
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystallophore: a versatile lanthanide complex for protein crystallography combining nucleating effects, phasing properties, and luminescence†
Sylvain
Engilberge‡
a,
François
Riobé‡
 b,
Sebastiano
Di Pietro
b,
Louise
Lassalle
a,
Nicolas
Coquelle
a,
Charles-Adrien
Arnaud
a,
Delphine
Pitrat
b,
Jean-Christophe
Mulatier
b,
Dominique
Madern
a,
Cécile
Breyton
a,
Olivier
Maury
*b and
Eric
Girard
*a
b,
Sebastiano
Di Pietro
b,
Louise
Lassalle
a,
Nicolas
Coquelle
a,
Charles-Adrien
Arnaud
a,
Delphine
Pitrat
b,
Jean-Christophe
Mulatier
b,
Dominique
Madern
a,
Cécile
Breyton
a,
Olivier
Maury
*b and
Eric
Girard
*a
aUniv. Grenoble Alpes, CEA, CNRS, IBS, F-38000 Grenoble, France. E-mail: eric.girard@ibs.fr
bUniv Lyon, Ens de Lyon, CNRS UMR 5182, Université Claude Bernard Lyon 1, Laboratoire de Chimie, F-69342 Lyon, France. E-mail: olivier.maury@ens-lyon.fr
First published on 6th June 2017
Abstract
Macromolecular crystallography suffers from two major issues: getting well-diffracting crystals and solving the phase problem inherent to large macromolecules. Here, we describe the first example of a lanthanide complex family named “crystallophore” (Xo4), which contributes to tackling both bottlenecks. This terbium complex, Tb–Xo4, is an appealing agent for biocrystallography, combining the exceptional phasing power of the Tb(III) heavy atom with powerful nucleating properties, providing ready-to-use crystals for structure determination. Furthermore, protein/Tb–Xo4 co-crystals can be easily detected and discriminated from other crystalline by-products using luminescence. We demonstrate the potential of this additive for the crystallisation and structure determination of eight proteins, two of whose structures were unknown.
Introduction
X-ray crystallography remains the method of choice to obtain high-precision structural information about biological macromolecules. This is illustrated by the fact that almost 90% of protein structures deposited in the Protein Data Bank were elucidated using this method. However, production of high-quality single crystals and phase determination are the two main issues inherent to X-ray crystallography that severely slow down the resolution of new protein structures.1 Phase determination has been partially solved with the increasing number of known structures and the development of sophisticated algorithms and programs for molecular replacement. When the lack of homologous structures hinders structure determination, de novo phasing using anomalous-based methods – Single-wavelength Anomalous Dispersion (SAD) and Multi-wavelength Anomalous Dispersion (MAD)2,3 – is used thanks to the development of tuneable high-energy synchrotron radiation sources. These methods rely on the presence of well-ordered heavy atoms within the crystal, and this can be achieved by the conventional seleno-methionine labelling of the protein4 or by the recent exploitation of the intrinsic proteins’ sulphur content.5,6 Thus, crystallization represents the major issue in the whole structure determination process. Commercially available crystallization screens associated with crystallization robotics have optimised the trial and error process. However, crystallization of biological macromolecules remains very time-consuming, without any guarantee of success, and has triggered the research for various additives to facilitate the nucleation process. In this context, heterogeneous nucleants have been developed and these include minerals,7,8 natural materials such as horse and human hair,9,10 homologous protein thin films,11 or carbon-nanotubes.12,13 The best results have been obtained with porous nucleating agents such as porous silicon,14 nanoporous gold,15 microporous zeolite,16 bioglass,17 and, more recently, molecularly imprinted polymers.18–20Over the last 15 years, lanthanide complexes have been widely used to solve the phase problem thanks to the large anomalous contribution of lanthanide ions.21 Insertion of these heavy ions into protein crystals has been achieved by (i) substitution of Ca2+ in calcium-binding proteins,22,23 (ii) covalent grafting of a lanthanide binding tag,24,25 and (iii) supramolecular co-crystallization of lanthanide complexes with protein. In this context, several complexes, including macrocyclic (DOTA, DO3A, and HPDO3A), polydentate (DTPA-BMA) or tris-dipicolinate lanthanide complexes (Fig. 1), have been evaluated on commercial proteins26–28 but have also allowed the structural determination of new proteins.29-35 Herein, we report a new terbium(III) complex that combines nucleating, phasing, and luminescence properties, allowing us to overcome the two major issues of X-ray macromolecular crystallography with a single tool (patent pending PCT/FR2016/053539). We called this type of complex “crystallophore” (Xo4) and illustrated its properties for the crystallisation and structure determination of a series of eight proteins, including two proteins of unknown structure.
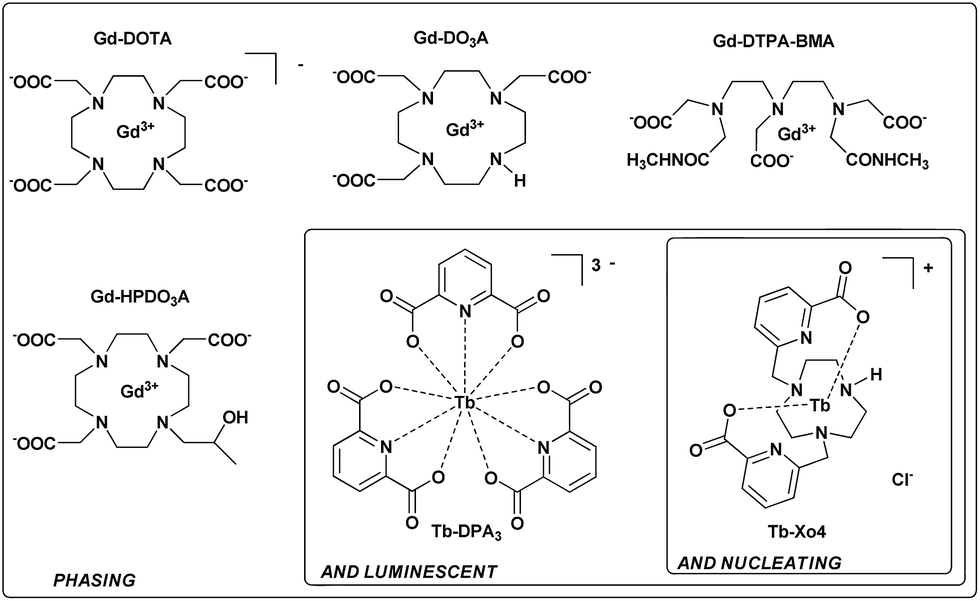 | ||
| Fig. 1 Three generations of complexes designed for protein structure determination. | ||
Results and discussion
Synthesis of Tb–Xo4
The complex was designed to be stable and highly soluble in aqueous media, and to be cationic with a non-saturated coordination sphere while remaining luminescent. It consists of a triazacyclononane macrocyclic scaffold bearing two picolinate arms, which ensure a good stability in the crystallization media and act as efficient antennae to sensitize terbium(III) luminescence (Fig. 1).36 This macrocyclic ligand strongly chelates the Tb(III) ion with a coordination number of seven; consequently two coordination sites remain accessible for further direct interaction with proteins. In particular, this cationic complex was designed to preferentially target protein carboxylate amino-acids like aspartate or glutamate. The compound was prepared and used as a chloride salt of the complex. Details of the synthesis and characterisation are provided in the ESI (Fig. S1–S8†).Nucleating properties of Tb–Xo4
The potentialities of this new complex were first evaluated with the benchmark hen egg white lysozyme (HEWL) protein. The standard conditions leading to the tetragonal crystal form, i.e. in the presence of NaCl as a precipitant, were used. First, the Tb–Xo4 optimal concentration was determined by evaluating different complex concentrations and by keeping the protein concentration constant at 20 mg mL−1 (Fig. 2). Interestingly, the former generation of complexes are generally used at high concentrations (50–100 mM), whereas crystals of HEWL have been obtained with lower concentrations of Tb–Xo4, with an optimal concentration of about 10 mM.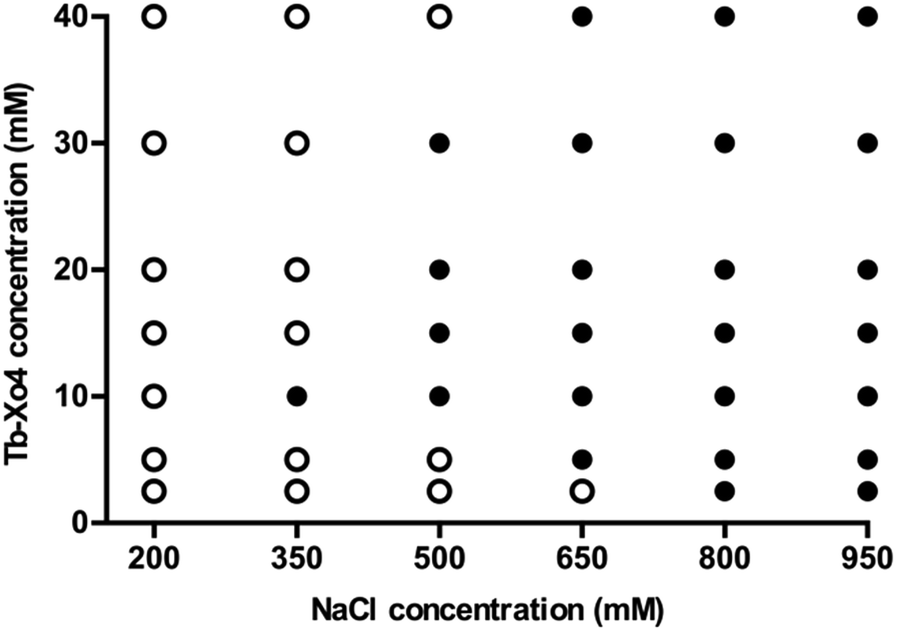 | ||
| Fig. 2 Crystallization diagrams of HEWL after two days with different Tb–Xo4 concentrations (0 to 100 mM). Black dots represent conditions where crystals are observed. HEWL crystallizes up to a Tb–Xo4 concentration of 100 mM. However, for clarity, results in the presence of 50 mM of the complex (similar to 40 mM) and 100 mM (similar to 2.5 mM) are not displayed. | ||
In order to get a deeper insight into this markedly different behaviour, the crystallization diagram of HEWL was determined (Fig. 3). The diagrams in the absence and in the presence of Tb–Xo4 are dramatically different, clearly indicating that the complex has a strong influence on the crystallisation process. The presence of Tb–Xo4 speeds up the appearance of crystals and promotes crystallization in a wider concentration range of the precipitant, here NaCl. Importantly, exploitable protein crystals can be obtained at very low precipitant concentrations as well as low protein concentrations. For example, HEWL tetragonal crystals could be grown at a concentration of NaCl as low as 200 mM, which has never been observed in either rational crystallogenesis studies37 or when using other nucleants.18 Furthermore, the protein concentration giving crystals after 2 days was reduced from 15 mg mL−1 under native conditions to 5 mg mL−1 in the presence of the terbium complex, an unprecedented result in the case of HEWL.18,37 This result clearly indicates that Tb–Xo4 presents a strong nucleating effect in the case of HEWL crystallisation.
 | ||
| Fig. 3 Crystallization diagrams determined for HEWL. White dots represent clear drops and black dots represent conditions giving crystals (hits). The presented diagrams correspond to the situation after 2 days (top) or 20 days (bottom) where no more evolution is observed. The left diagrams refer to the native proteins and the right ones to crystallization in the presence of Tb–Xo4 (10 mM). | ||
The general nucleating properties of Tb–Xo4 were then evaluated with several soluble proteins and under wide-ranging crystallization conditions. For this purpose, automated crystallization experiments were performed at the HTXlab (EMBL, Grenoble) with a set of 6 standard crystallization screens, corresponding to a total of 576 conditions. Six proteins of known structure, including three commercial ones (HEWL, thaumatin and proteinase K), as well as three in-house ones (protease1 from P. horikoshii, glyoxylate/hydroxypyruvate reductase (GRHPR) from P. furiosus and pb9 from the T5 phage tail) were tested (see ESI†). Two proteins of unknown structure were also tested: a halophilic tetrameric malate dehydrogenase (MDH) and a second protein of the T5 phage tail, pb6. Descriptions of the proteins and of the crystallization kits are provided in Tables S1 and S2† respectively. The nucleating properties of the complex were assessed using a comparative crystallization experiment. In the same crystallization plate, each protein was tested at a given concentration in the presence or in the absence of Tb–Xo4. The samples for automated crystallization were prepared by direct mixing of the complex in each protein solution to reach an optimal Tb–Xo4 concentration of 10 mM (Fig. 2). The use of this molecular additive is thus perfectly compatible with automated crystallization robots, in contrast with heterogeneous nucleants. In the following discussion, we have called a crystallization condition leading to the appearance of a crystal a “hit”. The results after a 34 days observation period are displayed in Fig. 4. We chose this period thanks to the recent analysis of the Oxford Structural Genomics Consortium showing that 98% of all crystals that led to deposited structures appeared within 30 days.38 However, it should also be noted that most of the hits are already present after 7 days (Fig. S9†). For all of the proteins tested, the presence of Tb–Xo4 in the crystallization drops provided additional hits compared to the native ones. This is also the case with the two proteins of unknown structure, MDH and pb6, with 2 and 6 unique hits out of 6 and 12 total hits, respectively. One should also note that, in the presence of the complex, some of the native hits are no longer preserved. Therefore, with the pragmatic goal of obtaining as many exploitable hits as possible, an optimal crystallization experiment would combine screening of the conditions of the protein both under native conditions and in the presence of Tb–Xo4.
 | ||
| Fig. 4 Details of the results of the automated crystallization screening (576 conditions) performed on the 8 proteins tested. The number of crystallization hits is depicted in grey for the native protein and with dots for protein supplemented with 10 mM of Tb–Xo4. As a result, the shared conditions are represented in grey with dots. Observation after 34 days. | ||
To further confirm the nucleating effect of Tb–Xo4, the crystallization diagrams of the unknown protein pb6 were determined similarly to those of HEWL (Fig. 3). The best amongst the 6 additional crystallisation conditions obtained after automated screening, i.e. in the presence of PEG 6000 as precipitant, was used (Fig. 5). As was already observed in the case of HEWL, crystals of pb6 appeared faster and in a broader range of precipitant concentrations in the presence of Tb–Xo4. Crystals were again obtained even at low protein concentrations. Consequently, such an additive opens the way to the crystallization of low concentration samples, which is a major benefit when considering precious protein samples. Beyond its nucleation properties, Tb–Xo4 also improves the quality of the crystals. This was illustrated for the crystals of both unknown proteins (pb6 and MDH) grown in the presence of the complex, which were clearly bigger and showed better morphologies than those of the native ones, making them ready-to-use for diffraction experiments (Fig. 5).
 | ||
| Fig. 5 (a) Crystallization diagrams determined for pb6 in the absence (top) and in the presence (bottom) of Tb–Xo4 (10 mM). Black dots represent conditions leading to crystals (hits). White dots represent clear drops. Crosses represent drops with protein precipitates. The presented diagrams correspond to the stable situation when no more evolution is observed. (b) Comparison of the crystals’ quality obtained under strictly similar crystallization conditions for the two proteins of unknown structures pb6 and MDH, in the absence and presence of 10 mM of Tb–Xo4 as indicated. | ||
The diffraction of all the crystals produced in the presence of Tb–Xo4 was tested, an unprecedented assessment for a new nucleating agent. Two different approaches were used to evaluate the diffraction quality of the crystals. Firstly, complete anomalous data sets were collected for all 8 proteins tested, and the diffraction resolution ranged from 2.85 to 1.80 Å using the best crystallization conditions (Tables 1 and S3†). In particular, for commercial proteins, the resolution is in the highest range of what is classically deposited in the Protein Data Bank. Secondly, we directly evaluated the diffraction quality of all of the crystals obtained on a high-throughput screening assay using a statistical approach. This in situ experiment was performed in the case of HEWL and the diffraction quality of the 113 hits obtained in the presence of Tb–Xo4 was compared to that of 20 native hits (Fig. 6). The diffraction quality was assessed using the number of Bragg peaks per frame as a function of the resolution, using the recently developed NanoPeakCell software.39 Compared to native crystals, the overall diffraction quality of the protein/Tb–Xo4 co-crystals, obtained under the same crystallization conditions, is slightly better in terms of the resolution and of number of detected Bragg peaks (Fig. 6). A similar analysis was performed on the crystals obtained only in the presence of the complex (i.e. unique hits, Fig. S11†). This led to a diffraction distribution equivalent to the one shown on Fig. 6. Consequently, the crystals grown only in the presence of the complex show identical diffraction quality compared to both native and Tb–Xo4 crystals obtained under shared crystallization conditions.
| Protein | Soaking in 100 mM Tb–Xo4 (soaking time) | Final Tb–Xo4 concentration for phasing (mM) | Phasing method | Resolution (Å) | Space group | FOM after SHARP | % solvent contenta | FOM after Solomon | Buccaneerb | U.A. content (molecules) |
|---|---|---|---|---|---|---|---|---|---|---|
| a As evaluated by AutoSHARP. b The values indicated correspond to the completeness of chains (%) as provided by Buccaneer. c Two-wavelength MAD experiment. | ||||||||||
| HEWL | No | 10 | SAD | 1.8 | P43212 | 0.42 | 41.3 | 0.88 | 97.2 | 1 |
| Thaumatin | No | 10 | SAD | 1.8 | P41212 | 0.40 | 45.6 | 0.92 | 75.4 | 1 |
| Proteinase K | No | 10 | SAD | 1.8 | P43212 | 0.31 | 45.8 | 0.86 | 0.0 | 1 |
| Proteinase K | Yes (120 s) | 100 | SAD | 1.8 | P43212 | 0.26 | 44.5 | 0.85 | 86.4 | 1 |
| Protease1 | No | 10 | SAD | 2.0 | P41212 | 0.21 | 61.9 | 0.95 | 62.7 | 3 |
| Protease1 | Yes (45 s) | 100 | SAD | 2.0 | P41212 | 0.35 | 60.1 | 0.96 | 87.8 | 3 |
| GRHPR | Yes (120 s) | 100 | SAD | 2.5 | I41 | 0.18 | 68.7 | 0.92 | 90.8 | 1 |
| pb9 | Yes (45 s) | 100 | SAD | 2.0 | P21 | 0.58 | 50.8 | 0.87 | 84.6 | 4 |
| pb6 | Yes (300 s) | 100 | MAD 2λc | 2.85 | P21 | 0.29 | 65.5 | 0.79 | 64.7 | 4 |
| MDH | No | 10 | SAD | 2.35 | R3 | 0.32 | 50.8 | 0.94 | 99.5 | 4 |
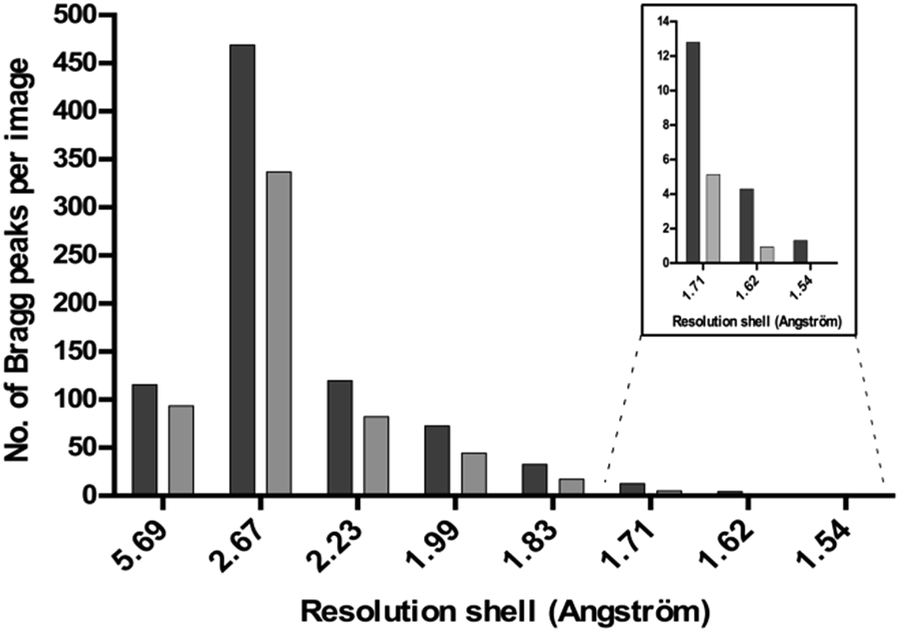 | ||
| Fig. 6 The diffraction quality (measured as the number of Bragg peaks per image) as a function of the resolution of native (light grey) and Tb–Xo4 (dark gray) HEWL crystals obtained under shared crystallization conditions. | ||
Overall, Tb–Xo4 exhibits a pronounced nucleating effect, increasing the number of crystallization conditions for all proteins tested, especially at lower protein and precipitant concentrations. Furthermore, it leads to the formation of larger crystals, ready-to-use for structure determination directly from the screening conditions.
Tb–Xo4 facilitates crystal detection
Under 280–370 nm excitation, Tb–Xo4 also presents the characteristic luminescence properties of terbium(III), i.e. a green emission arising from the 5D4 → 4FJ (J = 0–6) metal centered transitions (Fig. 7). This luminescence is very useful for the detection of the derived crystal within the crystallization drops, in particular allowing an easier discrimination between the protein and salt crystals. Besides, this luminescence detection can be an attractive feature for robotized crystallization drop imaging systems. Furthermore, with the appropriate adaptation of the instrumentation, the luminescence properties of Tb–Xo4 could be used for crystal centring in synchrotron beamlines.40,41 | ||
| Fig. 7 (a) Luminescence spectrum of Tb–Xo4 in aqueous solution (λex = 290 nm). MDH crystals obtained with 10 mM of the complex observed (b) in transmission mode and (c) under UV light irradiation (365 nm). | ||
Tb–Xo4 as a powerful phasing agent
Throughout the whole periodic table, lanthanide ions possess the largest anomalous contribution making them particularly suitable candidates for anomalous-based phasing of diffraction data.3,42 In addition, lanthanides may also be highly efficient for phasing with isomorphous techniques due to their high Z number. We evaluated the phasing properties of Tb–Xo4 by performing anomalous-based experiments for each aforementioned protein. The data were collected at the terbium LIII absorption edge by setting the incident beam energy to optimally exploit the large f′′ contribution. Either SAD or MAD phasing protocols were used to solve the phase problem (Table 1). All of the structures of the evaluated proteins were determined, leading to a percentage of the automatically built model that ranged from 46 to 100% (Table 1). All of the crystals were initially obtained under nucleating conditions (i.e. co-crystallized with 10 mM of Tb–Xo4). The structure determination can be achieved either directly from crystals obtained under nucleating conditions (HEWL, protease1, and MDH) or after a rapid soaking of the 10 mM co-crystallized crystals in a concentrated solution of the complex (i.e. at 100 mM of Tb–Xo4 for proteinase K, protease1, GRHPR, pb9, and pb6). A soaking time ranging from 30 s to 5 min was sufficient to obtain a strong anomalous signal (Table 1) without any degradation of the crystals. In particular, in the case of proteinase K and protease1, the soaking highly facilitated the phase determination, leading to 86% and 88% complete models (Table 1), respectively. These results unambiguously showed that Tb–Xo4 is an excellent phasing agent, and is fully compatible with conventional phasing methods. The combination of its nucleating and phasing properties brings major advantages compared to the tedious and time-consuming seleno-methionine labelling method or the method of heavy-atom incorporation.Application of the crystallophore for new protein structures determination
As a proof-of-concept, Tb–Xo4 was used to determine the structure of pb6 and MDH proteins as well as to resolve a difficult-to-phase protein, pb9.34 These structures range from 800 to 1800 ordered residues and have resolution limits from 2.85 to 2.0 Å (Table 1). A full description of the pb6 and MDH structures and associated biology will be published elsewhere. | ||
| Fig. 8 Experimentally-phased electron density maps for (a) pb6 at 2.85 Å resolution and (b) MDH at 2.35 Å resolution. The maps are contoured at 1.0 sigma. The Calpha trace corresponds to the automatically built model. (c) The packing of pb9 crystals (asymmetric unit content in black, symmetry-related molecules in grey). All of the Tb positions are shown with spheres. Details of the main Tb–Xo4 binding sites are shown in close-up views. pb9 PDB ID code 5MF2. | ||
Even though the structure of pb9 is already known, the resolving of the structure using the crystallophore fully illustrates the benefits of this agent. As for pb6 and MDH, we optimised one of the most promising additional hits (Table S3†). Based on these conditions, we confirmed that the optimal Tb–Xo4 concentration to obtain an optimal nucleant effect is between 5 and 10 mM (Fig. S10†). A rapid soaking of a crystal in a concentrated solution of the complex provided a successfully derived crystal for SAD phasing, as illustrated by the high figure of merit and by the 85% residues built by the automatic model building (Table 1). For the four pb9 molecules present in the asymmetric unit, 15 Tb–Xo4 binding sites were determined with refined occupancies ranging from 0.1 to 0.7 (Table S4†). A superimposition of our monomer (PDB entry 5MF2) on the PDB entry 4JMQ monomer34 led to a root mean square deviation of 0.55 Å, showing that the binding of Tb–Xo4 does not modify the overall structure of the protein.
As expected from the nucleating properties of Tb–Xo4, the analysis of the pb9 crystal packing showed the involvement of the complex at the interfaces between proteins (Fig. 8c). There is direct interaction between negatively charged residues, in particular glutamate, and the lanthanide atom at the main binding sites (Fig. 8c). Simultaneously, the crystallophore interacts through its hydrophobic macrocycle with the main-chain atoms of residues located on loops of symmetry-related proteins. In light of this binding mode, the nucleating properties of Tb–Xo4 could be explained by a reduction of surface entropy44,45 and/or by a slight modification of the surface topology,46 favouring contact between molecules. These results open fascinating perspectives for the study of the supramolecular interactions between the crystallophore and the proteins at the beginning of the crystallisation process.
Conclusion
In this paper, we describe the first example of a new class of lanthanide complexes, named crystallophore, that can act as powerful auxiliaries for protein crystallography. Tb–Xo4 presents multiple advantages as follows. (i) It is soluble and stable under all crystallisation conditions and consequently fully compatible with high throughput screening. (ii) It acts as a strong nucleating agent, providing new crystallisation conditions compared to the native ones and generally leads to the formation of bigger derived crystals, ready-to-use for X-ray diffraction. (iii) Under UV irradiation, the terbium(III) complex presents a green emission, enabling very easy detection of the derived crystal, and finally (iv) the crystallophore is a straightforward phasing agent thanks to the exceptional anomalous properties of the f-block elements facilitating crystal structure determination using SAD or MAD methods. Overall, the crystallophore is an all-in-one, easy-to-use tool that aims to overcome the two main bottlenecks of macromolecular crystallography: producing high quality single crystals and solving the phase problem. Future studies will be devoted to understanding the origin of such peculiar nucleating properties, with the aim of optimizing the design of this first compound and bringing new insight into the intricate issue of protein crystallization.Acknowledgements
Authors acknowledge the ANR agency for funding (Ln23 – ANR-13-BS07-0007-01) and for a grant to SE and SDP. LL and SDP are also grateful to the SATT Pulsalys (Lyon) for the maturation program (L-1382). We acknowledge the platforms of the Grenoble Instruct Center (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL), supported by the French Infrastructure for Integrated Structural Biology Initiative FRISBI (ANR-10-INSB-05-02) and by the Grenoble Alliance for Integrated Structural Cell Biology GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB).References
- S. Khurshid, E. Saridakis, L. Govada and N. E. Chayen, Nat. Protoc., 2014, 9, 1621–1633 CrossRef CAS PubMed.
- G. L. Taylor, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 325–338 CrossRef CAS PubMed.
- W. A. Hendrickson, Q. Rev. Biophys., 2014, 47, 49–93 CrossRef PubMed.
- S. Doublie, Methods Enzymol., 1997, 276, 523 CAS.
- Q. Liu, T. Dahmane, Z. Zhang, Z. Assur, J. Brasch, L. Shapiro, F. Mancia and W. A. Hendrickson, Science, 2012, 336, 1033–1037 CrossRef CAS PubMed.
- T. Weinert, V. Olieric, S. Waltersperger, E. Panepucci, L. Chen, H. Zhang, D. Zhou, J. Rose, A. Ebihara, S. Kuramitsu, D. Li, N. Howe, G. Schnapp, A. Pautsch, K. Bargsten, A. E. Prota, P. Surana, J. Kottur, D. T. Nair, F. Basilico, V. Cecatiello, S. Pasqualato, A. Boland, O. Weichenrieder, B.-C. Wang, M. O. Steinmetz, M. Caffrey and M. Wang, Nat. Methods, 2014, 12, 131–133 CrossRef PubMed.
- A. McPherson and P. Shlichta, Science, 1988, 239, 385–387 CAS.
- G. Falini, S. Fermani, G. Conforti and A. Ripamonti, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 1649–1652 CrossRef.
- A. D’Arcy, A. Mac Sweeney and A. Haber, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 1343–1346 CrossRef.
- D. G. Georgieva, M. E. Kuil, T. H. Oosterkamp, H. W. Zandbergen and J. P. Abrahams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 564–570 CrossRef CAS PubMed.
- E. Pechkova and C. Nicolini, J. Cell. Biochem., 2002, 85, 243–251 CrossRef CAS PubMed.
- L. Govada, H. S. Leese, E. Saridakis, S. Kassen, B. Chain, S. Khurshid, R. Menzel, S. Hu, M. S. P. Shaffer and N. E. Chayen, Sci. Rep., 2016, 6, 20053 CrossRef CAS PubMed.
- H. S. Leese, L. Govada, E. Saridakis, S. Khurshid, R. Menzel, T. Morishita, A. J. Clancy, E. R. White, N. E. Chayen and M. S. P. Shaffer, Chem. Sci., 2016, 7, 2916–2923 RSC.
- N. E. Chayen, E. Saridakis, R. El-Bahar and Y. Nemirovsky, J. Mol. Biol., 2001, 312, 591–595 CrossRef CAS PubMed.
- F. Kertis, S. Khurshid, O. Okman, J. W. Kysar, L. Govada, N. Chayen and J. Erlebacher, J. Mater. Chem., 2012, 22, 21928–21934 RSC.
- M. Sugahara, Y. Asada, Y. Morikawa, Y. Kageyama and N. Kunishima, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64, 686–695 CAS.
- N. E. Chayen, E. Saridakis and R. P. Sear, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 597–601 CrossRef CAS PubMed.
- E. Saridakis, S. Khurshid, L. Govada, Q. Phan, D. Hawkins, G. V. Crichlow, E. Lolis, S. M. Reddy and N. E. Chayen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11081–11086 CrossRef CAS PubMed.
- Y. Xing, Y. Hu, L. Jiang, Z. Gao, Z. Chen, Z. Chen and X. Ren, Cryst. Growth Des., 2015, 15, 4932–4937 CAS.
- S. Khurshid, L. Govada, H. F. El-Sharif, S. M. Reddy and N. E. Chayen, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 534–540 CAS.
- K. Djinovic-Carugo and O. Carugo, J. Inorg. Biochem., 2014, 143, 1–8 Search PubMed.
- R. Kahn, R. Fourme, R. Bosshard, M. Chiadmi, J. Risler, O. Dideberg and J. Wery, FEBS Lett., 1985, 179, 133–137 CrossRef CAS PubMed.
- W. I. Weis, R. Kahn, R. Fourme, K. Drickamer and W. A. Hendrickson, Science, 1991, 254, 1608–1615 CAS.
- N. R. Silvaggi, L. J. Martin, H. Schwalbe, B. Imperiali and K. N. Allen, J. Am. Chem. Soc., 2007, 129, 7114–7120 CrossRef CAS PubMed.
- K. N. Allen and B. Imperiali, Curr. Opin. Chem. Biol., 2010, 14, 247–254 CrossRef CAS PubMed.
- E. Girard, M. Stelter, P. L. Anelli, J. Vicat and R. Kahn, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 118–126 CrossRef CAS.
- G. Pompidor, A. D’Aléo, J. Vicat, L. Toupet, N. Giraud, R. Kahn and O. Maury, Angew. Chem., Int. Ed., 2008, 47, 3388–3391 CrossRef CAS PubMed.
- M. Stelter, R. Molina, S. Jeudy, R. Kahn, C. Abergel and J. A. Hermoso, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1506–1516 CAS.
- B. N. Chaudhuri, M. R. Sawaya, C. Y. Kim, G. S. Waldo, M. S. Park, T. C. Terwilliger and T. O. Yeates, Structure, 2003, 11, 753–764 CrossRef CAS PubMed.
- S. de Bono, L. Riechmann, E. Girard, R. L. Williams and G. Winter, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1396–1401 CrossRef CAS PubMed.
- P. Arnoux, T. Morosinotto, G. Saga, R. Bassi and D. Pignol, Plant Cell, 2009, 21, 2036–2044 CrossRef CAS PubMed.
- R. Molina, M. Stelter, R. Kahn and J. Hermoso, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 823–831 CAS.
- L. Larivière, C. Plaschka, M. Seizl, L. Wenzeck, F. Kurth and P. Cramer, Nature, 2012, 492, 448–451 CrossRef PubMed.
- A. Flayhan, F. M. D. Vellieux, R. Lurz, O. Maury, C. Contreras-Martel, E. Girard, P. Boulanger and C. Breyton, J. Virol., 2014, 88, 820–828 CrossRef PubMed.
- C. Eichmann, C. Tzitzilonis, T. Nakamura, W. Kwiatkowski, I. Maslennikov, S. Choe, S. A. Lipton and R. Riek, J. Mol. Biol., 2016, 428, 3737–3751 CrossRef CAS PubMed.
- G. Nocton, A. Nonat and C. Gateau, Helv. Chim. Acta, 2009, 92, 2257–2273 CrossRef CAS.
- P. Retailleau, A. Ducruix and M. Riès-Kautt, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 1576–1581 CrossRef.
- J. T. Ng, C. Dekker, P. Reardon and F. von Delft, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2016, 72, 224–235 CAS.
- N. Coquelle, A. S. Brewster, U. Kapp, A. Shilova, B. Weinhausen, M. Burghammer and J.-P. Colletier, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 1184–1196 CAS.
- X. Vernede, B. Lavault, J. Ohana, D. Nurizzo, J. Joly, L. Jacquamet, F. Felisaz, F. Cipriani and D. Bourgeois, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 253–261 CrossRef PubMed.
- L. M. G. Chavas, Y. Yamada, M. Hiraki, N. Igarashi, N. Matsugaki and S. Wakatsuki, J. Synchrotron Radiat., 2011, 18, 11–15 CrossRef CAS PubMed.
- E. Girard, M. Stelter, J. Vicat and R. Kahn, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 1914–1922 CrossRef.
- Y. Zivanovic, F. Confalonieri, L. Ponchon, R. Lurz, M. Chami, A. Flayhan, M. Renouard, A. Huet, P. Decottignies, A. R. Davidson, C. Breyton and P. Boulanger, J. Virol., 2014, 88, 1162–1174 CrossRef PubMed.
- Z. S. Derewenda, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 604–615 CrossRef CAS PubMed.
- L. Goldschmidt, D. Eisenberg and Z. S. Derewenda, Methods Mol. Biol., 2014, 1140, 201–209 CAS.
- Y. D. Devedjiev, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2015, 71, 157–162 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedures, Tb–Xo4 synthesis, Fig. S1–S11 and Tables S1–S4. See DOI: 10.1039/c7sc00758b |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2017 |