CG structural ensemble (capturing up/down and left/right rotational motion of NCT, Fig. S13†). The first order parameter describes the distance between the center of geometry of PS1 and the center of geometry of the large lobe of NCT (ddPS-NCT). Significant variation in the distribution of the ddPS-NCT distances indicates that a percentage of the γ-secretase structures simulated using the CG model exhibit an “up” conformation (with ddPS-NCT distances larger to 5.0 nm) while a fraction of the NCT domain adopts a “down” conformation (with a ddPS-NCT distance shorter than 4.5 nm). The second order parameter characterizes the rotational motion of NCT (dihNCT) and involves a dihedral angle formed by four points of the NCT structure: (1) the intracellular and (2) extracellular amino acids of the single TM helix of NCT, (3) the center of mass of the large lobe of NCT, and (4) the residues Arg583–Asp588 (located at the distal end of the NCT large lobe). Previous experimental structural studies of γ-secretase have proposed a possible interaction of the substrate N-terminal region with exposed Glu333 located at a lid opening.17 Taken together, those observations and our simulation analysis support the conjecture that NCT rotation (dihNCT) is essential to the γ-secretase substrate binding mechanism. The simulated dihNCT distribution indicates that the NCT large lobe rotates in the left/right directions, with preference for leftward rotation (dihNCT values of −50 deg). However, further analysis demonstrates that the identified NCT motion, described by the ddPS-NCT distance and the dihNCT rotation, does not modify the position of the NCT-lid (Fig. S14 and S15†). This suggests that Glu333 might not be involved in the substrate recognition mechanism. Furthermore, Wolfe and coworkers57 proposed that the NCT-ECD acts as a steric gatekeeper for substrate entry into the PS1 active site, instead of having a specific interaction with the substrate. This steric block mechanism could be associated with ECD up-down movement, left-right rotation, and interaction with the transmembrane subunits, as shown in the following section.
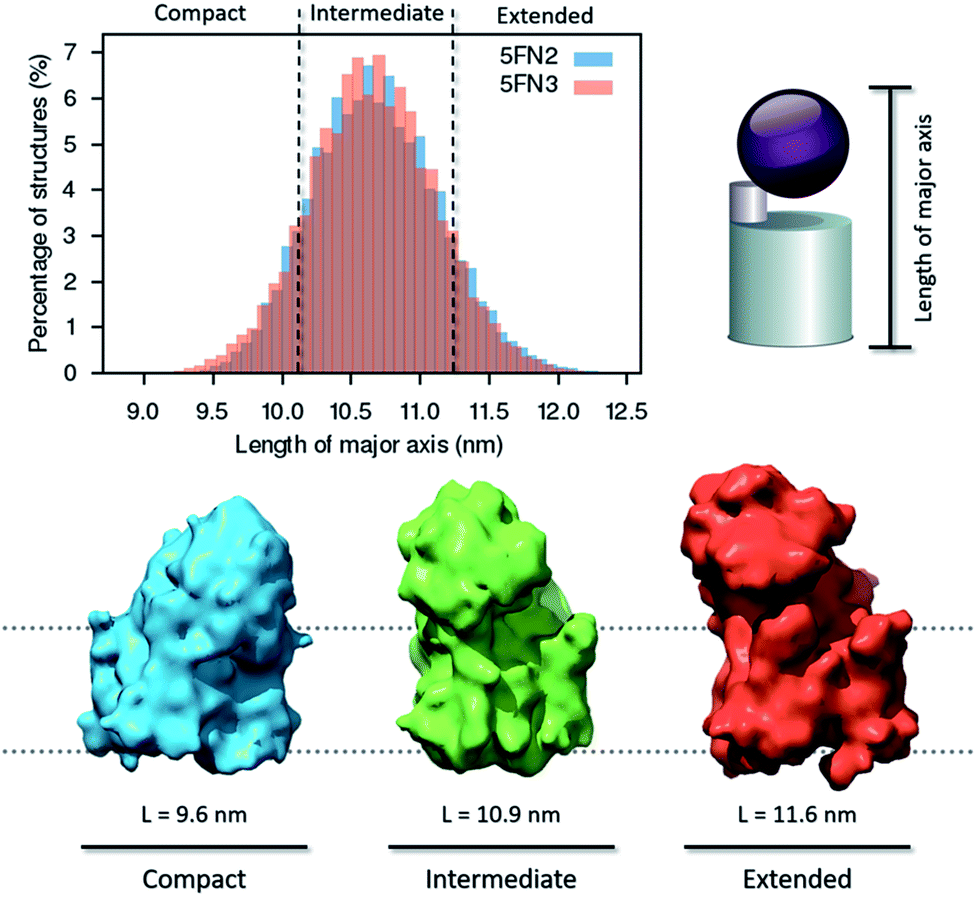 | ||
| Fig. 6 Distribution of major axis length and density map representation of the compact, intermediate, and extended conformations of the γ-secretase complex derived from 50 1 μs CG replica simulations of the γ-secretase complex. | ||
In further analysis, we extracted representative structures characterizing the three distinct conformational states in order to generate and analyze three density maps (8.0 Å resolution), employing UCSF Chimera software.42 The structural state transitions are observed to involve three distinctive interactions between the extracellular domain of NCT and key γ-secretase subunits: (1) in the extended state, the small lobe of NCT interacts with the C-terminal region of PEN2, (2) in the intermediate state, the large lobe of NCT interacts with the HL1 (hydrophilic TM1–TM2 loop) of PS1, and (3) in the compact state, the large lobe of NCT interacts with the APH-1A and PS1 extracellular domains. The broad distribution of structures in the intermediate state indicates that the second interaction, between the large lobe of NCT and HL1 of PS1, is essential for NCT ECD stabilization. Moreover, this conformation could be essential for NCT to act as a selective “gatekeeper” during peptide entry into the catalytic site.58 Experimental studies have demonstrated that when substrate or other agents (e.g. inhibitors and modulators) are bound to γ-secretase, the compact conformation of the complex is the most stable and favored state.20,22,56 Importantly, we found that although the NCT ECD interact with the hydrophilic PS1 HL1, APH-1A, and PEN2, no correlated motion was observed between the large NCT ECD and the PS1 TMs helices. These results suggest that the large NCT ECD motion is not critical to the activation or inactivation mechanism of the catalytic PS1 subunit (Fig. S16†).
Conclusions
Our multiscale simulation approach has elucidated the first detailed atomistic description of the structure and dynamics of the complete γ-secretase complex. Analysis of the structure and dynamics of our simulated γ-secretase models supports the hypothesis that PEN-2 and APH-1A play essential roles in stabilizing the PS1 catalytic subunit through interaction with the PS1 N-terminal and C-terminal fragments, respectively.Overall, the TMs of γ-secretase form a stable structural complex of relatively low mobility. However, we specifically observed that higher flexibility and correlated motion of PS1-TM2 and PS1-TM6 impacts the ddAsp distance essential for protease catalytic activity. Taken together, these findings suggest that correlated motion of key TMs helices, dependent on the ddAsp distance and TM tilt angles, is essential to the transitions between inactive to active states of PS1.
We have further demonstrated that the equilibrium between the state 1 (“inactive”) and state 2 (“active”) PS1 conformers is modulated by the protonation states of the catalytic residues Asp257 and Asp385. State 1 PS1 conformations are predominant in proteins having unprotonated catalytic Asp residues, while state 2 PS1 conformations are sampled when either catalytic Asp residue is protonated.
Analysis of global conformational changes in the γ-secretase complex identified significant mobility in the NCT ECD, characterized as up/down motion and left/right rotation of the large lobe of NCT ECD. Similar movement of the NCT ECD has been previously inferred from electron microscopy images.22 Based on these observations, it has been conjectured that γ-secretase function is controlled through modulation of relative populations of NCT ECD conformational states in the γ-secretase structural ensemble. Importantly, our data suggests that these movements are not correlated with NCT lid motion and the state 1/state 2 equilibrium of PS1, supporting that NCT-ECD only acts as steric gatekeeper for substrate entry into the active site, as proposed first by Wolfe and coworkers.57
This multiscale simulation analysis provides a detailed picture of the global structure and dynamics of the γ-secretase complex. The insight provided into the nature of the active and inactive state conformation, as well as the mechanism of transition between state 1 and state 2 of the PS1 catalytic domain, provides a foundation for future studies of the catalytic mechanism of substrate recognition and cleavage. It is our hope that the results of this study will contribute to a mechanistic understanding of the cleavage of APP-C99 by γ-secretase in the genesis of Aβ, critical to the structure-based design of AD therapeutics.
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
R. Aguayo-Ortiz (No. 510728/288862) and C. Chávez-García (No. 663225/583333) are grateful to CONACyT for the fellowships granted. The authors gratefully acknowledge the support of the Programa de Apoyo a la Investigación y el Posgrado (PAIP 5000-9155), Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT IA204716) and the Dirección General de Cómputo y de Tecnologías de Información (LANCAD-UNAM-DGTIC-306). JES acknowledges the generous support of the National Institutes of Health (R01 GM107703). The authors are very grateful to Sac N. I. Morales Betanzos for contributions to figures in this manuscript.Notes and references
- B. De Strooper and W. Annaert, Annu. Rev. Cell Dev. Biol., 2010, 26, 235–260 CrossRef CAS PubMed.
- F. M. LaFerla, K. N. Green and S. Oddo, Nat. Rev. Neurosci., 2007, 8, 499–509 CrossRef CAS PubMed.
- L. Mucke, Nature, 2009, 461, 895–897 CrossRef CAS PubMed.
- B. De Strooper, Neuron, 2003, 38, 9–12 CrossRef CAS PubMed.
- W. T. Kimberly, M. J. LaVoie, B. L. Ostaszewski, W. Ye, M. S. Wolfe and D. J. Selkoe, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6382–6387 CrossRef CAS PubMed.
- C. A. Hansson, S. Frykman, M. R. Farmery, L. O. Tjernberg, C. Nilsberth, S. E. Pursglove, A. Ito, B. Winblad, R. F. Cowburn, J. Thyberg and M. Ankarcrona, J. Biol. Chem., 2004, 279, 51654–51660 CrossRef CAS PubMed.
- L. Sun, L. Zhao, G. Yang, C. Yan, R. Zhou, X. Zhou, T. Xie, Y. Zhao, S. Wu, X. Li and Y. Shi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6003–6008 CrossRef CAS PubMed.
- X. Li, S. Dang, C. Yan, X. Gong, J. Wang and Y. Shi, Nature, 2013, 493, 56–61 CrossRef PubMed.
- T. Tomita and T. Iwatsubo, J. Biol. Chem., 2013, 288, 14673–14680 CrossRef CAS PubMed.
- S. Dang, S. Wu, J. Wang, H. Li, M. Huang, W. He, Y.-M. Li, C. C. L. Wong and Y. Shi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 201502150 CrossRef PubMed.
- X. Zhang, C. J. Yu and S. S. Sisodia, Mol. Neurodegener., 2015, 10, 39 CrossRef PubMed.
- X. Bai, C. Yan, G. Yang, P. Lu, D. Ma, L. Sun, R. Zhou, S. H. W. Scheres and Y. Shi, Nature, 2015, 525, 212–217 CrossRef CAS PubMed.
- Y. Li, L. S. Y. Liew, Q. Li and C. Kang, Sci. Rep., 2016, 6, 19522 CrossRef CAS PubMed.
- R. Francis, G. McGrath, J. Zhang, D. A. Ruddy, M. Sym, J. Apfeld, M. Nicoll, M. Maxwell, B. Hai, M. C. Ellis, A. L. Parks, W. Xu, J. Li, M. Gurney, R. L. Myers, C. S. Himes, R. Hiebsch, C. Ruble, J. S. Nye and D. Curtis, Dev. Cell, 2002, 3, 85–97 CrossRef CAS PubMed.
- N. Takasugi, T. Tomita, I. Hayashi, M. Tsuruoka, M. Niimura, Y. Takahashi, G. Thinakaran and T. Iwatsubo, Nature, 2003, 422, 438–441 CrossRef CAS PubMed.
- D. M. Bolduc and M. S. Wolfe, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14643–14644 CrossRef CAS PubMed.
- T. Xie, C. Yan, R. Zhou, Y. Zhao, L. Sun, G. Yang, P. Lu, D. Ma and Y. Shi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13349–13354 CrossRef CAS PubMed.
- P. Lu, X. C. Bai, D. Ma, T. Xie, C. Yan, L. Sun, G. Yang, Y. Zhao, R. Zhou, S. H. Scheres and Y. Shi, Nature, 2014, 512, 166–170 CrossRef CAS PubMed.
- X.-C. Bai, E. Rajendra, G. Yang, Y. Shi and S. H. Scheres, eLife, 2015, 4, e11182 Search PubMed.
- S. Shah, S. F. Lee, K. Tabuchi, Y. H. Hao, C. Yu, Q. LaPlant, H. Ball, C. E. Dann, T. Südhof and G. Yu, Cell, 2005, 122, 435–447 CrossRef CAS PubMed.
- L. Chávez-Gutiérrez, A. Tolia, E. Maes, T. Li, P. C. Wong and B. De Strooper, J. Biol. Chem., 2008, 283, 20096–20105 CrossRef PubMed.
- N. Elad, B. De Strooper, S. Lismont, W. Hagen, S. Veugelen, M. Arimon, K. Horre, O. Berezovska, C. Sachse and L. Chávez-Gutiérrez, J. Cell Sci., 2015, 128, 589–598 CrossRef CAS PubMed.
- R. Kong, S. Chang, W. Xia and S. T. C. Wong, J. Struct. Biol., 2015, 191, 120–129 CrossRef CAS PubMed.
- S. Li, W. Zhang and W. Han, Initial Substrate Binding of γ-Secretase: The Role of Substrate Flexibility, 2017 Search PubMed.
- M. L. Hekkelman, T. A. H. te Beek, S. R. Pettifer, D. Thorne, T. K. Attwood and G. Vriend, Nucleic Acids Res., 2010, 38, 719–723 CrossRef PubMed.
- M. A. Lomize, A. L. Lomize, I. D. Pogozheva and H. I. Mosberg, Bioinformatics, 2006, 22, 623–625 CrossRef CAS PubMed.
- J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. B. Iii, A. D. MacKerell Jr, B. Klauda and W. Im, J. Chem. Theory Comput., 2016, 12, 405–413 CrossRef CAS PubMed.
- E. L. Wu, X. Cheng, S. Jo, H. Rui, K. C. Song, E. M. Dávila-Contreras, Y. Qi, J. Lee, V. Monje-Galvan, R. M. Venable, J. B. Klauda and W. Im, J. Comput. Chem., 2014, 35, 1997–2004 CrossRef CAS PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páall, J. C. Smith, B. Hess and E. Lindah, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- R. B. Best, X. Zhu, J. Shim, P. E. M. Lopes, J. Mittal, M. Feig, J. MacKerell and D. Alexander, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS PubMed.
- R. Kumari, R. Kumar, O. Source, D. Discovery and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- N. Michaud-agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
- A. C. E. Dahl, M. Chavent and M. S. P. Sansom, Struct. Bioinf., 2012, 28, 2193–2194 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- W. L. DeLano, DeLano Scientific, San Carlos, CA, 2007, http://www.pymol.org Search PubMed.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. De Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- Y. Qi, H. I. Ingólfsson, X. Cheng, J. Lee, S. J. Marrink and W. Im, J. Chem. Theory Comput., 2015, 11, 4486–4494 CrossRef CAS PubMed.
- S. O. Yesylevskyy, L. V. Schäfer, D. Sengupta and S. J. Marrink, PLoS Comput. Biol., 2010, 6, 1–17 Search PubMed.
- X. Periole, M. Cavalli, S. J. Marrink and M. A. Ceruso, J. Chem. Theory Comput., 2009, 5, 2531–2543 CrossRef CAS PubMed.
- T. A. Wassenaar, K. Pluhackova, R. A. Böckmann, S. J. Marrink and D. P. Tieleman, J. Chem. Theory Comput., 2014, 10, 676–690 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- N. Watanabe, S. Takagi, A. Tominaga, T. Tomita and T. Iwatsubo, J. Biol. Chem., 2010, 285, 19738–19746 CrossRef CAS PubMed.
- S. Takagi-Niidome, T. Sasaki, S. Osawa, T. Sato, K. Morishima, T. Cai, T. Iwatsubo and T. Tomita, J. Neurosci., 2015, 35, 2646–2656 CrossRef CAS PubMed.
- A. Y. Kornilova, F. Bihel, C. Das and M. S. Wolfe, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3230–3235 CrossRef CAS PubMed.
- A. Tominaga, T. Cai, S. Takagi-Niidome, T. Iwatsubo and T. Tomita, J. Neurosci., 2016, 36, 1362–1372 CrossRef CAS PubMed.
- H. Steiner, M. Kostka, H. Romig, G. Basset, B. Pesold, J. Hardy, a. Capell, L. Meyn, M. L. Grim, R. Baumeister, K. Fechteler and C. Haass, Nat. Cell Biol., 2000, 2, 848–851 CrossRef CAS PubMed.
- D. B. Northrop, Acc. Chem. Res., 2001, 34, 790–797 CrossRef CAS PubMed.
- E. Erez, D. Fass and E. Bibi, Nature, 2009, 459, 371–378 CrossRef CAS PubMed.
- A. Tolia, L. Chávez-Gutiérrez and B. De Strooper, J. Biol. Chem., 2006, 281, 27633–27642 CrossRef CAS PubMed.
- M. Bentahir, O. Nyabi, J. Verhamme, A. Tolia, K. Horré, J. Wiltfang, H. Esselmann and B. De Strooper, J. Neurochem., 2006, 96, 732–742 CrossRef CAS PubMed.
- F. Sussman, M. C. Villaverde, J. L. Dominguez and U. E. Danielson, Curr. Pharm. Des., 2013, 19, 4257–4275 CrossRef CAS PubMed.
- M. H. M. Olsson, R. S. Chresten, M. Rostkowski and J. H. Jersen, J. Chem. Theory Comput., 2011, 2, 525–537 CrossRef PubMed.
- T. J. Dolinsky, J. E. Nielsen, J. A. McCammon and N. A. Baker, Nucleic Acids Res., 2004, 32, 665–667 CrossRef PubMed.
- A. Fukumori and H. Steiner, EMBO J., 2016, 35, 1628–1643 CrossRef CAS PubMed.
- Y. Li, S. H. J. Lu, C. J. Tsai, C. Bohm, S. Qamar, R. B. Dodd, W. Meadows, A. Jeon, A. McLeod, F. Chen, M. Arimon, O. Berezovska, B. T. Hyman, T. Tomita, T. Iwatsubo, C. M. Johnson, L. A. Farrer, G. Schmitt-Ulms, P. E. Fraser and P. H. St George-Hyslop, Structure, 2014, 22, 125–135 CrossRef CAS PubMed.
- D. M. Bolduc, D. R. Montagna, Y. Gu, D. J. Selkoe and M. S. Wolfe, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E509–E518 CrossRef CAS PubMed.
- S. Urban, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 201524151 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc00980a |
| This journal is © The Royal Society of Chemistry 2017 |