
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible on-surface wiring of resistive circuits†
Michael S.
Inkpen
 *ab,
Yann R.
Leroux
b,
Philippe
Hapiot
b,
Luis M.
Campos
c and
Latha
Venkataraman
*ac
*ab,
Yann R.
Leroux
b,
Philippe
Hapiot
b,
Luis M.
Campos
c and
Latha
Venkataraman
*ac
aDepartment of Applied Physics and Applied Mathematics, Columbia University, New York, NY 10027, USA. E-mail: msi2109@columbia.edu; lv2117@columbia.edu
bInstitut des Sciences Chimiques de Rennes (Equipe MaCSE), CNRS, Université de Rennes 1, Campus de Beaulieu, Bat 10C, Rennes Cedex, UMR 6226, France
cDepartment of Chemistry, Columbia University, New York, NY 10027, USA
First published on 7th April 2017
Abstract
Whilst most studies in single-molecule electronics involve components first synthesized ex situ, there is also great potential in exploiting chemical transformations to prepare devices in situ. Here, as a first step towards this goal, we conduct reversible reactions on monolayers to make and break covalent bonds between alkanes of different lengths, then measure the conductance of these molecules connected between electrodes using the scanning tunneling microscopy-based break junction (STM-BJ) method. In doing so, we develop the critical methodology required for assembling and disassembling surface-bound single-molecule circuits. We identify effective reaction conditions for surface-bound reagents, and importantly demonstrate that the electronic characteristics of wires created in situ agree with those created ex situ. Finally, we show that the STM-BJ technique is unique in its ability to definitively probe surface reaction yields both on a local (∼50 nm2) and pseudo-global (≥10 mm2) level. This investigation thus highlights a route to the construction and integration of more complex, and ultimately functional, surface-based single-molecule circuitry, as well as advancing a methodology that facilitates studies beyond the reach of traditional ex situ synthetic approaches.
Experimental techniques that enable the measurement of single-molecule properties have significantly advanced our understanding of how molecular structure relates to function, from charge transport1,2 and magnetism3–5 to mechanics.6,7 In part, this achievement results from the decoupling of properties and variables associated with multi-molecular characterizations,8,9 such as molecular packing effects,10,11 defect types and densities,12–14 or the absolute number of molecules being examined.15,16 Though the primary focus has so far been characterization, there is an increasing need to address the problem of how useful properties might ultimately be exploited at the single-molecule level, for example, using chemical principles.17–19 Investigations along these lines are of broad interest, seeking to explore fundamental questions regarding how molecular materials can be arranged with precision into varied and complex assemblies.
In single-molecule electronics, one such challenge concerns the organization of functional components into an addressable array of circuitry.20–23 Prospective methods would most likely utilize ‘bottom-up’, self-assembly processes (controllable at the molecular level),24–27 rather than exclusively rely upon classical, ‘top-down’, photo-lithographic techniques, although the path forward is not yet clear. One possible direction is the construction of surface-based molecular circuits using a multi-step ‘total synthesis’ approach. To date however, with the exception of irreversible, single-step reactions used to form new electrode–molecule contacts, almost all work in the area has involved the study of surface self-assembled components synthesized ex situ (prepared elsewhere before measurement).28–31 Though there have been numerous studies exploring in situ reactions on monolayers (where synthesis and measurement/use occur at the same position),32–40 few have subsequently probed modified substrates at a single-molecule level.41
In this work, we demonstrate how reversible synthetic methods can be used to manipulate surface-bound single-molecule circuits comprising different resistors ‘wired’ in series (Fig. 1). Here, components are represented by alkane chains of different lengths, and are assembled or dissembled in situ using esterification or hydrolysis to form or break covalent bonds. The success of surface-based reactions, first applied to a model system, were corroborated using a variety of methods including scanning tunneling microscope-based break junction (STM-BJ) measurements on ex situ synthesized materials, ex situ synthetic models, and X-ray photoelectron spectroscopy (XPS). Their subsequent application to a series of different surface-bound reagents facilitated conductance measurements of materials made entirely in situ. We find the STM-BJ method to be a highly sensitive tool for the characterization of surface-based reaction yields at both local (∼50 nm2) and pseudo-global (≥10 mm2) levels, where the reagent and product conductance peak intensities reflect their respective proportions in a mixed monolayer.
 | ||
| Fig. 1 Surface-based chemistry is used to effectively assemble or disassemble single-molecule circuits in situ (equivalent circuit diagrams depict the molecular components as ‘resistors’ only to illustrate the underlying concept). Modification of self-assembled monolayers is achieved using Steglich esterification and hydrolysis reactions (DIC = N,N′-diisopropylcarbodiimide, DMAP = 4-[dimethylamino]pyridine; n = 3 [4-OH/7-SMe], 5 [6-OH/9-SMe], 7 [8-OH/11-SMe]). | ||
Our approach also draws analogies to well-established techniques such as solid-phase peptide synthesis, bringing several advantages and opportunities (compared to solution-based synthetic methods) including facile purification of components and combinatorial “split-mix synthesis”.42,43 Importantly, surface bound products are in general not subject to any solubility requirements, removing the necessity to incorporate solubilizing groups or enabling measurements in solvents of different polarities. We note that solid-phase methodologies for the ex situ preparations of components were pioneered years ago,44 but have never previously been applied to conducting substrates for single-molecule measurements in situ. It is critical in this context to design systems following certain key principles: incorporating robust and conducting gold-binding groups, using appropriate reaction conditions, for example, not displacing the surface-bound species, and anticipating significant differences in the conductance of reagent and product for unambiguous determination of reaction success.
Results and discussion
The model surface-based molecular system developed here consists of discreet components of an electrical circuit that can be assembled or disassembled using in situ chemical reactions (structures shown in Fig. 1). 8-Mercaptooctanoic acid (8-OH) was chosen to function as surface-bound reactant (Fig. 1-left) given that similar α,ω-functionalized alkanes have been shown to form well-ordered, crystalline monolayers on gold.45,46 The strong Au–S bottom-contact could limit desorption, and the free, terminal carboxylic acid moiety could be amenable to a wide number of different modifications. We focused here on esterification reactions, given the large number of reported conditions available for testing, and also because any ester products could easily be hydrolyzed back to their parent carboxylic acids. Such reversible processes might enable reuse of functionalized substrates, as well as facilitating surface-based protecting group methodologies. Furthermore, by using simple alcohol reagents such as 2-(methylthio)ethanol we could form esters terminated with thiomethyl groups (e.g.11-SMe, Fig. 1-right). Both reagent and product would then be capable of forming junctions, as –SH, –COOH and –SMe are all known linkers for gold.47–50 The ester product 11-SMe, containing both the original alkane chain of 8-OH (resistor 1) plus an additional saturated bridge component (resistor 2), would function as the molecular part of a circuit comprising different resistors ‘wired’ in series (Fig. 1).Before studying the interconversion of 8-OH and 11-SMein situ we first characterized the ex situ synthesized components independently, enabling comparison of conductance measurements obtained in each case to establish that in situ and ex situ measurements yield the same results. Single-molecule conductance measurements were performed using the STM-BJ method as detailed in the Methods section,1,2 primarily to self-assembled monolayers, without altering the measurement technique except for having to periodically move the STM tip relative to the substrate to avoid depleting the contact area of molecules (Fig. 2a). Starting from commercially available 8-OH, 11-SMe was synthesized ex situ in two steps as the disulfide dimer (ESI, Scheme S1†). We note that disulfide formation offers a convenient ‘self-protecting group’ strategy for components comprising a single thiol group as the disulfide bond is thought to cleave in contact with gold to provide self-assembled monolayers similar in structure to those formed from analogous thiol-terminated precursors.25,51
 | ||
| Fig. 2 (a) Representation of a single-molecule junction formed between a gold STM tip and gold substrate functionalized with a self-assembled monolayer. Different surface-bound layers (inset) were studied following the modification of surface-bound materials in situ or by deposition of ex situ synthesized molecules onto clean gold substrates. (b) Overlaid one-dimensional logarithmically binned conductance histograms prepared from monolayer measurements of 8-OH (red, high G) and 11-SMe (blue, low G) synthesized ex situ (5000 traces, Vbias = 345 mV, 100 bins per decade). Conductance peaks for both acid (reagent, red) and ester (product, blue) are well separated, ultimately enabling the evaluation of surface-based reaction yields. Inset structures are the ex situ synthesized precursors used to form monolayers. | ||
Subsequent STM-BJ self-assembled monolayer measurements of both the acid reagent and ester product confirmed they form single-molecule junctions with well-separated conductance peaks (Fig. 2b, for two-dimensional histograms see ESI, Fig. S2†). Whilst junction formation probabilities inferred from conductance peak heights were generally lower for monolayers than for solution measurements (51% vs. 75% of traces contained plateau features, respectively, based on visual inspection of data for 8-OH), the most probable conductance values obtained were in good agreement (ESI, Fig. S1†). The observed differences in junction formation probabilities could be attributed to the limited number of molecules in the vicinity of an STM tip when probing a monolayer, compared to the continuous diffusion of new molecules from bulk to the tip when conducting measurements in solution. To confirm that the ester moiety of 11-SMe does not function as a linker group for gold, we also measured methyl 8-mercaptooctanoate (8-OMe) the methyl ester analogue of 8-OH. This compound did not form molecular junctions (ESI, Fig. S1†) in good agreement with previous work.49
In this work, we found that the choice of appropriate reaction conditions for surface-based syntheses required careful consideration. Alkanethiol monolayers have been reported to desorb upon exposure to strong acids/bases, oxidizing/reducing conditions, particular solvents or temperatures above ∼100 °C.52–54 Their stability is also a function of intermolecular interactions such as van der Waals forces55 or hydrogen bonding,56,57 not just the Au–S bonding motif. After testing different reaction conditions (e.g. Fischer esterification58), including unconventional esterification methods (using PPh3, I2, imidazole)59 that resulted in the apparent etching/dissolution of the gold surface, we found Steglich conditions (Fig. 1, top) to work well on monolayer films. These rapidly convert carboxylic acids to esters ex situ in the presence of an alcohol (HOR), a carbodiimide (dehydrating agent) and 4-(dimethylamino)pyridine (DMAP, catalyst).60 For the reverse reaction, we utilized lithium hydroxide (a mild base) in MeOH/H2O for hydrolysis of 11-SMe-functionalized substrates to 8-OH (Fig. 1, bottom).61
Fig. 3a shows 1D conductance histograms obtained from STM-BJ measurements of substrates comprising 8-OH monolayers before and after in situ reactions. Following exposure to Steglich esterification using N,N′-diisopropylcarbodiimide (DIC), the intensity of the conductance peak for 8-OH is significantly diminished in comparison to a new peak at lower conductance. The latter was readily attributed to junctions formed with 11-SMe through comparisons to conductance measurements of the ex situ synthesized material (Fig. 3b). Subsequent exposure of surface-bound 11-SMe to hydrolysis conditions restored the original 8-OH junction conductance peak. XPS measurements of the O 1s binding energies on representative substrates provided additional evidence for the on-surface esterification, corroborating the STM-BJ results (ESI, Fig. S6†). Though the relative intensities of conductance peaks before and after reactions demonstrate that high, if not complete, conversions for surface-based reactions are possible, we found that yields could vary from experiment to experiment (see ESI, Fig. S3† and discussion below).
 | ||
| Fig. 3 (a) Representative one-dimensional conductance histograms from monolayers measured before and after surface-based reactions. A pristine monolayer of 8-OH provides a peak at ∼10−4G0 (red). After in situ esterification this is significantly reduced in intensity with respect to a new peak at ∼10−6G0 (blue), corresponding to 11-SMe. Hydrolysis of this surface qualitatively restores the initial system (black). (b) A semi-log plot of conductance versus bridge length (where N is the number of atoms between surface linkers) for monolayer measurements of ex situ (crosses) and in situ (circles) synthesized species. Exponential fits to N-OH and N-SMe series (dashed lines) reveal comparable β-values. Note: the error in the conductance peak positions are smaller than the markers used to denote the values. | ||
Identical in situ Steglich conditions were next applied to synthesize 7-SMe and 9-SMe from monolayers of 4-OH and 6-OH (shorter alkane chains/smaller resistors, see Fig. 1 for molecular structures). STM-BJ measurements were then carried out on the reacted substrates. In all cases, we observed a new conductance peak at lower conductance that we attributed to the surface-bound product (for 1D and 2D histograms from monolayers measured before and after surface reactions see the ESI, Fig. S4†). We plot, in Fig. 3b, the conductance values of reactants (N-OH) and products (N-SMe). We find that for both series, the conductance decays exponentially with increasing number of carbon and oxygen atoms (N) in the backbone as G ∼ e−βN. The decay constants β are very similar for both series (0.93 per atom for the N-OH and 0.99 per atom for N-SMe), however, the thiomethyl-terminated ester products have a lower conductance for the same bridge length. This could result partly from the N-SMe series having an O within the backbone and partly due to a difference between the contact resistance of carboxylic acid group and a methylsulfide group.49 As demonstrated here with in situ synthesized 7-SMe and 9-SMe monolayers, the application of appropriate reactions to new systems (in this case Steglich esterification of carboxylic acids attached to different molecular backbones) enables conductance measurements of novel materials never previously isolated ex situ.
By varying the conditions used to convert monolayers of 8-OH to 11-SMe, we obtained additional insights into the surface-based esterification process. Somewhat surprisingly, if N,N′-dicyclohexylcarbodiimide (DCC) was used instead of DIC only poor conversions were observed (Fig. 4a-black line and ESI, Fig. S3b†). This is in stark contrast to the good or excellent isolated yields obtained from ex situ reactions using a model system, over much shorter timescales (ESI, Scheme S2†). We questioned whether the increased bulk of DCC (with cyclohexyl moieties) compared to DIC (isopropyl moieties) was limiting the surface-based reaction, where the close proximity of individual molecules in the monolayer could present a steric barrier. Similar effects have previously been noted for other monolayer-based reactions.25,62–65
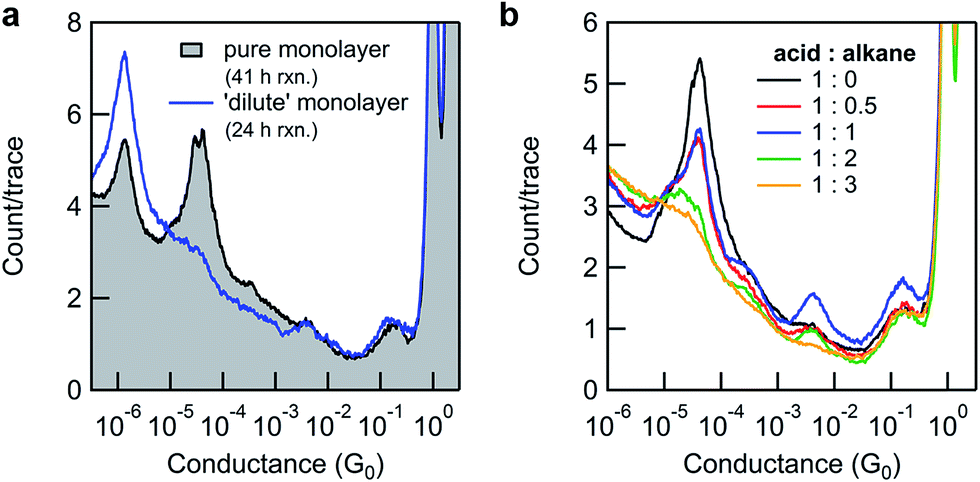 | ||
Fig. 4 (a) Overlaid one-dimensional conductance histograms (5000 traces) showing the effect of monolayer dilution on surface-based Steglich esterification yields using DCC. Black: incomplete conversion of a pure 8-OH monolayer to 11-SMe after 41 h reaction time. Blue: high conversion of 8-OH monolayer co-adsorbed with hexanethiol as a diluent (8-OH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexanethiol molar ratio of 1:0.5) after 24 h reaction time. (b) Overlaid one-dimensional conductance histograms showing the effect of monolayer dilution on the intensity of the molecular conductance peak. Every histogram is the average of three independent experiments (with 5000 traces measured for each substrate). :hexanethiol molar ratio of 1:0.5) after 24 h reaction time. (b) Overlaid one-dimensional conductance histograms showing the effect of monolayer dilution on the intensity of the molecular conductance peak. Every histogram is the average of three independent experiments (with 5000 traces measured for each substrate). | ||
To explore this hypothesis in the current context, we formed a series of 8-OH monolayers that were progressively diluted with (shorter) hexanethiol matrices through co-deposition of both components from solution. Whilst this should increase the average distance between reactive species on the surface (reducing intermolecular steric effects), it is important to note that the composition of monolayers formed in this way do not necessarily reflect the composition of the solution,66,67 and that surface-bound species may form discreet domains rather than mix perfectly.68 In any case, STM-BJ measurements clearly illustrated the expected trend. As the proportion of hexanethiol increases (from acid:alkane molar ratios of 1:0 to 1:3), the height of the 8-OH conductance peak decreases (Fig. 4b). Subsequent application of in situ reactions to selected semi-dilute monolayers showed it was now possible to achieve high conversions, even when using the sterically bulky DCC (Fig. 4a-blue line). This result supports the idea that steric effects imposed by adjacent adsorbates in carboxylic acid-terminated alkanethiol monolayers may strongly affect the success of surface-based reactions compared to otherwise identical transformations in solution.
The STM-BJ data collected from incomplete surface reactions was subjected to further analysis in an attempt to determine the spatial distribution of reactant and product on the surface. This was of interest to better understand the progression of reactions in two dimensions, where, for example, perfect mixing of species might suggest that the reaction rate is independent of molecular environment. If local surface concentrations of product and reagent varied significantly compared to the bulk average, it might instead indicate that the reaction rate is dependent on molecular environment or that the reaction proceeds via propagation from a nucleation point.65 The STM-BJ methodology used here in the measurement of monolayers facilitates just such an analysis, as by moving the tip relative to the substrate every ∼250 traces, we sample many different surface locations. In the absence of significant tip–surface drift, only molecules within a small area of around 50 nm2 will be measured at a given location.69 For mixed monolayers resulting from incomplete surface reactions, the ratio of product and reactant conductance peak intensities in a histogram prepared from traces collected at a single tip location then reflects the local surface reaction yield at that location. This is different from the pseudo-global surface reaction yields presented in histograms above (comprised of ≥5000 traces), as these are obtained by combining many tip–substrate positions (sometimes separated by relatively large distances, e.g. >1 mm). This reflects the remarkable and unique ability of the STM-BJ method to easily discriminate a surface monolayer modification (even as minor as a conversion from a –COOMe to a –COOH termination) at the nanometer scale. We note that whilst the presence or absence of a conductance peak readily signifies 0 or 100% yield (±∼10%), fully quantitative determinations of yield would require calibration of peak area and junction formation probability for each molecule studied (these are specific to molecular length, structure and linker groups).
Application of such an analysis to the incomplete reaction data shown in Fig. 4a (black line) reveals significant differences in the ratio of product and reactant peak intensities at different tip locations as shown in Fig. 5. Given the long reaction time (41 h) this substrate was exposed to, and the lack of reactivity of analogous surface layers on shorter timescales (24 h, see ESI, Fig. S3b†), the apparent inhomogeneity of bound surface species potentially indicates that breakdown of monolayer ordering (e.g. via desorption events) is a prerequisite for in situ esterification under Steglich conditions using DCC. This view is also consistent with the fact that we only observe high conversions under these reaction conditions after dilution of the reactant on the surface through co-adsorption with hexanethiol.
 | ||
| Fig. 5 (a) A plot of the conductance peak maximum (where Cmax = maximum count/trace at the most probable conductance) of product (blue triangles) and reactant peaks (red squares) against trace number for consecutive groups of 250 traces (intensities were obtained from linear background-subtracted Gaussian fits to 1D histograms, where 250 traces ∼ 1 tip position), for the data set presented in Fig. 4a (black line, 5000 traces, 20 tip positions). The same analysis is shown for a pure monolayer of 8-OH (black circles, from the data set shown in Fig. 2b), for comparison. (b) Two 250-trace histograms from the reacted monolayer data set used in (a), overlaid to show the extremes of variation/250 traces. | ||
This concept was further extended to exploit established surface-based reactions that have not yet been used to study materials at the single-molecule level. Under conditions introduced by van der Drift70 and developed by Frisbie,34 we reacted a self-assembled monolayer of 4-aminothiophenol with 4-(methylthio)benzaldehyde to form the corresponding imine (see ESI, Fig. S5a† and the Experimental section for further details). Whereas STM-BJ measurements on 4-aminothiophenol monolayers typically showed featureless conductance histograms, reacted monolayers yielded a peak at ∼10−3G0 that we attribute to the in situ synthesized product (ESI, Fig. S5†).
Conclusion
We have demonstrated how single and multi-step (reversible) surface-based reactions may be utilized within single-molecule electronics to assemble and disassemble simple resistive circuits in situ and characterized these circuits through STM-BJ measurements. In principle, many other components can be ‘wired’ up using analogous methods, through application of any number of different reactions, even exploiting recent advances in ‘on-surface’ synthesis,71 to potentially create circuits with broader functionality. By modifying the conditions used to effect surface-based reactions, we demonstrate further the impact of intermolecular steric effects on in situ synthesized product yields, a factor not typically important to consider when conducting reactions in solution. As robust in situ methodologies become available, the ability to construct and characterize new components without isolating them first ex situ also offers many possibilities for rapid component screening in the search for novel properties. The unique sensitivity of the STM-BJ method to definitively probe surface reaction yields at the nanometer scale is particularly useful in this context, being complementary to existing analysis techniques such as surface electrochemistry, XPS and infrared reflection-absorbance spectroscopy, and offering additional insights into the nature of chemical reactions in two-dimensions. Though this study has focused attention on single-molecule electronics, we suggest the application of chemical reactions in situ is equally relevant to other fields (from catalysis72 to molecular machinery73,74) where the ability to pattern surface-based systems at the single-molecule level is a desired goal.Methods
STM-BJ
Here a bias voltage (Vbias) is applied between a freshly cut gold STM tip and a gold substrate, and the junction current (I) is recorded as a function of tip–substrate displacement. By driving the tip in and out of the substrate, an initial large area gold contact with a conductance G = I/Vbias much larger than the quantum of conductance, G0 = 2e2h, is first created. The tip is then retracted and the contact is thinned to a single-atom contact (G ∼ G0) before breaking completely. This process forms a sub-nm gap between two atomically sharp electrodes where, in the absence of any bridging molecules, increasing tip–substrate displacement results in a rapid drop of the measured conductance to the electrical noise floor. In the presence of bridging molecules, the conductance first drops to an interim value (characteristic of charge transport through the molecule), before ultimately dropping to noise as increasing tip–substrate displacement ruptures the molecular junction. After repeatedly forming thousands of junctions in this way, the conductance data is analyzed using statistical methods (see the ESI,† Experimental section for further details).Monolayer preparation
For experiments involving self-assembled monolayers template stripped Au (TSAu) substrates were used. Following methodologies introduced by others,75,76 a >100 nm Au layer was evaporated onto 4′′ Prime Grade 〈100〉 1–10 Ω cm−1 500–550 μm single side polished silicon wafers (Nova Electronic Materials, LLC, Texas, USA). Glass tiles (∼1 cm2, cleaned using oxygen plasma to improve adhesion) were then bonded to the Au layer (forming a Si–Au–glass assembly) using either NOA 61 optical adhesive (Norland Products Inc., New Jersey, USA), or EP41S-5 two-part epoxy (Masterbond, New Jersey, USA). The former was cured through 15 min exposure to a Dymax 400 Watt UV Curing System then aged for ≥12 h at 50 °C, the latter cured for ≥24 h at room temperature. Immediately prior to use, individual Au–adhesive–glass ensembles were cleaved from the silicon wafer using a razor blade, to reveal a clean, flat Au surface. Glassware in contact with solutions used for monolayer preparation was first cleaned with oxygen plasma for at least 10 min. Monolayers of carboxylic acid functionalized thioalkanes were formed through immersion of freshly cleaved TSAu substrates into 0.02 mM solutions in ethanol:acetic acid (9:1 v/v) for 20–26 h.45 Monolayers of 4-aminothiophenol were prepared by immersion of freshly cleaved TSAu substrates in a 10 mM ethanolic solution for 2 h.34,70 Monolayers of 11-SMe were formed by immersion of freshly cleaved TSAu substrates in a 0.5 mM ethanolic solution for 20–26 h.25 After emersion, substrates were rinsed copiously with ethanol (>4 × 2 mL), and dried in a stream of N2. STM-BJ measurements on monolayers were performed in air, with the tip repeatedly moved laterally relative to the substrate by a distance of ≥200 nm after measuring ∼250 traces at a given tip–substrate position. This ensured there was always enough molecules to form junctions, as well as helping to average out local variations by sampling a greater surface area. Without moving, we observed a steady decrease in the molecular conductance peak intensity over 1000–2000 measurements, presumably due to local dilution of the monolayer by clean gold pulled out of the surface with repeated tip–substrate contact.69
Typical in situ synthetic protocols
:8 v/v) was prepared, then added to the vial and stirred at room temperature for 1 day. The substrates were subsequently removed and washed sequentially with H2O, 1 M HCl (60 s immersion), H2O and EtOH, before drying in a stream of N2.
Conflict of interest
The authors declare no competing financial interest.Abbreviations
| STM-BJ | Scanning tunneling microscope-based break junction |
| XPS | X-ray photoelectron spectroscopy |
| DIC | N,N′-Diisopropylcarbodiimide |
| DMAP | 4-(Dimethylamino)pyridine |
| DCC | N,N′-Dicyclohexylcarbodiimide |
Acknowledgements
This research was supported primarily by a Marie Skłodowska Curie Global Fellowship (M.S.I., MOLCLICK: 657247) within the Horizon 2020 Programme. This work was supported in part by the National Science Foundation grant DMR-1507440.References
- B. Xu and N. J. Tao, Science, 2003, 301, 1221–1223 CrossRef CAS PubMed.
- L. Venkataraman, J. E. Klare, C. Nuckolls, M. S. Hybertsen and M. L. Steigerwald, Nature, 2006, 442, 904–907 CrossRef CAS PubMed.
- W. Liang, M. P. Shores, M. Bockrath, J. R. Long and H. Park, Nature, 2002, 417, 725–729 CrossRef CAS PubMed.
- J. Park, A. N. Pasupathy, J. I. Goldsmith, C. Chang, Y. Yaish, J. R. Petta, M. Rinkoski, J. P. Sethna, H. D. Abruña, P. L. McEuen and D. C. Ralph, Nature, 2002, 417, 722–725 CrossRef CAS PubMed.
- S. Thiele, F. Balestro, R. Ballou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Science, 2014, 344, 1135–1138 CrossRef CAS PubMed.
- M. Rief, M. Gautel, F. Oesterhelt, J. M. Fernandez and H. E. Gaub, Science, 1997, 276, 1109–1112 CrossRef CAS PubMed.
- S. V. Aradhya, M. Frei, M. S. Hybertsen and L. Venkataraman, Nat. Mater., 2012, 11, 872–876 CrossRef CAS PubMed.
- R. L. McCreery and A. J. Bergren, Adv. Mater., 2009, 21, 1–20 CrossRef PubMed.
- L. Cademartiri, M. M. Thuo, C. A. Nijhuis, W. F. Reus, S. Tricard, J. R. Barber, R. N. S. Sodhi, P. Brodersen, C. Kim, R. C. Chiechi and G. M. Whitesides, J. Phys. Chem. C, 2012, 116, 10848–10860 CAS.
- Z. J. Donhauser, B. A. Mantooth, K. F. Kelly, L. A. Bumm, J. D. Monnell, J. J. Stapleton, D. W. Price Jr, A. M. Rawlett, D. L. Allara, J. M. Tour and P. S. Weiss, Science, 2001, 292, 2303–2307 CrossRef CAS PubMed.
- F.-R. F. Fan, J. Yang, L. Cai, D. W. Price, S. M. Dirk, D. V. Kosynkin, Y. Yao, A. M. Rawlett, J. M. Tour and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 5550–5560 CrossRef CAS PubMed.
- B. C. Haynie, A. V. Walker, T. B. Tighe, D. L. Allara and N. Winograd, Appl. Surf. Sci., 2003, 203–204, 433–436 CrossRef CAS.
- H. B. Akkerman, P. W. M. Blom, D. M. de Leeuw and B. de Boer, Nature, 2006, 441, 69–72 CrossRef CAS PubMed.
- P. N. Nirmalraj, H. Schmid, B. Gotsmann and H. Riel, Langmuir, 2013, 29, 1340–1345 CrossRef CAS PubMed.
- V. B. Engelkes, J. M. Beebe and C. D. Frisbie, J. Am. Chem. Soc., 2004, 126, 14287–14296 CrossRef CAS PubMed.
- C. Chu, J.-S. Na and G. N. Parsons, J. Am. Chem. Soc., 2007, 129, 2287–2296 CrossRef CAS PubMed.
- X. Guo, J. P. Small, J. E. Klare, Y. Wang, M. S. Purewal, I. W. Tam, B. H. Hong, R. Caldwell, L. Huang, S. O'Brien, J. Yan, R. Breslow, S. J. Wind, J. Hone, P. Kim and C. Nuckolls, Science, 2006, 311, 356–359 CrossRef CAS PubMed.
- Y. Okawa, S. K. Mandal, C. Hu, Y. Tateyama, S. Goedecker, S. Tsukamoto, T. Hasegawa, J. K. Gimzewski and M. Aono, J. Am. Chem. Soc., 2011, 133, 8227–8233 CrossRef CAS PubMed.
- J. Zhu, J. McMorrow, R. Crespo-Otero, G. Ao, M. Zheng, W. P. Gillin and M. Palma, J. Am. Chem. Soc., 2016, 138, 2905–2908 CrossRef CAS PubMed.
- J. R. Heath, P. J. Kuekes, G. S. Snider and R. S. Williams, Science, 1998, 280, 1716–1721 CrossRef CAS.
- J. C. Ellenbogen and J. C. Love, Proc. IEEE, 2000, 88, 386–426 CrossRef CAS.
- J. M. Tour, W. L. V. Zandt, C. P. Husband, S. M. Husband, L. S. Wilson, P. D. Franzon and D. P. Nackashi, IEEE Trans. Nanotechnol., 2002, 1, 100–109 CrossRef.
- M. Forshaw, R. Stadler, D. Crawley and K. Nikolić, Nanotechnology, 2004, 15, S220 CrossRef CAS.
- J. A. Theobald, N. S. Oxtoby, M. A. Phillips, N. R. Champness and P. H. Beton, Nature, 2003, 424, 1029–1031 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- M. E. Cañas-Ventura, W. Xiao, D. Wasserfallen, K. Müllen, H. Brune, J. V. Barth and R. Fasel, Angew. Chem., Int. Ed., 2007, 46, 1814–1818 CrossRef PubMed.
- R. Madueno, M. T. Raisanen, C. Silien and M. Buck, Nature, 2008, 454, 618–621 CrossRef CAS PubMed.
- Z. L. Cheng, R. Skouta, H. Vazquez, J. R. Widawsky, S. Schneebeli, W. Chen, M. S. Hybertsen, R. Breslow and L. Venkataraman, Nat. Nanotechnol., 2011, 6, 353–357 CrossRef CAS PubMed.
- W. Hong, H. Li, S.-X. Liu, Y. Fu, J. Li, V. Kaliginedi, S. Decurtins and T. Wandlowski, J. Am. Chem. Soc., 2012, 134, 19425–19431 CrossRef CAS PubMed.
- A. Batra, G. Kladnik, N. Gorjizadeh, J. Meisner, M. Steigerwald, C. Nuckolls, S. Y. Quek, D. Cvetko, A. Morgante and L. Venkataraman, J. Am. Chem. Soc., 2014, 136, 12556–12559 CrossRef CAS PubMed.
- C. Huang, S. Chen, K. Baruël Ørnsø, D. Reber, M. Baghernejad, Y. Fu, T. Wandlowski, S. Decurtins, W. Hong, K. S. Thygesen and S.-X. Liu, Angew. Chem., Int. Ed., 2015, 54, 14304–14307 CrossRef CAS PubMed.
- T. P. Sullivan and W. T. S. Huck, Eur. J. Org. Chem., 2003, 2003, 17–29 CrossRef.
- J. Li, P. S. Thiara and M. Mrksich, Langmuir, 2007, 23, 11826–11835 CrossRef CAS PubMed.
- S. Ho Choi, B. Kim and C. D. Frisbie, Science, 2008, 320, 1482–1486 CrossRef PubMed.
- Y. R. Leroux, H. Fei, J.-M. Noël, C. Roux and P. Hapiot, J. Am. Chem. Soc., 2010, 132, 14039–14041 CrossRef CAS PubMed.
- L. Lee, P. A. Brooksby, Y. R. Leroux, P. Hapiot and A. J. Downard, Langmuir, 2013, 29, 3133–3139 CrossRef CAS PubMed.
- L. Santos, A. Mattiuzzi, I. Jabin, N. Vandencasteele, F. Reniers, O. Reinaud, P. Hapiot, S. Lhenry, Y. Leroux and C. Lagrost, J. Phys. Chem. C, 2014, 118, 15919–15928 CAS.
- L. Lee, H. Ma, P. A. Brooksby, S. A. Brown, Y. R. Leroux, P. Hapiot and A. J. Downard, Langmuir, 2014, 30, 7104–7111 CrossRef CAS PubMed.
- L. Lee, Y. R. Leroux, P. Hapiot and A. J. Downard, Langmuir, 2015, 31, 5071–5077 CrossRef CAS PubMed.
- S. Y. Sayed, A. Bayat, M. Kondratenko, Y. Leroux, P. Hapiot and R. L. McCreery, J. Am. Chem. Soc., 2013, 135, 12972–12975 CrossRef CAS PubMed.
- I. Díez-Pérez, J. Hihath, Y. Lee, L. Yu, L. Adamska, M. A. Kozhushner, I. I. Oleynik and N. J. Tao, Nat. Chem., 2009, 1, 635–641 CrossRef PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- Á. Furka, F. Sebestyén, M. Asgedom and G. Dibó, Int. J. Pept. Protein Res., 1991, 37, 487–493 CrossRef PubMed.
- L. Jones, J. S. Schumm and J. M. Tour, J. Org. Chem., 1997, 62, 1388–1410 CrossRef CAS.
- R. Arnold, W. Azzam, A. Terfort and C. Wöll, Langmuir, 2002, 18, 3980–3992 CrossRef CAS.
- A. W. Snow, G. G. Jernigan and M. G. Ancona, Analyst, 2011, 136, 4935–4949 RSC.
- F. Chen, X. Li, J. Hihath, Z. Huang and N. J. Tao, J. Am. Chem. Soc., 2006, 128, 15874–15881 CrossRef CAS PubMed.
- Y. S. Park, A. C. Whalley, M. Kamenetska, M. L. Steigerwald, M. S. Hybertsen, C. Nuckolls and L. Venkataraman, J. Am. Chem. Soc., 2007, 129, 15768–15769 CrossRef CAS PubMed.
- S. Ahn, S. V. Aradhya, R. S. Klausen, B. Capozzi, X. Roy, M. L. Steigerwald, C. Nuckolls and L. Venkataraman, Phys. Chem. Chem. Phys., 2012, 14, 13841–13845 RSC.
- M. S. Inkpen, M. Lemmer, N. Fitzpatrick, D. C. Milan, R. J. Nichols, N. J. Long and T. Albrecht, J. Am. Chem. Soc., 2015, 137, 9971–9981 CrossRef CAS PubMed.
- C. D. Bain, H. A. Biebuyck and G. M. Whitesides, Langmuir, 1989, 5, 723–727 CrossRef CAS.
- D. Khobragade, E. S. Stensrud, M. Mucha, J. R. Smith, R. Pohl, I. Stibor and J. Michl, Langmuir, 2010, 26, 8483–8490 CrossRef CAS PubMed.
- C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H.-B. Kraatz, N. J. Mosey, T. Seki, E. C. Keske, J. D. Leake, A. Rousina-Webb and G. Wu, Nat. Chem., 2014, 6, 409–414 CrossRef CAS PubMed.
- L. Srisombat, A. C. Jamison and T. R. Lee, Colloids Surf., A, 2011, 390, 1–19 CrossRef CAS.
- M. D. Porter, T. B. Bright, D. L. Allara and C. E. D. Chidsey, J. Am. Chem. Soc., 1987, 109, 3559–3568 CrossRef CAS.
- S.-W. Tam-Chang, H. A. Biebuyck, G. M. Whitesides, N. Jeon and R. G. Nuzzo, Langmuir, 1995, 11, 4371–4382 CrossRef CAS.
- J. C. Thomas, D. P. Goronzy, K. Dragomiretskiy, D. Zosso, J. Gilles, S. J. Osher, A. L. Bertozzi and P. S. Weiss, ACS Nano, 2016, 10, 5446–5451 CrossRef CAS PubMed.
- H. M. Saavedra, C. M. Thompson, J. N. Hohman, V. H. Crespi and P. S. Weiss, J. Am. Chem. Soc., 2009, 131, 2252–2259 CrossRef CAS PubMed.
- S. P. Morcillo, L. Álvarez de Cienfuegos, A. J. Mota, J. Justicia and R. Robles, J. Org. Chem., 2011, 76, 2277–2281 CrossRef CAS PubMed.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed., 1978, 17, 522–524 CrossRef.
- B. Dayal, G. Salen, B. Toome, G. S. Tint, S. Shefer and J. Padia, Steroids, 1990, 55, 233–237 CrossRef CAS PubMed.
- D. A. Hutt and G. J. Leggett, Langmuir, 1997, 13, 2740–2748 CrossRef CAS.
- B. Dordi, H. Schönherr and G. J. Vancso, Langmuir, 2003, 19, 5780–5786 CrossRef CAS.
- M. R. Anderson and R. Baltzersen, J. Colloid Interface Sci., 2003, 263, 516–521 CrossRef CAS PubMed.
- K. Rajalingam, A. Bashir, M. Badin, F. Schröder, N. Hardman, T. Strunskus, R. A. Fischer and C. Wöll, ChemPhysChem, 2007, 8, 657–660 CrossRef CAS PubMed.
- C. D. Bain and G. M. Whitesides, J. Am. Chem. Soc., 1988, 110, 6560–6561 CrossRef CAS.
- C. D. Bain and G. M. Whitesides, Science, 1988, 240, 62–63 CAS.
- S. J. Stranick, A. N. Parikh, Y. T. Tao, D. L. Allara and P. S. Weiss, J. Phys. Chem., 1994, 98, 7636–7646 CrossRef CAS.
- J. He, O. Sankey, M. Lee, N. Tao, X. Li and S. Lindsay, Faraday Discuss., 2006, 131, 145–154 RSC.
- J. J. W. M. Rosink, M. A. Blauw, L. J. Geerligs, E. van der Drift, B. A. C. Rousseeuw, S. Radelaar, W. G. Sloof and E. J. M. Fakkeldij, Langmuir, 2000, 16, 4547–4553 CrossRef CAS.
- R. Lindner and A. Kühnle, ChemPhysChem, 2015, 16, 1582–1592 CrossRef CAS PubMed.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed.
- U. G. E. Perera, F. Ample, H. Kersell, Y. Zhang, G. Vives, J. Echeverria, M. Grisolia, G. Rapenne, C. Joachim and S. W. Hla, Nat. Nanotechnol., 2013, 8, 46–51 CrossRef CAS PubMed.
- I. Aprahamian, Nat. Chem., 2016, 8, 97–99 CAS.
- M. Hegner, P. Wagner and G. Semenza, Surf. Sci., 1993, 291, 39–46 CrossRef CAS.
- E. A. Weiss, G. K. Kaufman, J. K. Kriebel, Z. Li, R. Schalek and G. M. Whitesides, Langmuir, 2007, 23, 9686–9694 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and STM-BJ data, synthetic procedures, X-ray photoelectron and NMR spectra. Raw data used in figure plots is available at http://zenodo.org (10.5281/zenodo.496177). See DOI: 10.1039/c7sc00599g |
| This journal is © The Royal Society of Chemistry 2017 |