
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the separation and coupling of corannulene trianion-radicals through sizable alkali metal belts†
Sarah N.
Spisak
a,
Andrey Yu.
Rogachev
*b,
Alexander V.
Zabula
ac,
Alexander S.
Filatov
a,
Rodolphe
Clérac
de and
Marina A.
Petrukhina
 *a
*a
aDepartment of Chemistry, University at Albany, State University of New York, Albany, NY 12222, USA. E-mail: mpetrukhina@albany.edu
bDepartment of Chemistry, Illinois Institute of Technology, Chicago, IL 60616, USA. E-mail: arogache@iit.edu
cDepartment of Chemistry, University of Pennsylvania, Philadelphia, PA 19104, USA
dCNRS, CRPP, UPR 8641, F-33600, Pessac, France
eUniv. Bordeaux, CRPP, UPR 8641, F-33600, Pessac, France
First published on 23rd February 2017
Abstract
The first heterobimetallic sandwich-type aggregate formed by bowl-shaped corannulene trianion-radicals, C20H10˙3−, has been synthesized using mixed-metal reduction of C20H10. The product was crystallographically characterized to reveal the self-assembly of [Cs+//(C20H103−)/4K+/(C20H103−)//Cs+], in which two triply-charged corannulene decks encapsulate a rectangle of four potassium ions (the K⋯K separations are 4.212(4) and 5.185(4) Å), with the exterior concave bowl cavities being selectively filled by one cesium ion each. In order to provide insights into the geometrical features and electronic structure of this novel mixed-metal organometallic self-assembly, an in-depth theoretical investigation has been carried out. Specifically, the influence of internal metal binding on the geometry and magnetic coupling of C20H10˙3− radicals is investigated for Group 1 metals. This study reveals that replacement of the sandwiched potassium ions with larger (Cs) and smaller (Li) ions allows variation of the size of the encapsulated metal belts, and thus enables tuning of the coupling of C20H10˙3− radicals.
Introduction
Multi-electron reduction of planar and non-planar polycyclic aromatic hydrocarbons (PAHs) has been the focus of broad attention over many years from both fundamental and applied viewpoints.1–4 The discoveries of curved and bent π-conjugated molecules, such as fullerenes, nanotubes and their fragments,5 further reinvigorated this field with a focus on geometry perturbation, aromaticity, electronic properties, and magnetism of the resulting charged carbon-rich species.6–9 Special interest in non-planar radicals with extended π-surfaces10 has been driven by their unique magnetic properties11 and interesting coupling pathways,12 which open up their applications in organic microelectronics and energy storage.13 Furthermore, carbon-rich PAHs with pre-designed structures and substitution patterns are used as unique redox-active scaffolds for the generation of biradical14 and tetraradicaloid species15 and for the preparation of novel metal–organic frameworks.16Our interests focus on bowl-shaped PAHs (often called buckybowls or π-bowls)17 which are known to readily uptake multiple electrons in stepwise reduction processes. For example, corannulene (C20H10, Scheme 1), which maps onto 1/3 of the C60-fullerene surface, can acquire up to four electrons, owing to the doubly degenerate nature of its LUMO.18 By now, the products of all reduction steps, the mono-,19 di-,19b tri-,20 and tetraanions21 of corannulene (C20H10n−, n = 1–4), have been isolated and crystallographically characterized. These structural studies revealed the remarkable ability of very electron-rich tetrareduced corannulene (which bears one electron per five carbon atoms thus is more electron-rich than the hexaanion of fullerene, C606−) to form unique supramolecular products with high numbers of encapsulated alkali metal ions, including unprecedented heterobimetallic combinations.22 The binding properties of the transient triply-reduced corannulene remained unknown until our recent report on the first structural characterization of the corannulene trianion isolated with cesium counterions.20 X-ray crystallographic analysis revealed that two C20H10˙3− anions self-assemble into a novel sandwich-type aggregate having four interior and two exterior cesium ions, [Cs+//(C20H103−)/4Cs+/(C20H103−)//Cs+]. These structural results20–22 clearly differentiate the highly negatively charged tri- and tetra-reduced anions, that show tendencies to self-assemble with multiple metal ions, from mono- and doubly-reduced corannulene, which can be isolated in their “naked” forms.19,23 Moreover, the aforementioned homometallic cesium sandwich exhibits several special features, such as a large axial shift of two bowl-shaped decks and the absence of magnetic coupling between two trianion-radicals within the sandwich, as well as an unexpectedly high curvature of corannulene bowls. These experimental observations raised interesting questions about the role of alkali metals in the geometry perturbation of triply-reduced corannulene and its supramolecular aggregation processes. To provide further insights into the unique self-aggregates formed by C20H10˙3−, we set out to investigate the bimetallic reduction of corannulene using K and Cs metals. Herein, we isolated the first mixed-metal product with triply-reduced corannulene and used a combination of experimental and computational techniques to understand its geometric and electronic structures. This task required an in-depth theoretical analysis of the role of encapsulated alkali metal ions, therefore all Group 1 metals ranging in size from Li to Cs were included in the calculations.
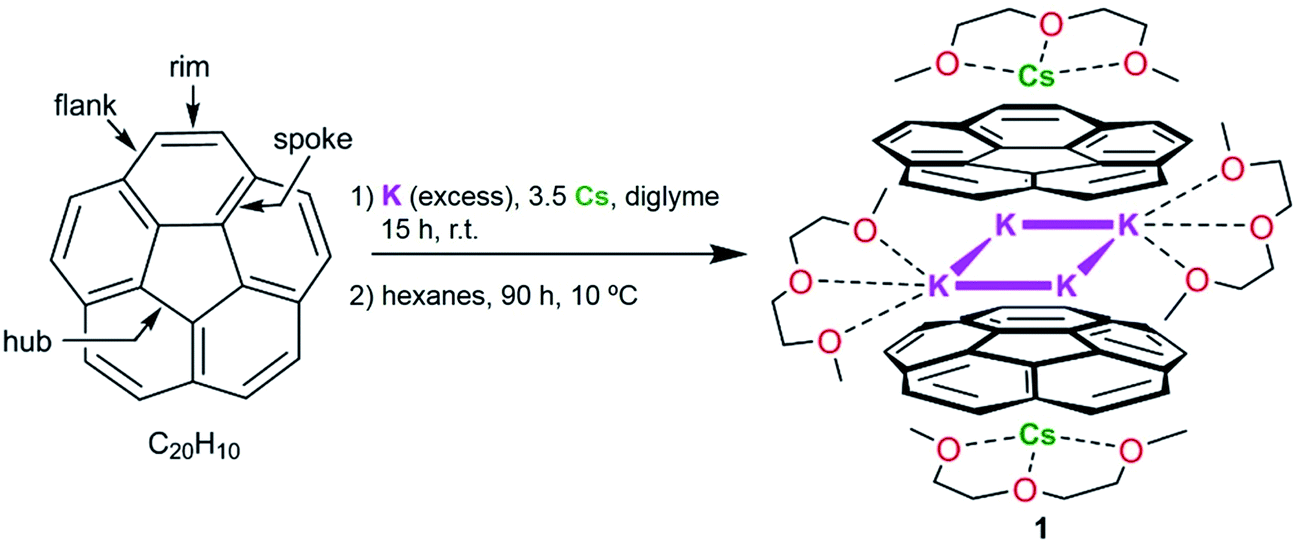 | ||
| Scheme 1 Preparation of 1. | ||
Results and discussion
The first isolation of the corannulene trianion in the form of its crystalline supramolecular product with cesium counterions prompted us to investigate whether other alkali metals are capable of reducing corannulene to the trianion stage and whether these reactions allow its bulk solid-state isolation. Previously, paramagnetic C20H10˙3− radicals were detected in situ by ESR spectroscopy using a reduction reaction of corannulene with lithium metal in THF.24 We have observed that Li-induced reduction of corannulene quickly proceeds to the final reduction state, C20H104−, and thus is impractical for the preparation of products based on transient trianions. However, the outcome of using other Group 1 metals in these reactions remained unclear.Herein, we found that reduction of C20H10 in diglyme using an excess of K metal (ca. 10 eq.) and a controlled amount of metallic Cs (ca. 3.5 eq. in respect to C20H10) affords a deep-red coloured solution (characteristic of the third reduction step) after 15 hours of stirring at room temperature. Notably, the reduction proceeds significantly faster compared with the cesium-only reaction, requiring more than 60 hours in order to reach the same stage. The UV-vis spectrum of the resulting reaction mixture exhibits a broad absorbance band with λmax around 514 nm and a very intense band with λmax = 388 nm, which is characteristic of the C20H10˙3− anion (see ESI, Fig. S1 and S2†). These values are slightly shifted compared to the trianion generated by the Cs-only reduction (λmax = 386 nm and 495 nm).
After reaching the third reduction stage, the reaction mixture was filtered to afford a deep-red solution which was layered with hexanes and kept at 10 °C. Dark-red blocks of [K2Cs(diglyme)2(C20H103−)] (1) were present after 90 hours in ca. 50–60% yield (Scheme 1). Crystals of 1 are extremely air- and moisture-sensitive and show very limited solubility in THF.
Notably, further reduction of C20H10˙3− to the C20H104− state was not observed in this work, even when an excess of K and Cs metals was used over an extended reaction time period (one month). It should be mentioned that the reduction of corannulene with potassium metal only, in diglyme, also results in the in situ formation of C20H10˙3− as the major product (Fig. S3†). However, our numerous attempts to crystallize the homometallic potassium analogue of 1 have been unsuccessful so far. In contrast, the crystals of 1 are obtained reproducibly in good yield, thus illustrating the important role of Cs+ ions in the formation and crystallization of the title product.
According to an X-ray diffraction study (see ESI† for more details), the asymmetric unit of 1 consists of one corannulene trianion, two potassium ions, one cesium ion, and two diglyme molecules (Fig. 1a). In the molecular structure, four potassium ions are held between the two triply-reduced corannulene bowls to yield the supramolecular aggregate, [(C20H103−)/4K+/(C20H103−)]2− (Fig. 1b). The triple-decker sandwich in 1 is similar to that found in the all-cesium product, [(C20H103−)/4Cs+/(C20H103−)]2−. However, four potassium ions are now sandwiched between two C20H10˙3− anions instead of the larger cesium ions, thus bringing the two corannulene decks notably closer together (5.87 Å vs. 6.73 Å). As observed in the cesium analogue, a convex-to-convex arrangement of the two bowls is also found in 1. The [(C20H103−)/4K+/(C20H103−)]2− aggregate demonstrates a staggered conformation of two C20H10˙3− anions that are slipped in respect to each other by 2.093(4) Å (vs. 1.843(5) Å in the Cs-only sandwich).
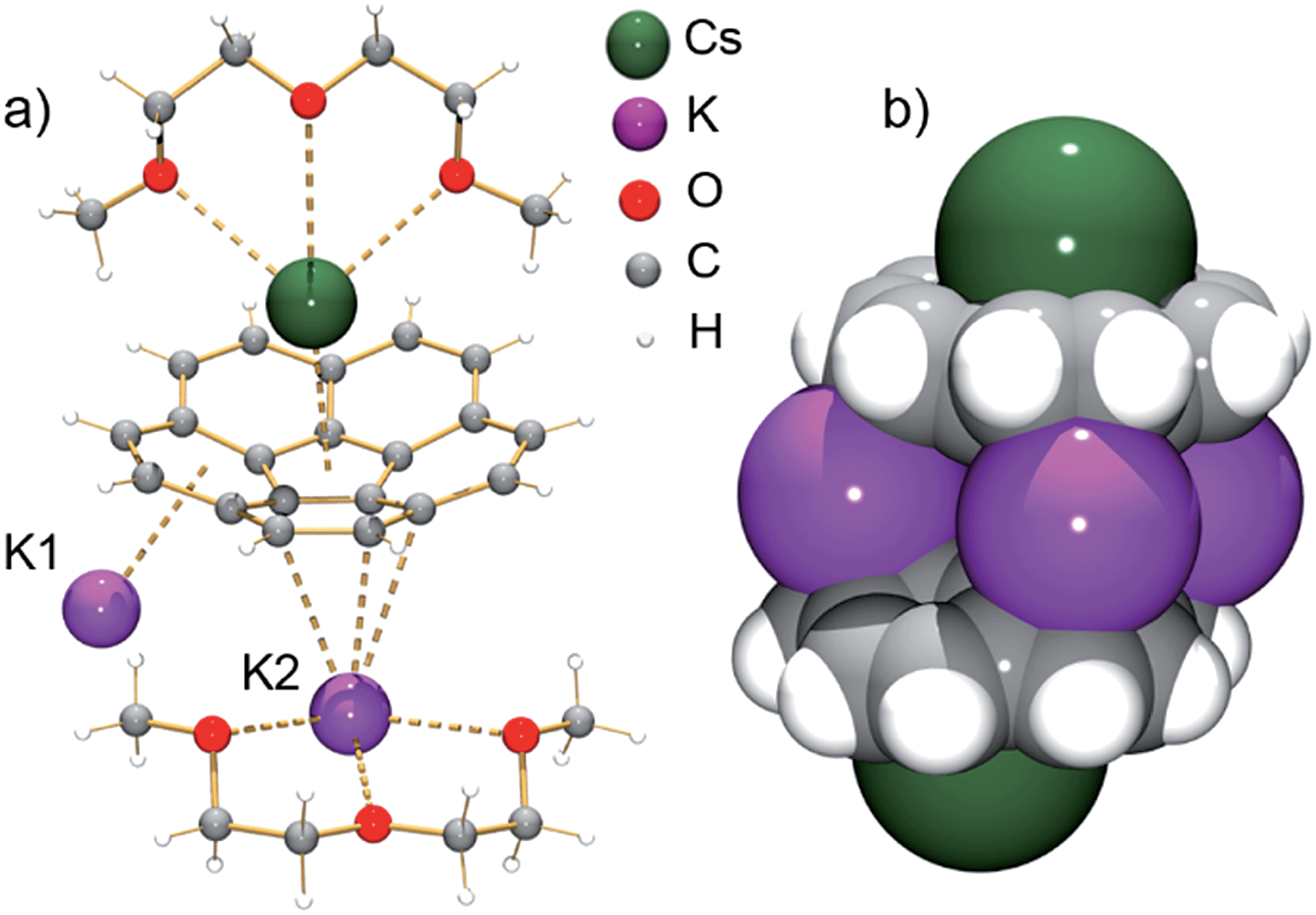 | ||
| Fig. 1 Asymmetric unit (a, ball-and-stick model) and sandwich view (b, space-filling model, diglyme molecules are removed) of 1. | ||
The sandwiched K1, K2, K1′ and K2′ ions form a rectangle with K⋯K separations of 4.212(4) Å and 5.185(4) Å (compared to those in elemental potassium of 4.54 Å).25 Notably, the ionic radius of the K+ ion is 1.33 Å vs. that of 1.69 Å for Cs+.26 The K1 ions are bound to both anionic bowls in an η6-mode with K1⋯C interatomic distances of 2.950(3)–3.634(4) Å (K1⋯C6(centroid): 2.792(3) and 3.041(4) Å). In contrast, the K2 ions are η3- and η6-bound to the bowls with K2⋯C distances of 2.945(3)–3.441(3) Å and 2.978(3)–3.354(4) Å (K2⋯C6(centroid) is 2.818(4) Å). The coordination sphere of the encapsulated K2 ion is completed by chelating diglyme, with the K⋯O distances ranging from 2.690(3)–2.859(3) Å.
As observed with the cesium-generated C20H10˙− and C20H102− anions,19a,27 the concave cavities of the charged corannulene bowls are always occupied by large Cs+ ions. In 1, similar to the [Cs+//(C20H103−)/4Cs+/(C20H103−)//Cs+] aggregate (2), the external concave cavities of the two bowls are filled by one cesium ion each, thus confirming the remarkable selectivity of the Cs+ ion towards endo-binding, even in the presence of prevalent K+ ions. The cesium ion is asymmetrically coordinated to the endo surface of C20H10˙3− in an η5-fashion (Cs⋯C distance of 3.151(3)–3.338(3) Å) with a distance to the center of the five-membered ring of 3.02 Å. The Cs⋯C distances between the Cs+ ion and the benzene rings of C20H10˙3− are significantly longer (3.374(3)–3.969(4) Å). Additionally, the external cesium ion is bound to three oxygen atoms of diglyme (Cs⋯O of 3.111(3)–3.469(3) Å) and one oxygen atom of a neighboring diglyme molecule (3.343(3) Å). As a result, the [Cs(diglyme)]+ cations serve as building blocks in the formation of a 1D polymer in the crystal structure of 1 (Fig. 2).
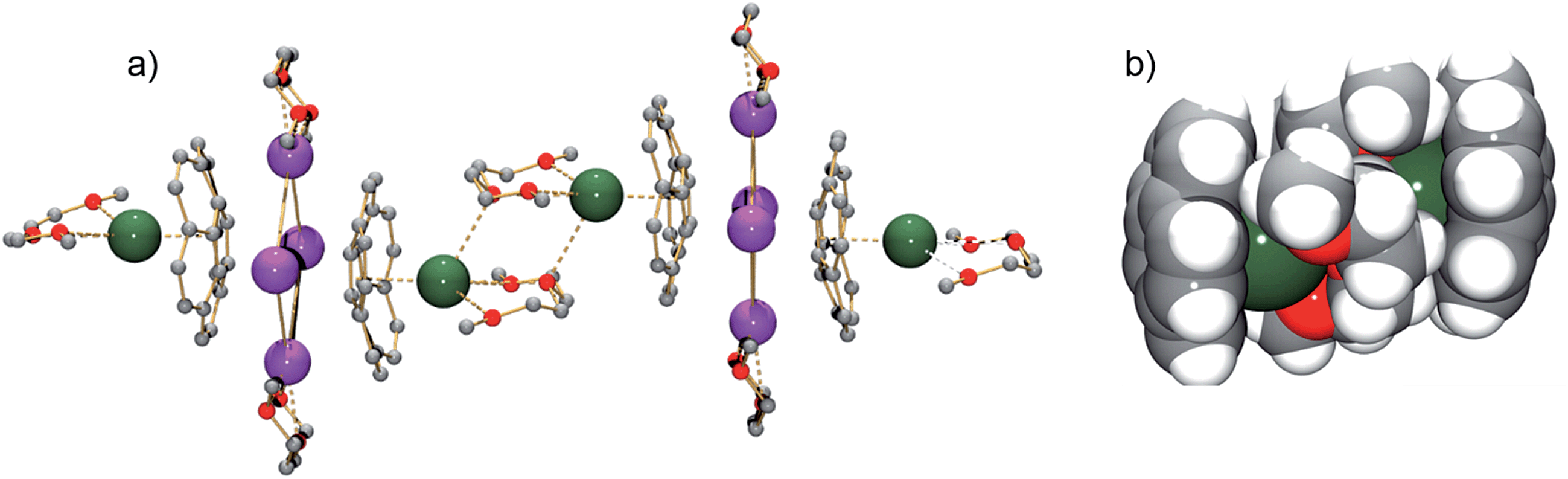 | ||
| Fig. 2 A fragment of the polymeric chain in 1 (a, ball-and-stick, all H-atoms are removed) and a space-filling model showing the [(C20H103−)/[Cs(diglyme)]2/(C20H103−)]4− connection (b). | ||
In the solid state, additional contacts between K1 and the adjacent C20H10˙3− moieties (3.284(4)–3.430(4) Å) are observed, propagating the structure in the a-direction (Fig. 3) and explaining the observed very limited solubility of 1.
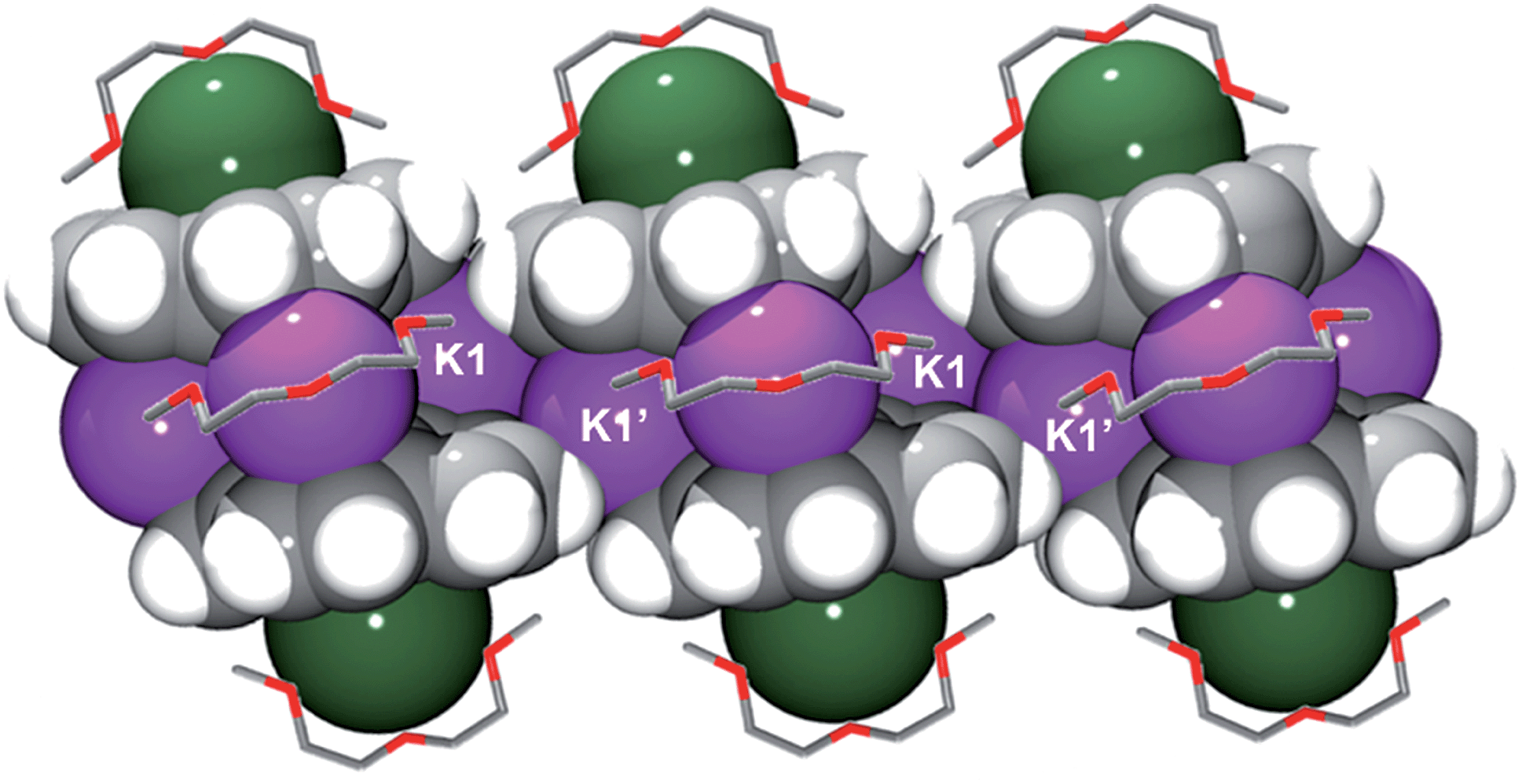 | ||
| Fig. 3 Space filling model of three adjacent sandwiches with the diglyme molecules shown as capped sticks. The hydrogen atoms of diglyme are omitted for clarity. | ||
The structural characterization of the mixed-metal sandwich 1 allows its direct comparison with the all-cesium analogue (2). Generally, the geometric perturbations of the C20H10˙3− core in the two products are quite similar. In both cases, the hub C–C bond lengths of C20H10˙3− are comparable to those in neutral C20H10 (Table 1),28 whereas the spoke and rim C–C bond distances are elongated. The acquisition of three electrons by C20H10 and the subsequent aggregation with multiple alkali metal ions do not induce any significant flattening of the resulting trianion compared to the parent bowl. The bowl depth of C20H10˙3− in 1 is 0.808(4) Å vs. 0.875(2) Å for neutral C20H10.28 Notably, the curvature of the corannulene trianion separated by the potassium belt in 1 is reduced compared to that in 2 (0.81 vs. 0.85 Å), reflecting the greater effect of large encapsulated cesium ions on the bowl depth.
Magnetic behavior of Cs-only vs. K/Cs sandwich products
We have previously found that the sandwich formed by C20H10˙3− with Cs+ ions, [Cs+//(C20H103−)/4Cs+/(C20H103−)//Cs+], shows weak antiferromagnetic coupling of the corannulene trianion-radicals through the large alkali metal belt, as confirmed by magnetic measurements and high-level theoretical modeling.20 The extreme air- and moisture-sensitivity of the product 2 allowed only a rough estimation of the exchange coupling within the sandwich, with an interaction constant in between −5 and −10 K (with H = −2JS1S2) and a gap, Δ = |2J|, between the singlet ground state and the triplet excited state of about 10–20 K.Multiple crystalline samples of 1 prepared from different reaction batches have been measured in this work, all showing product decomposition with time even under strictly anaerobic conditions (see ESI and Fig. S5†). Nevertheless, all the magnetic data clearly point to the diamagnetic ground state of the new heterometallic product, [Cs+//(C20H103−)/4K+/(C20H103−)//Cs+], and indicate weak antiferromagnetic interaction between two trianion-radicals through the potassium belt in the sandwich. As expected due to a closer proximity of the radicals, this magnetic exchange is slightly greater than in the analogous cesium-only compound, reaching −11.5(5) K (−8.0(3) cm−1 and Δ = |2J| = 16 cm−1).
Computational study
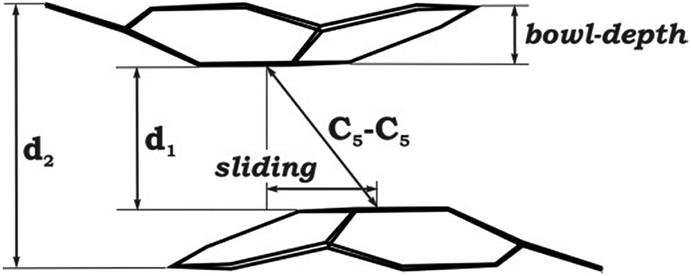
Considering the observed experimental limitations of magnetic measurements caused by the extremely high sensitivity of 1 towards air and moisture, we turned to theoretical investigations. Previously, we have demonstrated that the PBE0/def2-TZVP+ECP(K,Cs)//cc-pVDZ(C,H,O) level of theory provides an adequate balance between accuracy and computational effort (see details in ESI†). Similar to the Cs-only system,20 four different models were considered initially: (i) the simplest fully optimized [Cs+//(C20H103−)/4K+/(C20H103−)//Cs+] model (1-K-small); (ii) the same model, but with the core structure taken from the X-ray experiment and kept unchanged, while the positions of the hydrogen atoms were optimized (1H-K-small); (iii) the fully optimized model, in which all solvent molecules were considered explicitly (1-K-full); (iv) the same model as 1-K-full, but with only the hydrogen atom positions optimized (1H-K-full), whereas the rest was taken from the crystal structure and kept frozen (see further details in ESI†).|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | Model | |||||||
| 1-M-small | 1H-K-small | 1H-K-full | 1-K-full | |||||
| M = Li | M = Na | M = K | M = Rb | M = Cs | ||||
| a The J-coupling constant (in cm−1) is calculated using the Yamaguchi formula (broken-symmetry BS(1,1) solution)29 at the PBE0/TZVP/ZORA level of theory as implemented in the ORCA program suite.30 | ||||||||
| Bowl-depth | 0.510 | 0.731 | 0.768 | 0.814 | 0.826 | 0.808 | 0.808 | 0.758 |
| d 1 | 3.180 | 3.723 | 4.348 | 4.629 | 4.865 | 4.247 | 4.247 | 4.417 |
| d 2 | 4.252 | 5.204 | 5.952 | 6.283 | 6.541 | 5.872 | 5.872 | 5.967 |
| Bowl sliding | 1.601 | 1.171 | 0.840 | 0.750 | 0.453 | 2.091 | 2.091 | 1.510 |
| C5–C5 | 3.560 | 3.904 | 4.430 | 4.691 | 4.891 | 4.735 | 4.735 | 4.675 |
| M⋯M (min) | 3.576 | 3.844 | 4.265 | 4.481 | 4.668 | 4.212 | 4.212 | 4.340 |
| M⋯M (max) | 4.516 | 4.210 | 4.468 | 4.630 | 4.804 | 5.185 | 5.185 | 4.701 |
| Δ(M⋯M) | 0.940 | 0.366 | 0.203 | 0.149 | 0.136 | 0.973 | 0.973 | 0.361 |
| 2Ja | +32.32 | −3.00 | −6.44 | −24.99 | −2.39 | +1.50 | −0.69 | −2.62 |
Previously, our calculations revealed that the large Cs+ ions jammed between bowl-shaped triply-reduced corannulene anions prevent any significant magnetic coupling in the sandwich-like aggregates.20 The electronic structure of such an assembly was found to be best described as a system with two uncoupled radicals. Replacing Cs+ ions with much smaller K+ ions gave a 2J-coupling constant of −0.69 cm−1 for the 1H-K-full model calculated using a broken-symmetry approach (see details in ESI†). Relaxation of the geometry during the optimization procedure led to a slight increase in the anti-ferromagnetic coupling between the trianion-radicals (2J = −2.62 cm−1) in the 1-K-full system. Interestingly, the removal of coordinated solvent molecules from the full models showed some influence on the magnetic coupling. The corresponding 2J-constants were computed to be +1.50 cm−1 and −6.44 cm−1 for the 1H-K-small and 1-K-small models, respectively, thus illustrating that at this level of theory it is difficult to make unambiguous conclusions about the ground state and magnetic coupling in the target systems. Therefore, we turned to the highly accurate multireference Møller–Plesset perturbation theory of the second order (MRMP2).31 Since the application of this technique to the full systems is not feasible, we mainly focused on using the 1H-K-small and 1-K-small models. The active space for the reference CASSCF wavefunction included 14 electrons over 8 orbitals, CASSCF(14,8). Calculations performed at the MRMP2 level of theory revealed that the energy gap, Δ, between the open-shell singlet and triplet states is equal to −6.46 cm−1 for 1H-K-small and −54.43 cm−1 for 1-K-small. Notably, a good agreement between the J-constant values for the 1H-K-small model and the experimentally measured one (−8.0 cm−1) was observed. There is also an agreement with the experiment-based conclusion that the anti-ferromagnetic coupling in 1 is stronger than that in 2 (−0.01 cm−1 calculated at the same level of theory).20 Relaxation of the geometry increased the magnitude of the coupling as it is exemplified by the 1-K-small model. The same trend was observed in the full models, for which the coupling parameters were calculated at the BS-DFT level of theory. This clearly shows the applicability of the broken-symmetry DFT approach to the modeling of magnetic coupling in such complicated systems. Although it showed some deficiency in providing the exact numbers, the qualitative trends are reproduced correctly.
Interestingly, due to weak magnetic coupling between the bowl-shaped fragments, the charge and spin distributions in the sandwich systems remain similar to those of the isolated C20H10˙3− species (see ESI† for details). Only a slight polarization of charge distribution was found in the 1H-K-small model due to the axial shift of the bowls with respect to each other. The spin density of the unpaired electrons is exclusively localized on the negatively-charged corannulene bowls. Moreover, the topology of the spin density is not notably influenced by the presence of solvent molecules and/or by crystal packing effects (Fig. 4).
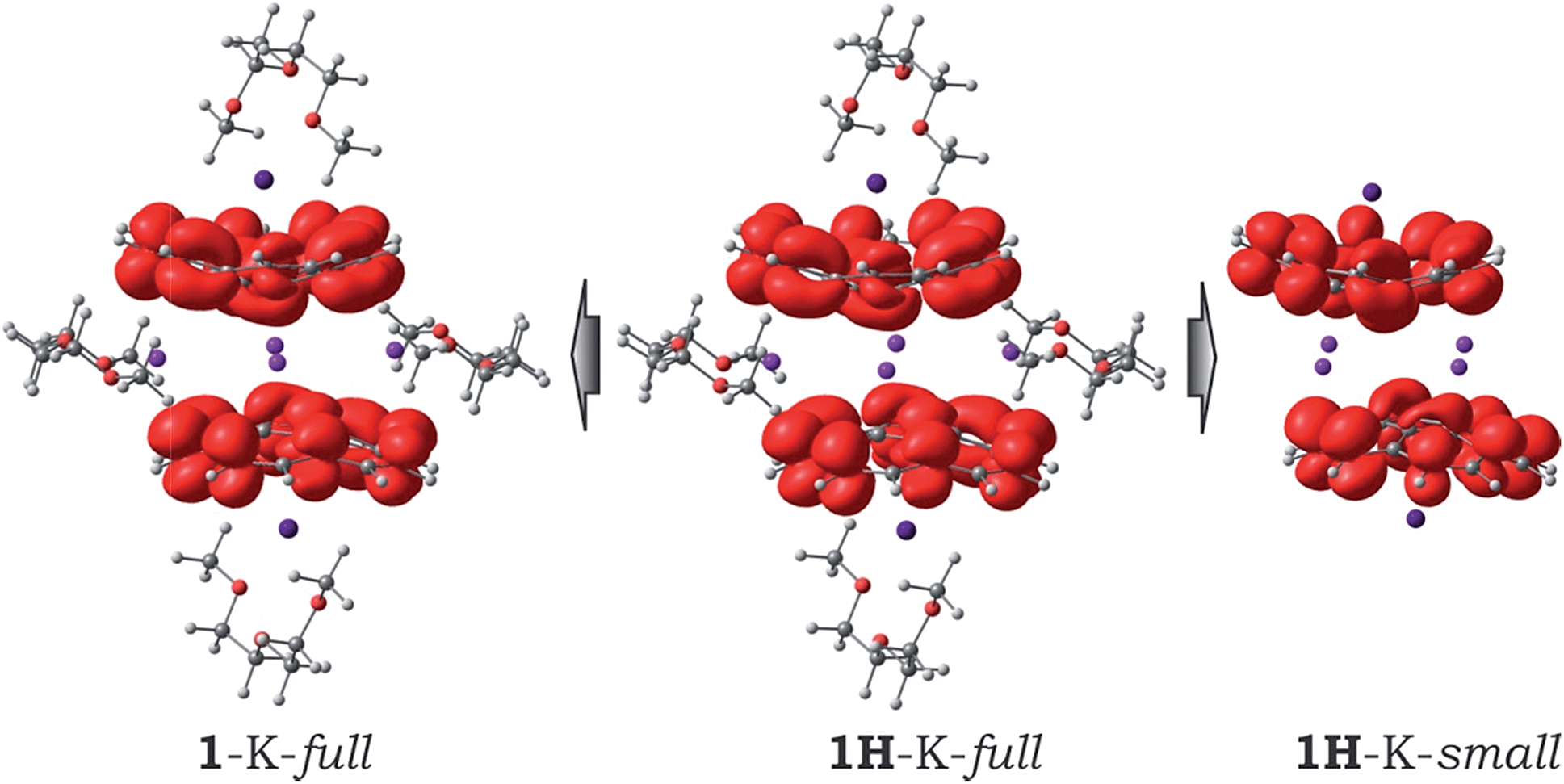 | ||
| Fig. 4 Spin density distribution in the 1H-K-full, 1-K-full, and 1H-K-small models. | ||
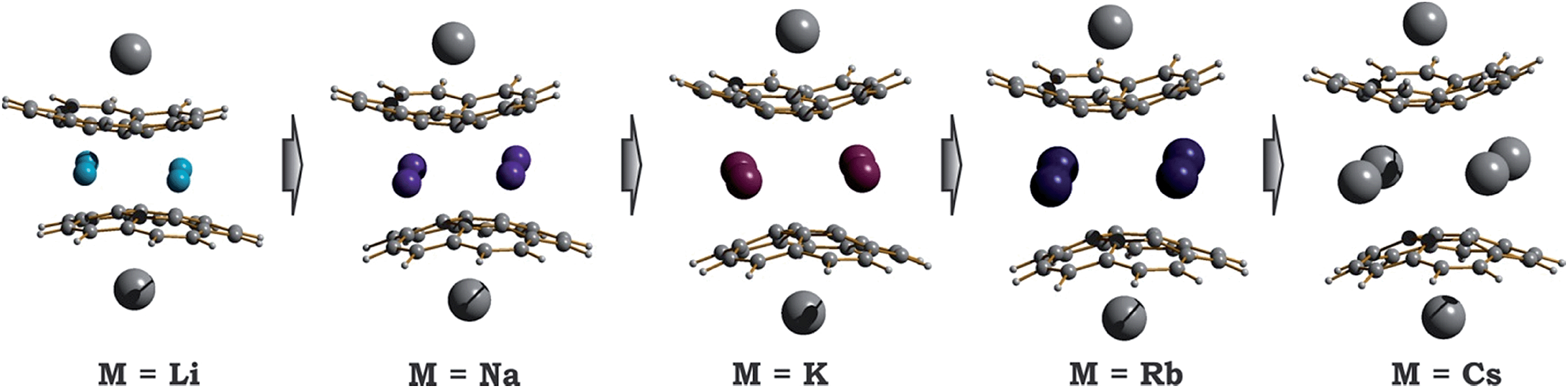 | ||
| Fig. 5 Equilibrium geometry configurations for 1-M-small systems, where M = Li, Na, K, Rb and Cs (PBE0/def2-TZVP(M)//cc-pVDZ). | ||
As expected, the downsizing of the positively-charged tetranuclear alkali metal belts held between the C20H10˙3− bowls results in the decreasing of the supramolecular aggregate height, as can be illustrated by the d2 parameter (Table 2). The distance between the two corannulene bowls (d1) also decreases from 4.417 Å to 3.180 Å going from Cs to Li, which may result in the increased coupling between the C20H10˙3− radicals. In addition, the effect of the alkali metal size on the curvature of the bowl-shaped decks is observed. The calculated bowl-depth of C20H10˙3− increases when going from the 1-Li-small (0.510 Å) to 1-Cs-small (0.826 Å) system. This variation shows the remarkable flexibility of the corannulene framework and provides an argument that its curvature in the sandwich-like aggregates depends not only on the charge state (as predicted earlier32) but, importantly, on the nature and size of the encapsulated metal ions.
Interestingly, decreasing the M+ ion size in the calculated systems also led to a pronounced shift of one bowl with respect to another (Table 2). The largest bowl slip was computed for the 1-Li-small system (1.601 Å), whereas the smallest one was found for its Cs-derivative (0.453 Å). Previously, such a shift was attributed exclusively to the influence of coordinated solvent molecules and crystal packing effects. Herein, the calculated shifts for all 1-M-small systems unambiguously reveal that the size and nature of the alkali metals, constituting the positively charged belts, play a very important role. As a consequence, the shape of the encapsulated tetranuclear metal unit undergoes a smooth transformation from almost square-planar in the 1-Cs-small system (Δ(M⋯M) = 0.136 Å) to a very pronounced rectangular shape in the 1-Li-small system (Δ(M⋯M) = 0.940 Å).
As a result of this bowl sliding clearly affecting the product geometry, one can detect a noticeable polarization in the charge distribution along the series of sandwich-like aggregates (as exemplified by the molecular electrostatic potential (MEP) maps in Fig. 6). While the charge state of the corannulene fragments remains −3, the negative charge on their π-surface is becoming more and more localized in order to minimize the electrostatic repulsion between the bowls. For instance, in the case of the 1-Cs-small system having the greatest separation between the two bowls (Table 2) and minimal sliding, the charge distribution is almost symmetrical. At the same time, for the 1-Li-small system (with the smallest d1 and the largest axial shift between the bowls), the localization of negative charges was found to be the most pronounced in the series.
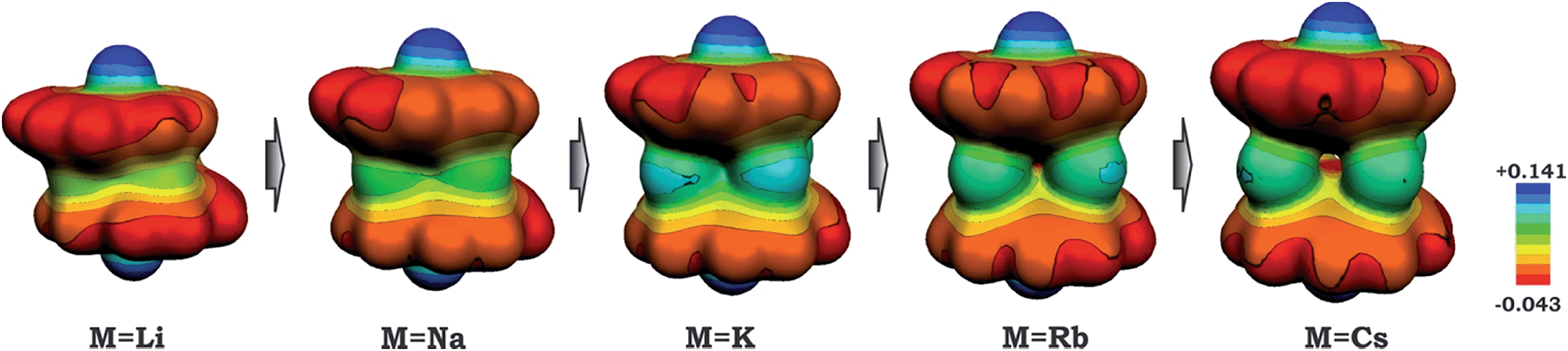 | ||
| Fig. 6 Molecular electrostatic potential maps for 1-M-small systems, where M = Li, Na, K, Rb and Cs (PBE0/def2-TZVP(M)//cc-pVDZ). | ||
Considering the significant variations in geometry when going from Cs to Li (Table 2), one may expect accompanied changes in the spin density distribution and, consequently, in the magnetic coupling of the two C20H10˙3− radicals. The increased shift of the corannulene bowls with respect to each other (parameter “bowl sliding” in Table 2) notably stabilizes the localization of the unpaired electrons on opposite sides of the supramolecular aggregate, as exemplified by the spin density distribution (Fig. 7). This localization is almost negligible in the 1-Cs-small system but reaches a maximum in the 1-Li-small system. As expected, the associated stabilization results in an increasing ferromagnetic interaction between the bowl-shaped radicals and in a decreasing anti-ferromagnetic component. This can be clearly illustrated by comparison of the 1H-K-small (sliding = 2.09 Å) and 1-K-small (sliding = 0.840 Å) systems, for which the magnetic coupling constants were calculated to be 2J = +1.50 cm−1 and −6.44 cm−1 (at the BS-DFT level of theory), respectively. The same trend was observed for the 1H-Cs-small and 1-Cs-small models (sliding = 1.84 Å and 0.453 Å; J = +3.84 cm−1 and −2.39 cm−1, respectively).20 Eventually, this ferromagnetic interaction is maximized in the case of the 1-Li-small system (2J = +32.32 cm−1), where the axial sliding is the most pronounced (1.601 Å).
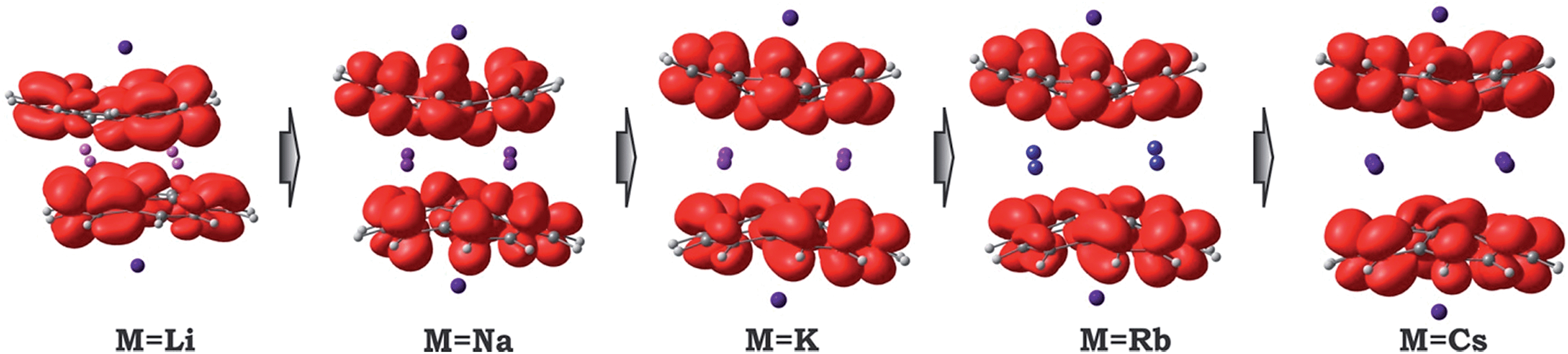 | ||
| Fig. 7 Spin density distribution in 1-M-small systems, where M = Li, Na, K, Rb and Cs (PBE0/def2-TZVP(M)//cc-pVDZ). | ||
At the same time, another tendency in the magnetic coupling between the curved corannulene radicals as a function of the product geometry was also observed, namely an increase in anti-ferromagnetic coupling with decreasing distance d1 (Table 2). In order to provide further evidence for this trend, theoretical modeling of the modified 1-Li-small model (hereafter called 1-Li-small-iso) was performed at the same level of theory. In this new model, two bowl fragments were placed on top of each other in a non-sliding fashion, whereas the distance d1 was kept exactly the same as in the fully relaxed 1-Li-small system (see details in ESI†). Thus, the influence of the bowl shift on the magnetic coupling between the corannulene radicals was excluded. The 2J-constant for the 1-Li-small-iso model was calculated to be −42.50 cm−1 (in contrast to 2J = +32.32 cm−1 calculated for the 1-Li-small model), which clearly illustrates the strengthening of an anti-ferromagnetic component.
Hence, the actual magnetic coupling between two corannulene radicals is a result of the interplay between these two trends in the product geometry change induced by the downsizing of the encapsulated alkali metal belt from the largest Cs+ ions to the smallest Li+ ions. As one might expect for the two opposing trends, the correlation between alkali metal size and magnetic coupling is not linear and may have a maximum/minimum. This is what was observed in the case of M = Rb, where the anti-ferromagnetic coupling was found to be maximized.
In order to complete the description of the magnetic coupling for the series, an analysis of the interactions between the bowl-shaped corannulene fragments and the positively-charged alkali metal belt was performed using the Energy Decomposition Analysis (EDA) approach. The accepted fragmentation scheme was the same as that proposed in a recent study.20 Three interacting fragments were considered (Fig. S11†), namely two [(Cs)(C20H10)]2− and one [M4]4+ species (M = Li, Na, K, Rb and Cs). Such fragmentation allows one to tentatively evaluate the interactions in the supramolecular aggregates as well as to estimate the coupling between the negatively-charged bowls. The results of the EDA analysis are collected in Table 3.
| Parameter | Compound | ||||
|---|---|---|---|---|---|
| 1-Li-small | 1-Na-small | 1-K-small | 1-Rb-small | 1-Cs-small | |
| ΔEint | −1230.57 | −1133.72 | −1024.70 | −981.60 | −942.57 |
| ΔEorb | −383.30 | −298.56 | −256.84 | −239.17 | −241.89 |
| ΔEelstat | −977.79 | −946.44 | −892.07 | −867.31 | −853.45 |
| ΔEPauli | +130.52 | +111.27 | +124.22 | +124.88 | +152.77 |
As shown in Table 3, the coupling between corannulene bowls gets significantly stronger when going from Cs to Li, as exemplified by the total interaction energy ΔEint (which changes from −1230.57 kcal mol−1 to −942.57 kcal mol−1, respectively). This finding is in excellent agreement with previous conclusions, based on charge and spin density distributions as well as on magnetic coupling in the target systems. Importantly, both of the attractive components of ΔEint, namely the orbital (ΔEorb) and electrostatic (ΔEelstat) components, follow exactly the same trends. At the same time, the repulsive Pauli interaction (ΔEPauli) shows an opposite tendency in the series, again making systems with small alkali metal ions sandwiched between the corannulene bowls more stable. However, even the high stability of the 1-Li-small system with triply-reduced bowls does not make it as stable as the system based on tetrareduced corannulene. The total interaction energy in such a 14−-small system was previously calculated to be −1536.03 kcal mol−1.20 The main difference should arise from the dramatically stronger electrostatic component in the latter (−977.79 kcal mol−1 in the 1-Li-small system vs. −1452.97 kcal mol−1 in the 14−-small system).
Conclusions
In this work, the isolation and X-ray structural characterization of the first heterobimetallic sandwich-type aggregate formed by triply-reduced corannulene, [Cs+//(C20H103−)/4K+/(C20H103−)//Cs+], has been accomplished. The successful synthesis of this mixed-metal assembly based on the selectivity of the concave and convex binding of the corannulene trianion towards potassium and cesium ions provides a remarkable experimental illustration of the fascinating coordinating properties of highly charged π bowls.Furthermore, analysis of the structural and magnetic data for the title product has opened up the first discussion on the role of encapsulated metals on the perturbation of molecular geometry and magnetic coupling of bowl-shaped radicals within sandwich-type architectures. The variation of tetranuclear alkali metal belts from Li to Cs in the [Cs+//(C20H103−)/4M+/(C20H103−)//Cs+] aggregates revealed unique structural and electronic transformations within the series. Remarkably, the replacement of the sandwiched K ions with larger (Cs and Rb) and smaller (Li) ions is accompanied by notable geometrical changes of the sandwich architectures, as reflected by the different axial shifts and separations of the corannulene trianion radicals. Notably, the downsizing of the sandwiched alkali metal belts allows for the tuning of the coupling of C20H10˙3− radicals from anti-ferromagnetic to ferromagnetic in nature.
Acknowledgements
The financial support for this work from the National Science Foundation (CHE-1608628 and CHE-1337594) is gratefully acknowledged by M. A. P. Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund for the partial support of this research through 54967ND3 grant. The start-up funding from the Illinois Institute of Technology (IIT) is greatly acknowledged by A. Yu. R. Finally, R. C thanks the University of Bordeaux, the ANR, the Conseil Régional d'Aquitaine, the CNRS and the GdR MCM-2 for their financial support.References
- (a) M. Rabinovitz, I. Willner and A. Minsky, Acc. Chem. Res., 1983, 16, 298–304 CrossRef CAS; (b) M. Rabinovitz, Top. Curr. Chem., 1988, 146, 99–169 CrossRef CAS; (c) R. Benshafrut, E. Shabtai, M. Rabinovitz and L. T. Scott, Eur. J. Org. Chem., 2000, 1091–1106 CrossRef CAS.
- (a) H. Bock, T. Hauck, C. Nather and Z. Havlas, Angew. Chem., Int. Ed. Engl., 1997, 36, 638–639 CrossRef CAS; (b) H. Bock, Z. Havlas, D. Hess and C. Nather, Angew. Chem., Int. Ed., 1998, 37, 502–504 CrossRef CAS; (c) H. Bock, Z. Havlas, K. Gharagozloo-Hubmann, S. Holl and M. Sievert, Angew. Chem., Int. Ed., 2003, 42, 4385–4389 CrossRef CAS PubMed.
- (a) J. D. Smith, Adv. Organomet. Chem., 1999, 43, 267–348 CrossRef; (b) A. V. Zabula and M. A. Petrukhina, Adv. Organomet. Chem., 2013, 61, 375–462 CrossRef CAS.
- D. Eisenberg and R. Shenhar, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 527–545 CrossRef.
- Fragments of Fullerenes and Carbon Nanotubes: Designed Synthesis, Unusual Reactions, and Coordination Chemistry, ed. M. A. Petrukhina and L. T. Scott, John Wiley & Sons, New Jersey, 2012, pp. 1–413 Search PubMed.
- I. Hirosawa, K. Prassides, J. Mizuki, K. Tanigaki, M. Gevaert, A. Lappas and J. K. Cockcroft, Science, 1994, 264, 1294–1297 CAS.
- S. Aoyagi, Y. Sado, E. Nishibori, H. Sawa, H. Okada, H. Tobita, Y. Kasama, R. Kitaura and H. Shinohara, Angew. Chem., Int. Ed., 2012, 51, 3377–3381 CrossRef CAS PubMed.
- K. Y. Amsharov, Y. Krämer and M. Jansen, Angew. Chem., Int. Ed., 2011, 50, 11640–11643 CrossRef CAS PubMed.
- (a) A. V. Zabula, A. S. Filatov, J. Xia, R. Jasti and M. A. Petrukhina, Angew. Chem., Int. Ed., 2013, 52, 5033–5036 CrossRef CAS PubMed; (b) S. N. Spisak, J. Li, A. Yu. Rogachev, Z. Wei, O. Papaianina, K. Amsharov, A. V. Rybalchenko, A. A. Goryunkov and M. A. Petrukhina, Organometallics, 2016, 35, 3105–3111 CrossRef CAS.
- (a) Y. Morita, A. Ueda, S. Nishida, K. Fukui, T. Ise, D. Shiomi, K. Sato, T. Takui and K. Nakasuji, Angew. Chem., Int. Ed., 2008, 47, 2035–2038 CrossRef CAS PubMed; (b) S. Nishida, Y. Morita, A. Ueda, T. Kobayashi, K. Fukui, K. Ogasawara, K. Sato, T. Takui and L. Nakasuji, J. Am. Chem. Soc., 2008, 130, 14954–14955 CrossRef CAS PubMed; (c) A. Ueda, S. Nishida, K. Fukui, T. Ise, D. Shiomi, K. Sato, T. Takui, K. Nakasuji and Y. Morita, Angew. Chem., Int. Ed., 2010, 49, 1678–1682 CrossRef CAS PubMed; (d) A. Ueda, K. Ogasawara, S. Nishida, T. Ise, T. Yoshino, S. Nakazawa, K. Sato, T. Takui, K. Nakasuji and Y. Morita, Angew. Chem., Int. Ed., 2010, 49, 6333–6337 CrossRef CAS PubMed.
- (a) Y. Takabayashi, A. Y. Ganin, M. J. Rosseinsky and K. Prassides, Chem. Commun., 2007, 870–872 RSC; (b) D. Arcon, A. Y. Ganin, Y. Takabayashi, M. J. Rosseinsky and K. Prassides, Chem. Mater., 2008, 20, 4391–4397 CrossRef CAS; (c) A. Y. Ganin, Y. Takabayashi, P. Jeglic, D. Arcon, A. Potocnik, P. J. Baker, Y. Ohishi, M. T. McDonald, M. D. Tzirakis, A. McLennan, G. R. Darling, M. Takata, M. J. Rosseinsky and K. Prassides, Nature, 2010, 466, 221–225 CrossRef CAS PubMed; (d) N. V. Kozhemyakina, K. Y. Amsharov, J. Nuss and M. Jansen, Chem.–Eur. J., 2011, 17, 1798–1805 CrossRef CAS PubMed; (e) D. V. Konarev, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, New J. Chem., 2011, 35, 1829–1835 RSC.
- (a) T. Kato and T. Yamabe, J. Chem. Phys., 2002, 117, 2324–2331 CrossRef CAS; (b) I. Aprahamian, R. E. Hoffman, T. Sheradsky, D. V. Preda, M. Bancu, L. T. Scott and M. Rabinovitz, Angew. Chem., Int. Ed., 2002, 41, 1712–1715 CrossRef CAS; (c) H. Phan, K. Lekin, S. M. Winter, R. T. Oakley and M. Shatruk, J. Am. Chem. Soc., 2013, 135, 15674–15677 CrossRef CAS PubMed; (d) Z. Mou, K. Uchida, T. Kubo and M. Kertesz, J. Am. Chem. Soc., 2014, 136, 18009–18022 CrossRef CAS PubMed; (e) M. Yamada, H. Kurihara, M. Suzuki, M. Saito, Z. Slanina, F. Uhlik, T. Aizawa, T. Kato, M. M. Olmstead, A. L. Balch, Y. Maeda, S. Nagase, X. Lu and T. Akasaka, J. Am. Chem. Soc., 2015, 137, 232–238 CrossRef CAS PubMed.
- (a) J. P. Lemmon, Nature, 2015, 525, 447–449 CrossRef CAS PubMed; (b) K. E. Moore, D. D. Tune and B. S. Flavel, Adv. Mater., 2015, 27, 3105–3137 CrossRef CAS PubMed; (c) F. Meng, W. Lu, Q. Li, J.-H. Byun, Y. Oh and T.-W. Chou, Adv. Mater., 2015, 27, 5113–5131 CrossRef CAS PubMed.
- G. E. Rudebusch, G. L. Espejo, J. L. Zafra, M. Peńa-Alvarez, S. N. Spisak, K. Fukuda, Z. Wei, M. Nakano, M. A. Petrukhina, J. Casado and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 12648–12654 CrossRef CAS PubMed.
- S. Nobusue, H. Miyoshi, A. Shimizu, I. Hisaki, K. Fukuda, M. Nakano and Y. Tobe, Angew. Chem., Int. Ed., 2015, 54, 2090–2094 CrossRef CAS PubMed.
- W. B. Fellows, A. M. Rice, D. E. Williams, E. A. Dolgopolova, A. K. Vannucci, P. J. Pellechia, M. D. Smith, J. A. Krause and N. B. Shustova, Angew. Chem., Int. Ed., 2016, 55, 2195–2199 CrossRef CAS PubMed.
- (a) V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868–4884 CrossRef CAS PubMed; (b) Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed; (c) A. Sygula, Eur. J. Org. Chem., 2011, 1611–1625 CrossRef CAS; (d) Y.-T. Wu, T.-C. Wu, M.-K. Chen and H.-J. Hsin, Pure Appl. Chem., 2014, 86, 539–544 CAS.
- A. Ayalon, M. Rabinovitz, P.-C. Cheng and L. T. Scott, Angew. Chem., Int. Ed. Engl., 1992, 31, 1636–1637 CrossRef.
- (a) S. N. Spisak, A. V. Zabula, A. S. Filatov, A. Yu. Rogachev and M. A. Petrukhina, Angew. Chem., Int. Ed., 2011, 50, 8090–8094 CrossRef CAS PubMed; (b) A. V. Zabula, S. N. Spisak, A. S. Filatov, V. M. Grigoryants and M. A. Petrukhina, Chem.–Eur. J., 2012, 18, 6476–6484 CrossRef CAS PubMed; (c) S. N. Spisak, N. J. Sumner, A. V. Zabula, A. S. Filatov, A. Yu. Rogachev and M. A. Petrukhina, Organometallics, 2013, 32, 3773–3779 CrossRef CAS.
- A. V. Zabula, S. N. Spisak, A. S. Filatov, A. Yu. Rogachev, R. Clérac and M. A. Petrukhina, Chem. Sci., 2016, 7, 1954–1961 RSC.
- (a) A. V. Zabula, A. S. Filatov, S. N. Spisak, A. Yu. Rogachev and M. A. Petrukhina, Science, 2011, 333, 1008–1011 CrossRef CAS PubMed; (b) A. V. Zabula, S. N. Spisak, A. S. Filatov and M. A. Petrukhina, Organometallics, 2012, 31, 5541–5545 CrossRef CAS; (c) A. V. Zabula, S. N. Spisak, A. S. Filatov and M. A. Petrukhina, Angew. Chem., Int. Ed., 2012, 51, 12194–12198 CrossRef CAS PubMed.
- (a) A. S. Filatov, A. V. Zabula, S. N. Spisak, A. Yu. Rogachev and M. A. Petrukhina, Angew. Chem., Int. Ed., 2014, 53, 140–145 CrossRef CAS PubMed; (b) A. S. Filatov, S. N. Spisak, A. V. Zabula, J. McNeely, A. Yu. Rogachev and M. A. Petrukhina, Chem. Sci., 2015, 6, 1959–1966 RSC.
- S. N. Spisak, A. V. Zabula, M. V. Ferguson, A. S. Filatov and M. A. Petrukhina, Organometallics, 2013, 32, 538–543 CrossRef CAS.
- (a) M. Baumgarten, J. L. Gherghel, M. Wagner, A. Weitz, M. Rabinovitz, P.-C. Cheng and L. T. Scott, J. Am. Chem. Soc., 1995, 117, 6254–6257 CrossRef CAS; (b) G. Zilber, V. Rozenshtein, P.-C. Cheng, L. T. Scott, M. Rabinovitz and H. Levanon, J. Am. Chem. Soc., 1995, 117, 10720–10725 CrossRef CAS.
- Handbook of Chemistry and Physics, ed. D. R. Lide, Chemical Rubber Co., Cleveland, 1972 Search PubMed.
- R. J. Collin and B. C. Smith, Dalton Trans., 2005, 702–705 RSC.
- S. N. Spisak, A. V. Zabula, A. S. Filatov and M. A. Petrukhina, J. Organomet. Chem., 2015, 784, 69–74 CrossRef CAS.
- M. A. Petrukhina, K. W. Andreini, J. Mack and L. T. Scott, J. Org. Chem., 2005, 70, 5713–5716 CrossRef CAS PubMed.
- K. Yamaguchi, Y. Takahara and T. Fueno, in Applied Quantum Chemistry, ed. V. H. Smith, Reidel, Dordrecht, 1986, p. 155 Search PubMed.
- F. Neese, ORCA, University of Bonn, Bonn, Germany, 2009 Search PubMed.
- A. A. Granovsky, J. Chem. Phys., 2011, 134, 214113 CrossRef PubMed.
- C. Bruno, R. Benassi, A. Passalacqua, F. Paolucci, C. Fontanesi, M. Marcaccio, E. A. Jackson and L. T. Scott, J. Phys. Chem. B, 2009, 113, 1954–1962 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of preparation, characterization, X-ray diffraction study, and theoretical calculations. CCDC 1516728. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05370j |
| This journal is © The Royal Society of Chemistry 2017 |