
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insights on the Pd-catalyzed addition of C–X bonds across alkynes – a combined experimental and computational study†
Theresa
Sperger
 a,
Christine M.
Le
b,
Mark
Lautens
*b and
Franziska
Schoenebeck
*a
a,
Christine M.
Le
b,
Mark
Lautens
*b and
Franziska
Schoenebeck
*a
aRWTH Aachen University, Institute of Organic Chemistry, Landoltweg 1, 52074 Aachen, Germany. E-mail: franziska.schoenebeck@rwth-aachen.de
bUniversity of Toronto, Davenport Laboratories, Department of Chemistry, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: mlautens@chem.utoronto.ca
First published on 27th January 2017
Abstract
The Pd-catalyzed intramolecular addition of carbamoyl chlorides and aryl halides across alkynes is investigated by means of DFT calculations and mechanistic test experiments. The data suggest a mechanistic pathway that involves oxidative addition, alkyne insertion, cis → trans isomerization and reductive elimination. Our data indicate that oxidative addition is the reactivity limiting step in the addition of aryl chlorides and bromides across alkynes. However, for the corresponding addition of carbamoyl chlorides, alkyne insertion is found to be limiting. Full energetic reaction pathways for the intramolecular additions across alkynes are presented herein and the role of ligands, alkyne substituents and tether moieties are discussed. Notably, the calculations could rationalize a pronounced effect of the alkyne substituent, which accounts for the exceptional reactivity of TIPS-substituted alkynes. In particular, the bulky silyl moiety is shown to significantly destabilize the formed Pd(II)-intermediates, thus facilitating both cis → trans isomerization and reductive elimination, which overall results in a flatter energetic landscape and a therefore increased catalytic efficiency.
Introduction
Methylene oxindoles are a highly relevant motif in many biologically active molecules and natural products.1–8 Despite their use in medicinal chemistry and natural product synthesis, methods to access methylene oxindoles in a highly stereoselective manner are limited.9–15 In this context, Lautens and co-workers have developed the synthesis of methylene oxindoles via Pd(0)-catalyzed carbohalogenation of alkynes, which involves an unusual and remarkable reductive elimination of C(sp2)–X (X = I, Br, Cl) from Pd(II) as key step.16While oxidative additions of aryl and vinyl halides to Pd(0) are widely studied and a relatively well understood step in Pd-catalysis, the corresponding back reaction, i.e. the reductive elimination of C–X bonds from Pd(II) has had significantly less precedence.17 A reason for this is that reductive elimination from Pd(II) is generally slow and disfavored. Therefore, chemists have turned to alternative protocols, e.g. oxidatively accessing the reductive elimination from higher oxidation state Pd intermediates, such as Pd(III) and Pd(IV).18–27 However, C–X bond formation via reductive elimination from Pd(II) may become feasible under sterically demanding conditions (bulky ligands and substituents) and if potential side-reactions are suppressed (substrate control, e.g. avoiding the β-H elimination).28–32 Advances in substrate design as well as catalyst development by the Lautens group recently allowed for the reductive elimination of C(sp2)–X (X = I, Br, Cl) from Pd(II) in the carbohalogenation of alkynes (Scheme 1).16 Interestingly, the synthetic protocol was compatible with ether, alkyl and amine tethers (Scheme 1, 1), but the use of an amide tether (4) required an alternative synthetic route via the chlorocarbamoylation of alkynes.15 This complementary synthesis utilizes the reactivity of carbamoyl chlorides, a relatively underexplored class of substrates. In contrast to the original synthetic method which employs aryl halides (1) in combination with bulky phosphines, such as Q-Phos and PtBu3, the use of carbamoyl chlorides (3) required a less bulky aryl phosphaadamantane ligand (PA-Ph = 1,3,5,7-tetramethyl-2,4,8-trioxa-6-phenyl-6-phosphaadamantane) in order to reach good conversions. Furthermore, the chlorocarbamoylation reaction (Scheme 1b) exclusively allows for a bulky tri-iso-propylsilyl (TIPS) alkyne substituent, but is not effective using less bulky silyls or bulky aryl moieties (such as Mes = mesityl). This is in contrast to the carbohalogenation reaction (Scheme 1a) that displays good yields for a number of aryl halides with either bulky silyl or aryl alkyne substituents (e.g. TIPS, TBS = tert-butyldimethylsilyl, Mes, 1-naphthyl, 9-anthracenyl).
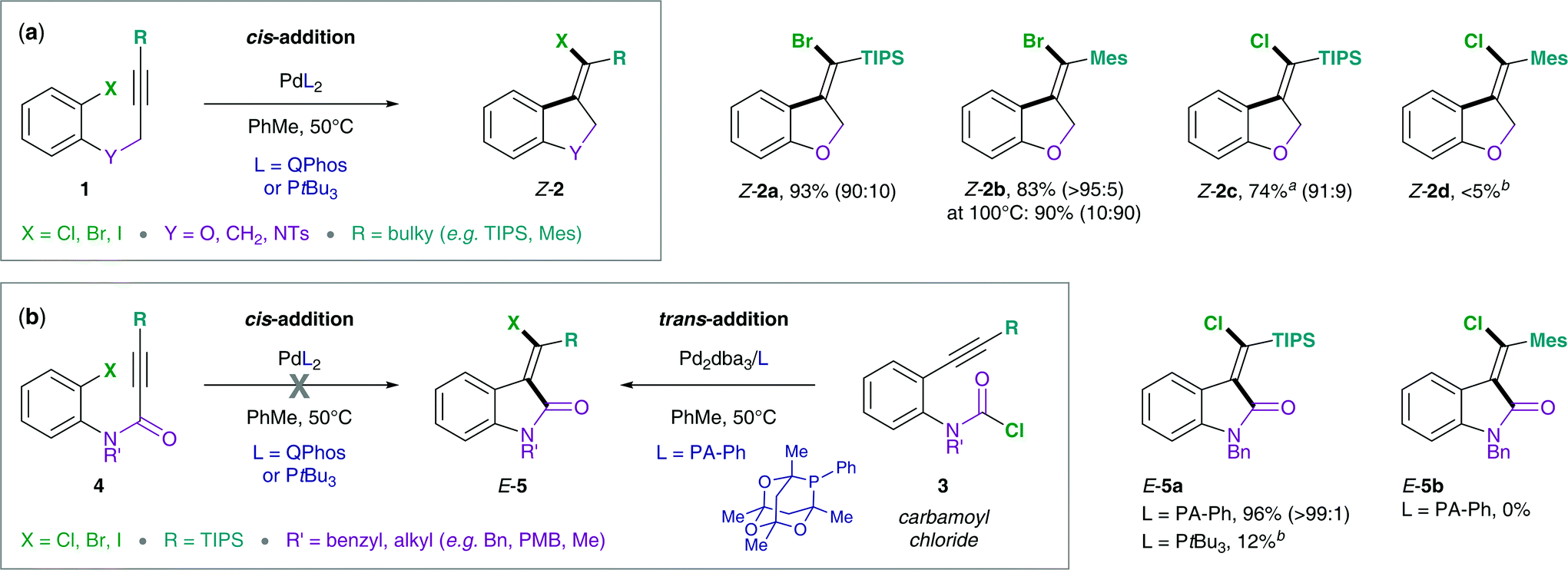 | ||
| Scheme 1 Experimental data by Lautens and co-workers: (a) intramolecular addition of aryl halides (1) across alkynes,16 (b) intramolecular addition of carbamoyl chlorides (3) across alkynes.15aReaction was performed at 110 °C using 1,2,2,6,6-pentamethylpiperidine (PMP, 0.25 equiv.) as an additive.16,33bYield was determined by 1H NMR analysis of the crude reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. | ||
Herein we present a combined computational and experimental study in an effort to understand the underlying mechanism of these transformations and its implication on reactivity. More specifically, we aim to shed light on selectivity- and reactivity-controlling factors of ligand and substrate and the implicit requirements on alkyne substituent as well phosphine ligand. We hope these results will aid future substrate and catalyst design to further expand the scope of these atom-economic and synthetically relevant Pd-catalyzed intramolecular transformations.
Computational methods
DFT calculations were performed using Gaussian 09, Revision D.01.34 Geometry optimizations and frequency calculations were conducted in the gas-phase at the B3LYP/6-31G(d) level of theory, employing LANL2DZ as an ECP for Pd. All stationary points were verified as either minima or transition states. Additionally, transition states (TSs) were confirmed by following the intrinsic reaction coordinate (IRC) to the corresponding intermediates. Energies were calculated at the M06L/def2-TZVP level of theory, employing the CPCM solvation model to account for toluene as the solvent.35 All energies were converted to 1 M standard state.Results and discussion
General mechanism
The proposed mechanism of the intramolecular addition of aryl halides and carbamoyl chlorides is shown in Scheme 2. On the basis of our calculations, we propose that initial oxidative addition of either aryl halide 1 or carbamoyl chloride 3 to monophosphine Pd(0) is followed by insertion of the alkyne, i.e. cis-carbopalladation. Direct reductive elimination (for aryl halide substrates 1) or rapid cis → trans isomerization and successive reductive elimination (in the case of carbamoyl chloride substrates 3) then yields the observed methylene oxindole products Z-2 and E-5, respectively.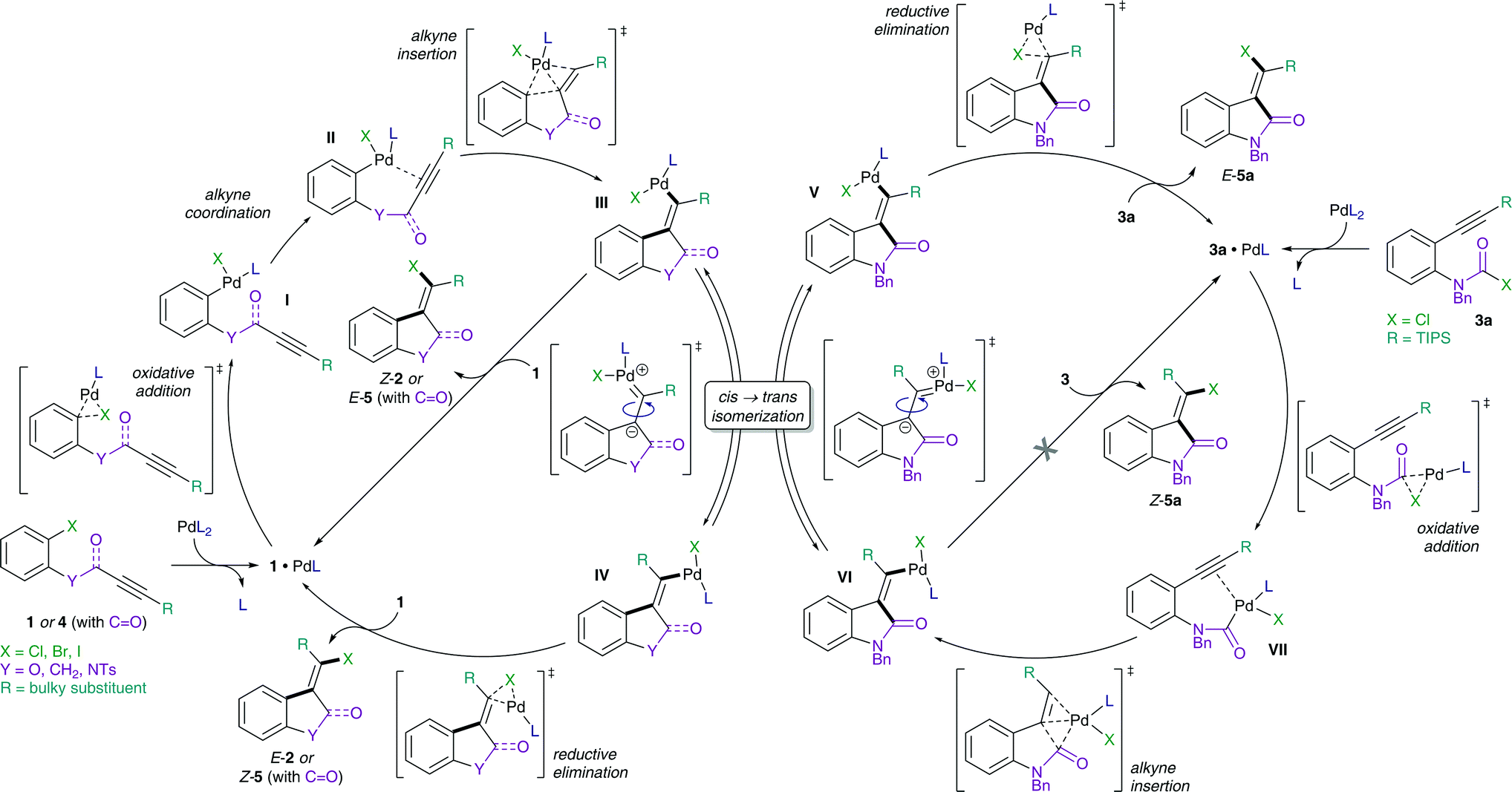 | ||
| Scheme 2 Proposed mechanisms for the addition of aryl halides (1 and 4, left) and carbamoyl chlorides (3, right) across alkynes. | ||
However, while oxidative additions to aryl halides have been subject to extensive computational and mechanistic studies,38–45 little is known on the nature of the transition state for reactions with carbamoyl chlorides. The high electrophilicity of these species may imply an ionic/electron transfer or formal nucleophilic substitution reaction. By means of computations it is challenging to unambiguously distinguish between these charged and neutral pathways due to the applied computational approximations.46,47
We therefore designed a test experiment and performed a competitive Suzuki cross-coupling using substrate 6, possessing both aryl chloride and carbamoyl chloride moieties (Scheme 3).48 An exclusive activation of carbamoyl chloride over aryl chloride was observed experimentally.49
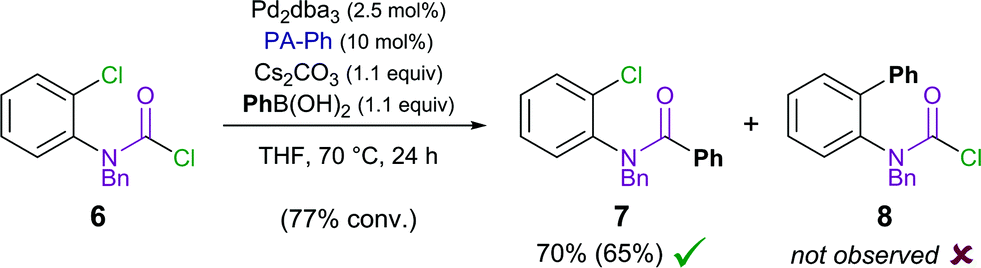 | ||
| Scheme 3 Competitive Suzuki cross-coupling of aryl versus carbamoyl chloride (isolated yield shown in parenthesis). | ||
However, despite the clear preference for oxidative addition of carbamoyl chloride over aryl chloride, the arguably fast oxidative addition step (i.e. fast compared to oxidative addition to aryl chloride) may still be the rate-determining TS of the reaction. Therefore, a resting state analysis of the reaction of 3a and 4a with catalytic Pd/PA-Ph was performed.48 Almost instantaneous conversion of carbamoyl chloride 3a with concomitant formation of new phosphine-containing species and free ligand was observed by 31P NMR. In contrast, aryl chloride 4a only yielded traces of product under the same reaction conditions and only one major P-containing species was observed, which is most likely the result of catalyst decomposition. In addition, two species (6.8 and 6.7 ppm by 31P NMR) were observed for both substrates and are likely to be cis- and trans-Pd(II) intermediates, Va and VIa for substrate 3a as well as IIIa and IVa for substrate 4a, respectively. Furthermore, in the reaction of carbamoyl chloride 3a, a species at 5.2 ppm in the 31P NMR spectrum was formed, which decreased over time and might be the oxidative addition intermediate VIIa. With this information in hand, oxidative addition of carbamoyl chloride 3a is unlikely to be the elementary step with the highest activation barrier. Instead, the observation of potential oxidative addition intermediate VIIa (species at 5.2 ppm in 31P NMR) suggests alkyne insertion, i.e. carbopalladation to be the turnover-determining transition state (TDTS). Thus, oxidative addition of carbamoyl chloride 3a is suspected to proceed via a fast, possibly ionic, nucleophilic substitution TS rather than a concerted, 3-membered oxidative addition TS.50 Based on the experimental results, we assume oxidative addition of 3a to be fast and alkyne insertion to be the TDTS for the addition of carbamoyl chlorides across alkynes. Since catalytic turnover depends on both activation barriers (ΔG‡) and driving force (i.e. reaction free energy, ΔrG) of the reaction, the free energy difference between the rate-determining intermediate (TDI) and transition state (TDTS), commonly referred to as the energetic span (δE),51,52 determines the efficiency and speed of the catalytic cycle. Therefore, in order to assess and compare the reactivities of carbamoyl and aryl chlorides in their addition reactions across alkynes, full reaction pathways and their corresponding energetic spans were calculated (Scheme 4).
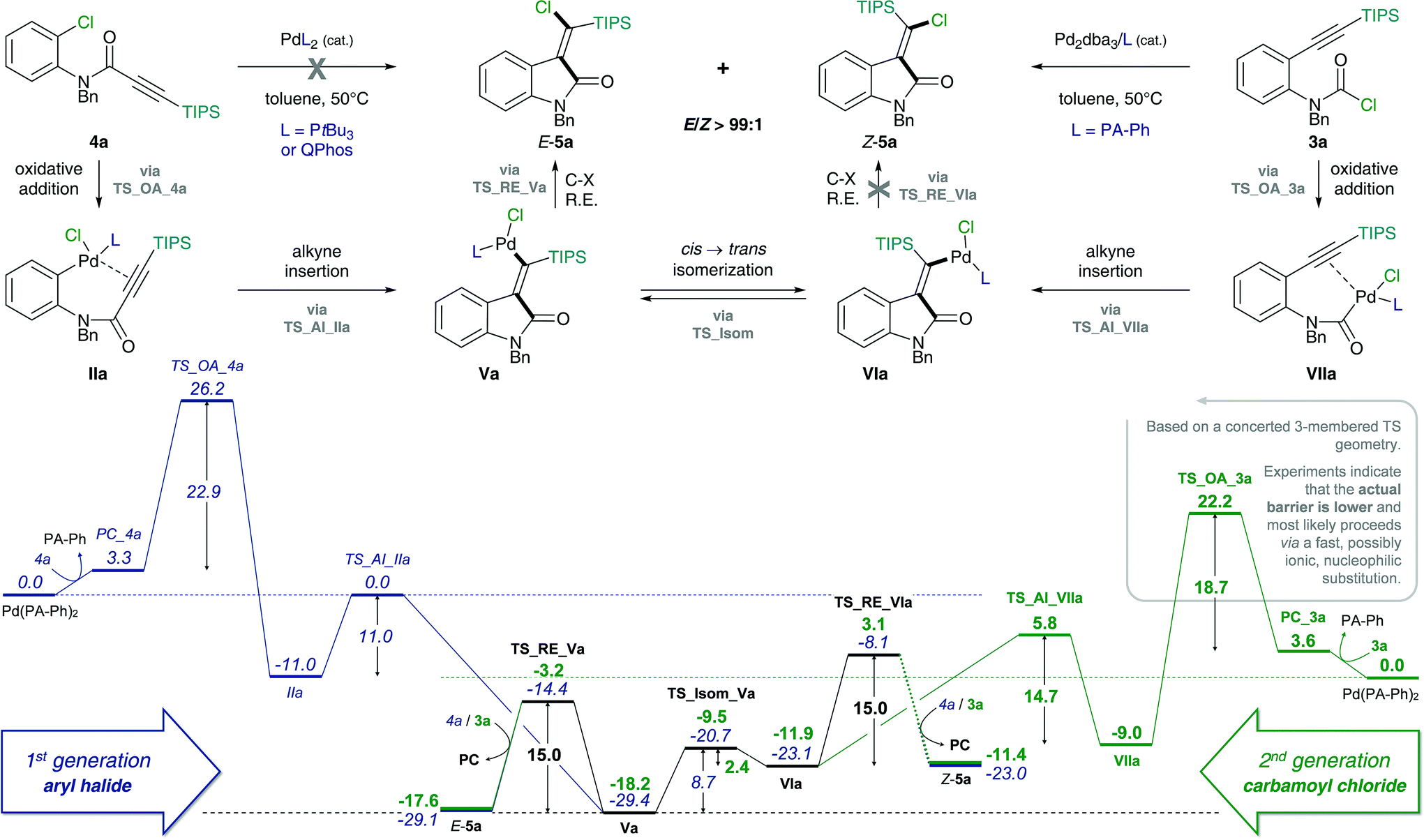 | ||
| Scheme 4 Calculated Gibbs free energy pathways of first and second generation substrates 3a and 4a possessing an amide tether. Energies (in kcal mol−1) were calculated at the CPCM (toluene) M06L/def2-TZVP//B3LYP/6-31G(d)(LANL2DZ) level of theory. Abbreviations: oxidative addition (OA), alkyne insertion (AI), cis → trans isomerization (Isom) and reductive elimination (RE). | ||
Table 1 shows the calculated energetic spans for the addition of aryl halides (TDTS = oxidative addition) and carbamoyl chlorides (TDTS = alkyne insertion) across alkynes. Further analysis of calculated Gibbs free energy pathways and energetic spans of substrates 3 and 4 revealed insights on the effects of ligand, halide and alkyne substituent on the reaction outcome – the results of which are discussed in detail in the following sections.
| Entry | Ligand | Substrate | X | R | ΔrG | δE | Yield of 5 (ref. 15) |
|---|---|---|---|---|---|---|---|
| a Energetic spans (in kcal mol−1) were calculated according to δE = G(TDTS) − G(TDI) + ΔrG (with G(TDTS) = Gibbs free energy of turnover-determining TS, G(TDI) = Gibbs free energy of turnover-determining intermediate, ΔrG = Gibbs free energy of reaction), at the CPCM (toluene) M06L/def2-TZVP//B3LYP/6-31G(d)(LANL2DZ) level of theory. b Reaction was conducted using stoichiometric amounts of Pd and was heated at 100 °C for 24 h. | |||||||
| 1 | PtBu3 | 4a | Cl | TIPS | −21.9 | 28.9 | 0% (16%)b |
| 2 | PtBu3 | 4b | Cl | Mes | −15.5 | 45.0 | — |
| 3 | PA-Ph | 4a | Cl | TIPS | −25.8 | 29.8 | — |
| 4 | PA-Ph | 3a | Cl | TIPS | −14.0 | 10.0 | 99% |
| 5 | PtBu3 | 3a | Cl | TIPS | −10.8 | 16.4 | 12% |
| 6 | PtBu3 | 3b | Cl | Mes | −2.9 | 38.9 | 0% |
Stoichiometric studies of 4a (R = TIPS) employing Pd(PtBu3)2 showed no conversion at 50 °C, but when heated at 100 °C for 24 h yielded 16% of E-5a, along with 64% of recovered starting aryl chloride 4a. This result indicates that reaction of 4a to form 5a is possible, at least in a stoichiometric manner. However, the higher reaction temperatures lead to catalyst decomposition and thus prevent a catalytic reaction.52 The observed decomposition of catalyst may be facilitated by the amide moiety. Hence, possible deactivation/side reaction pathways have been investigated computationally (Fig. 1). More specifically, transition states for the oxidative insertion of Pd(PA-Ph) to potentially activated bonds of substrates 3a and 4a were calculated and the corresponding activation barriers compared. In the case of the second generation carbamoyl chloride substrate 3a, all bond activations are of higher barrier than oxidative addition via a concerted, 3-membered TS. Notably, this is despite the fact that experiments indicate an even lower barrier for oxidative addition to the carbamoyl chloride via a rapid, possibly ionic, nucleophilic substitution (vide supra). In contrast, for the first generation aryl chloride substrate 4a, which did not react under analogous reaction conditions, calculations suggest that an activation of the C–Si bond of the alkyne is competing with oxidative addition to the C–Cl bond. This observation is most likely due to an increase in reactivity of the C–Si bond as a result of the conjugation of the alkyne with the amide.
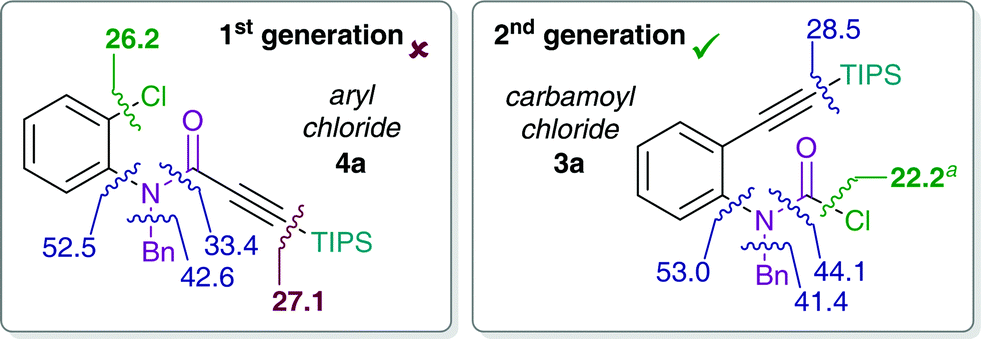 | ||
| Fig. 1 Activation free energies for insertion of Pd(PA-Ph) to potentially activated bonds of substrates 4a (left) and 3a (right). Values (in kcal mol−1) refer to Gibbs free energies calculated at the CPCM (toluene) M06L/def2-TZVP//B3LYP/6-31G(d)(LANL2DZ) level of theory. aCalculated barrier for oxidative addition of carbamoyl chloride via a concerted 3-membered TS. Experiments however indicate that the actual barrier is lower and most likely proceeds via a fast, possibly ionic, nucleophilic substitution. | ||
This activation of the alkyne substituent is not present for all other tether moieties (amine, ether, alkyl), therefore providing a potential explanation for the observed difference in reactivity.
 | ||
| Scheme 5 Ligand effect on alkyne chlorocarbamoylation of 3a: calculated Gibbs free energy pathways employing L = PtBu3 (blue, italics) and L = PA-Ph (green, bold). Energies (in kcal mol−1) were calculated at the CPCM (toluene) M06L/def2-TZVP//B3LYP/6-31G(d)(LANL2DZ) level of theory. | ||
Silyl effect
Experimentally, only substrates bearing a silyl-substituent on the alkyne were reactive in the chlorocarbamoylation reaction, whereas substrates with a mesityl-substituted alkyne did not cyclize under analogous reaction conditions. Next, we investigated the role of the alkyne substituent computationally. A significant increase in energetic span for mesityl-substituted substrates 4b and 3b (Table 1, entries 2 and 6, respectively) was observed compared to substrates 4a and 3a bearing a TIPS-substituent (entries 1 and 5, respectively). When comparing the energetic pathways for substrates 3a (R = TIPS) and 3b (R = Mes), an effect of the TIPS-moiety was observed on (i) cis → trans isomerization and (ii) reductive elimination (Scheme 6). More specifically, the TIPS-group leads to a significant destabilization of Pd(II) intermediates VI and V, along with a stabilization of the transition state for cis → trans isomerization, which causes a substantial decrease in the barrier of isomerization and overall results in a flatter energetic pathway and thus a smaller energetic span.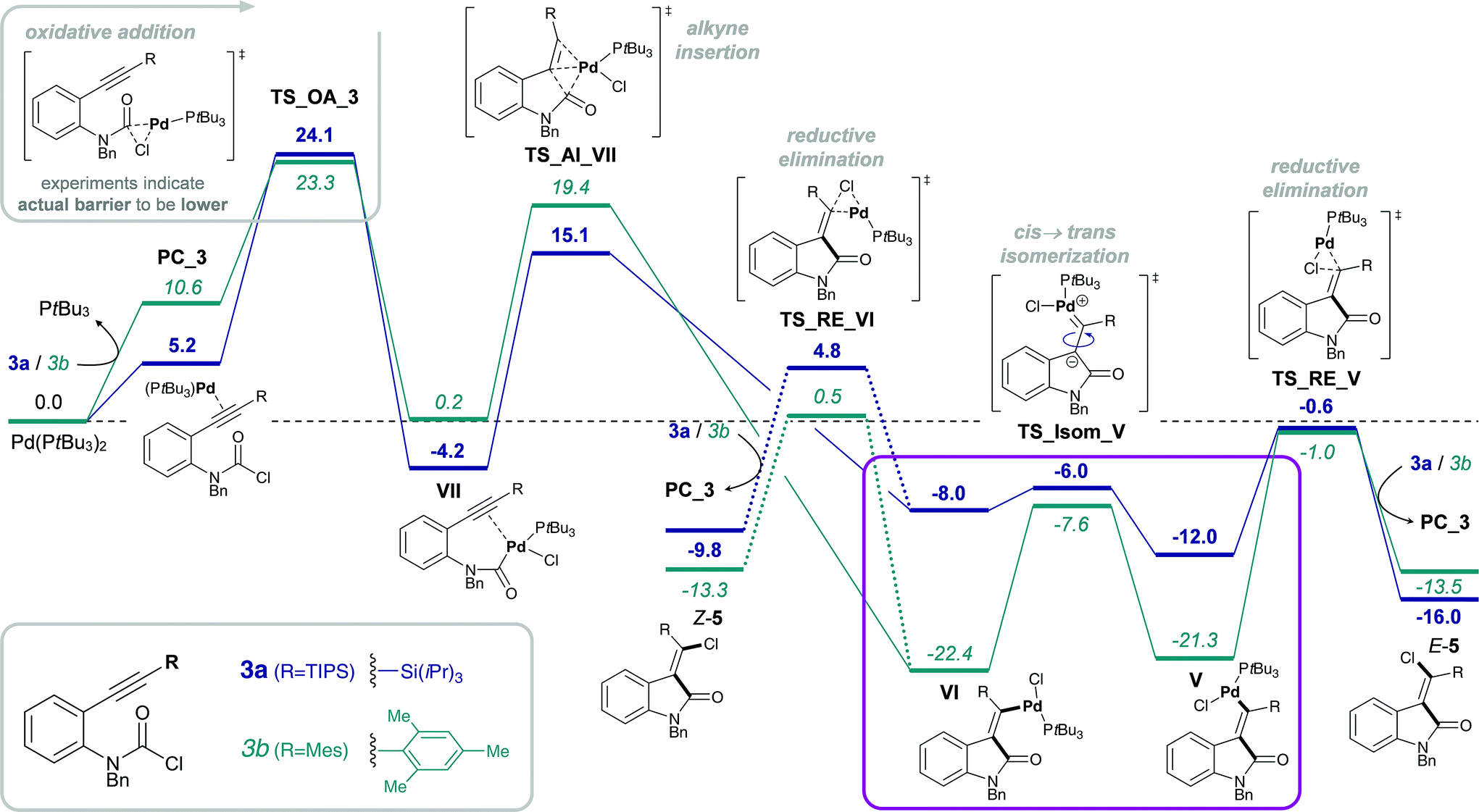 | ||
| Scheme 6 Effect of silyl substituent on the alkyne: calculated Gibbs free energy pathways of 3a (R = TIPS, blue, bold) and 3b (R = Mes, teal, italics) at the CPCM (toluene) M06L/def2-TZVP//B3LYP/6-31G(d)(LANL2DZ) level of theory (energies are given in kcal mol−1). | ||
 | ||
| Fig. 2 Effect of alkyne substituent on Pd(II) intermediates, VI and V, and cis → trans isomerization: analysis of charges, bond lengths and bond orders. | ||
This result is in agreement with the known effect of silyl groups to be able to stabilize carbocations in α,59 β60,61 and γ62 positions. In addition, analyzing the changes in bond lengths during the isomerization from cis- to trans-Pd(II) intermediate (VIa to Va) shows a slight elongation of the double bond and concomitant shortening of both C–Si and C–Pd bonds in the TS, suggesting a delocalization of positive charge between Pd, C and Si.48 This corresponds with the calculated Mayer bond orders,63,64 which indicate a decrease in bond order of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond during the TS for both R = TIPS and R = Mes, although more pronounced for the latter. At the same time, the C–R (R = TIPS or Mes) bond order increases during isomerization (indicating delocalization of charge into the R-substituent). This increase is only minor for the silyl moiety (increase of 8%), but more evident for mesityl (increase of 20%) as expected due to the bigger required changes in order to reach the TS.48
C double bond during the TS for both R = TIPS and R = Mes, although more pronounced for the latter. At the same time, the C–R (R = TIPS or Mes) bond order increases during isomerization (indicating delocalization of charge into the R-substituent). This increase is only minor for the silyl moiety (increase of 8%), but more evident for mesityl (increase of 20%) as expected due to the bigger required changes in order to reach the TS.48
Overall, the silyl moiety exerts a combination of effects on several intermediates and steps in the chlorocarbamoylation reaction, giving rise to its unique reactivity.
Origins of observed divergence in selectivity
In the reaction of carbamoyl chlorides an exclusive E-selectivity is reached by means of a rapid cis → trans isomerization (see Scheme 1b). However, in the corresponding reaction of aryl halides medium to good levels of Z-selectivity are observed as a result of direct reductive elimination. In the case of mesityl substituted alkynes, a switch in selectivity (i.e. to obtain E-2b) can be reached at elevated reaction temperatures (see Scheme 1a). Additionally, the substrate scope is not limited to TIPS-substituted alkynes, tolerating other bulky groups, such as aryls for example.Analysis of the calculated free energy pathways revealed a pronounced effect of the alkyne substituent on the reactivity profile (Scheme 7). Analogous to reactions of the amide-tethered substrates 3 and 4 (Schemes 4–6), a TIPS-substituent favors the cis-Pd(II) intermediate IIIa, but the corresponding trans-intermediate IV is more stable when possessing a mesityl-substituent (IVb). In addition, the barrier towards isomerization is significantly lower for R = TIPS compared to R = Mes, i.e. ΔG‡ = 5.8 and 13.1 kcal mol−1 for substrates 1a (R = TIPS) and 1b (R = Mes), respectively. Moreover, the stability of the pre-complexes PC_1, where the active monophosphine Pd(0) (PtBu3) species coordinates to the alkyne group of substrate 1, is more pronounced when R = TIPS, indicating that the silyl group increases the electrophilic character of the alkyne moiety. This would suggest that the reductive elimination from the vinyl Pd(II) intermediates, III and IV, is reversible for substrates 1b and 1d bearing a mesityl-substituent. In combination with the trans-Pd(II) intermediate IV being more stable than the cis-intermediate III, a reversal of the observed Z-selectivity for R = Mes (at 50 °C) can be reached at elevated reaction temperatures (at 100 °C).16,65
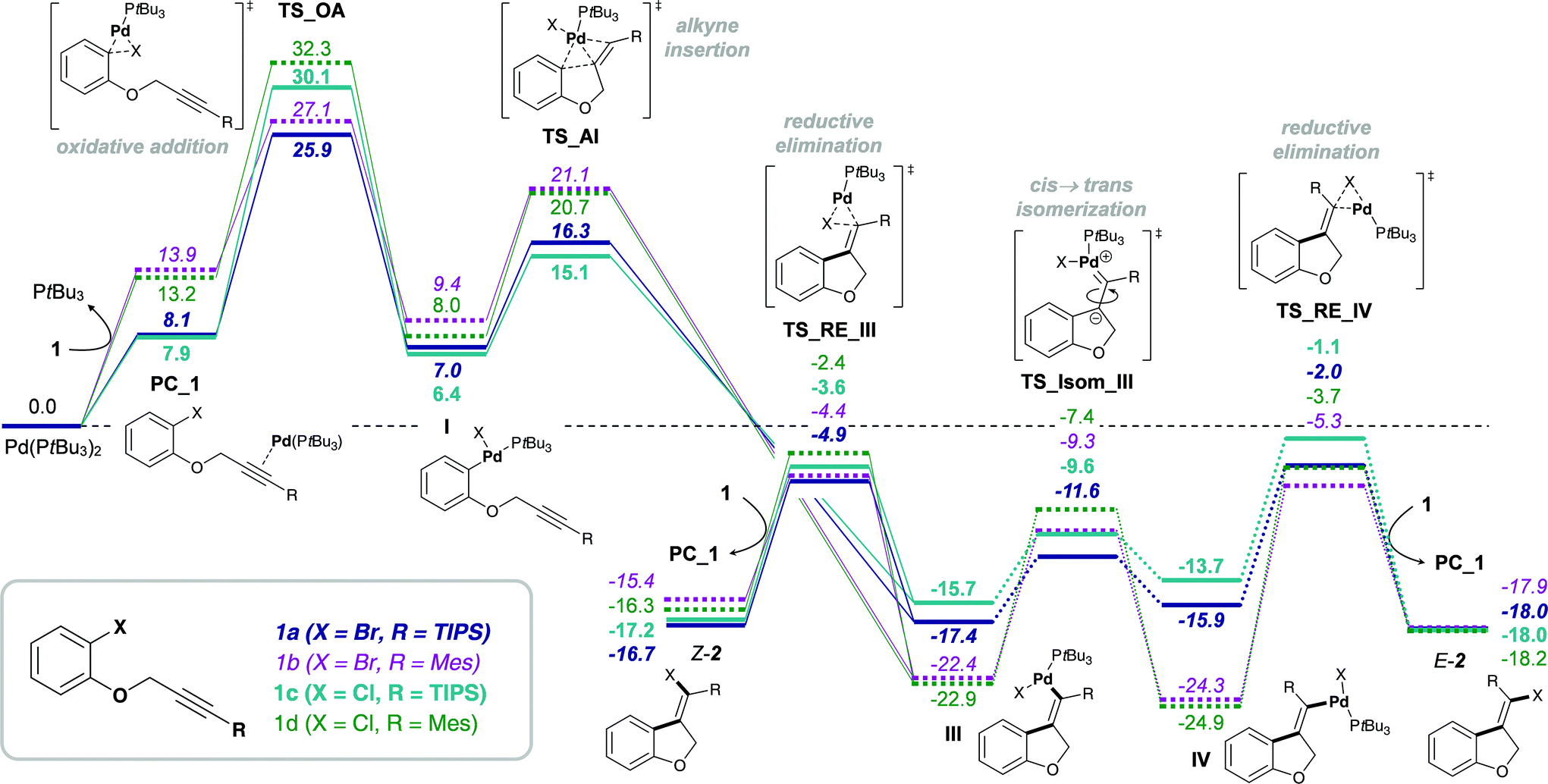 | ||
| Scheme 7 Gibbs free energy pathway of the Pd(PtBu3)2-mediated alkyne carbohalogenation of 1a–d, calculated at the CPCM (toluene) M06L/def2-TZVP//B3LYP/6-31G(d)(LANL2DZ) level of theory (values are given in kcal mol−1). | ||
Conclusions
The mechanisms of alkyne carbohalogenation and chlorocarbamoylation have been investigated by means of DFT calculations and experiments. Catalytic pathways involving oxidative addition, alkyne insertion, cis → trans isomerization and reductive elimination are proposed. Oxidative addition is suggested to be reactivity limiting in the case of addition of aryl halides across alkynes: in the corresponding reaction of carbamoyl chlorides, oxidative addition was however shown to be fast and our data indicated that instead alkyne insertion, i.e. carbopalladation was reactivity limiting. Furthermore, the effects of halide, phosphine ligand and alkyne substituent on reactivity were investigated.While the bulky PtBu3 was vital for reactivity in the intramolecular addition of aryl halides across alkynes due to a lowering of the barriers for reductive elimination, the less bulky phosphaadamantane ligand PA-Ph is uniquely suited for the corresponding addition reaction of carbamoyl chlorides. Calculations indicate that this is due to a significant decrease in the barrier for the reactivity limiting alkyne insertion with the less bulky PA-Ph ligand compared to PtBu3. Notably, a pronounced effect of the alkyne substituent on reactivity was unravelled, which accounts for the exceptional reactivity of substrates bearing a TIPS-substituent. More specifically, the bulky TIPS-group was shown to cause a significant destabilization of Pd(II) intermediates VI and V, along with a stabilization of the cis → trans isomerization TS. This overall results in a smaller energetic span and thus significantly increases catalytic turnover.
Acknowledgements
F. S. and T. S. wish to thank the RWTH Aachen, MIWF NRW and the Evonik Foundation (doctoral scholarship to T. S.) for financial support and greatly appreciate the granted computing time on the RWTH Bull Cluster (grant number jara0091). M. L. thanks NSERC, the University of Toronto and Alphora Inc. for funding. C. M. L. thanks NSERC for a postgraduate scholarship.Notes and references
- S. T. Davis, S. H. Dickerson, S. V. Frye, P. A. Harris, R. N. Hunter, L. F. Kuyper, K. E. Lackey, M. J. Luzzio, J. M. Veal and D. H. Walker, Patent WO9915500, 1999.
- A. Heckel, G. J. Roth, J. Kley, S. Hoerer and I. Uphues, Patent WO2005087727 A1, 2005.
- S. Hauf, R. W. Cole, S. LaTerra, C. Zimmer, G. Schnapp, R. Walter, A. Heckel, J. van Meel, C. L. Rieder and J.-M. Peters, J. Cell Biol., 2003, 161, 281–294 CrossRef CAS PubMed.
- C. A. Grandinetti and B. R. Goldspiel, Pharmacotherapy, 2007, 27, 1125–1144 CrossRef CAS PubMed.
- F. Hilberg, G. J. Roth, M. Krssak, S. Kautschitsch, W. Sommergruber, U. Tontsch-Grunt, P. Garin-Chesa, G. Bader, A. Zoephel, J. Quant, A. Heckel and W. J. Rettig, Cancer Res., 2008, 68, 4774–4782 CrossRef CAS PubMed.
- B. M. Trost, N. Cramer and H. Bernsmann, J. Am. Chem. Soc., 2007, 129, 3086–3087 CrossRef CAS PubMed.
- B. M. Trost, N. Cramer and S. M. Silverman, J. Am. Chem. Soc., 2007, 129, 12396–12397 CrossRef CAS PubMed.
- S. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 1967–1970 CrossRef CAS PubMed.
- G. Cantagrel, B. de Carné-Carnavalet, C. Meyer and J. Cossy, Org. Lett., 2009, 11, 4262–4265 CrossRef CAS PubMed.
- S. Tang, Q.-F. Yu, P. Peng, J.-H. Li, P. Zhong and R.-Y. Tang, Org. Lett., 2007, 9, 3413–3416 CrossRef CAS PubMed.
- M. Sassatelli, É. Debiton, B. Aboab, M. Prudhomme and P. Moreau, Eur. J. Med. Chem., 2006, 41, 709–716 CrossRef CAS PubMed.
- M. S. C. Pedras, J. L. Sorensen, F. I. Okanga and I. L. Zaharia, Bioorg. Med. Chem. Lett., 1999, 9, 3015–3020 CrossRef CAS PubMed.
- E. M. Beccalli and A. Marchesini, Tetrahedron, 1995, 51, 2353–2362 CrossRef CAS.
- E. M. Beccalli, A. Marchesini and T. Pilati, Tetrahedron, 1994, 50, 12697–12712 CrossRef CAS.
- C. M. Le, X. Hou, T. Sperger, F. Schoenebeck and M. Lautens, Angew. Chem., Int. Ed., 2015, 54, 15897–15900 CrossRef CAS PubMed.
- C. M. Le, P. J. C. Menzies, D. A. Petrone and M. Lautens, Angew. Chem., Int. Ed., 2015, 54, 254–257 CrossRef CAS PubMed.
- X. Jiang, H. Liu and Z. Gu, Asian J. Org. Chem., 2012, 1, 16–24 CrossRef CAS.
- Higher Oxidation State Organopalladium and Platinum Chemistry, ed. A. J. Canty, Springer-Verlag, Berlin Heidelberg, 2011 Search PubMed.
- A. J. Canty, Acc. Chem. Res., 1992, 25, 83–90 CrossRef CAS.
- A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177–185 CrossRef CAS PubMed.
- K. M. Engle, T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478–1491 CrossRef CAS PubMed.
- D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302–309 CrossRef CAS PubMed.
- D. C. Powers, D. Benitez, E. Tkatchouk, W. a. Goddard and T. Ritter, J. Am. Chem. Soc., 2010, 132, 14092–14103 CrossRef CAS PubMed.
- D. C. Powers, E. Lee, A. Ariafard, M. S. Sanford, B. F. Yates, A. J. Canty and T. Ritter, J. Am. Chem. Soc., 2012, 134, 12002–12009 CrossRef CAS PubMed.
- D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Org. Lett., 2006, 8, 2523–2526 CrossRef CAS PubMed.
- K. J. Stowers and M. S. Sanford, Org. Lett., 2009, 11, 4584–4587 CrossRef CAS PubMed.
- P. L. Arnold, M. S. Sanford and S. M. Pearson, J. Am. Chem. Soc., 2009, 131, 13912–13913 CrossRef CAS PubMed.
- A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 1232–1233 CrossRef CAS PubMed.
- A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 13944–13945 CrossRef CAS PubMed.
- A. H. Roy and J. F. Hartwig, Organometallics, 2004, 23, 1533–1541 CrossRef CAS.
- D. A. Watson, M. Su, G. Teverovskiy, Y. Zhang, J. García-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661–1664 CrossRef CAS PubMed.
- X. Shen, A. M. Hyde and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14076–14078 CrossRef CAS PubMed.
- D. A. Petrone, H. Yoon, H. Weinstabl and M. Lautens, Angew. Chem., Int. Ed., 2014, 53, 7908–7912 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- For appropriateness of computational method, see: T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532–9586 CrossRef CAS PubMed.
- Y. Lan, P. Liu, S. G. Newman, M. Lautens and K. N. Houk, Chem. Sci., 2012, 3, 1987–1995 RSC.
- S. G. Newman and M. Lautens, J. Am. Chem. Soc., 2011, 133, 1778–1780 CrossRef CAS PubMed.
- C. L. McMullin, N. Fey and J. N. Harvey, Dalton Trans., 2014, 43, 13545–13556 RSC.
- C. L. McMullin, J. Jover, J. N. Harvey and N. Fey, Dalton Trans., 2010, 39, 10833–10836 RSC.
- K. Vikse, T. Naka, J. S. McIndoe, M. Besora and F. Maseras, ChemCatChem, 2013, 5, 3604–3609 CrossRef CAS.
- M. Besora, C. Gourlaouen, B. Yates and F. Maseras, Dalton Trans., 2011, 40, 11089–11094 RSC.
- F. Barrios-Landeros, B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 8141–8154 CrossRef CAS PubMed.
- M. Ahlquist and P.-O. Norrby, Organometallics, 2007, 26, 550–553 CrossRef CAS.
- M. Ahlquist, P. Fristrup, D. Tanner and P.-O. Norrby, Organometallics, 2006, 25, 2066–2073 CrossRef CAS.
- H. M. Senn and T. Ziegler, Organometallics, 2004, 23, 2980–2988 CrossRef CAS.
- T. Sperger, H. C. Fisher and F. Schoenebeck, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 226–242 CrossRef CAS.
- F. Proutière and F. Schoenebeck, Angew. Chem., Int. Ed., 2011, 50, 8192–8195 CrossRef PubMed.
- Please see ESI† for further details.
- Calculations are in line with the experiment and predict a difference in activation barriers of oxidative addition of ΔΔG‡ = 8.3 kcal mol−1 in favor of carbamoyl chloride activation.
- An ionic, nucleophilic addition TS could not be located owing to the difficulties associated with the description of charges, which represent a well-known challenge. For further information, please refer to ref. 46.
- C. Amatore and A. Jutand, J. Organomet. Chem., 1999, 576, 254–278 CrossRef CAS.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- Notably, catalyst decomposition was observed via31P NMR, no Pd(PtBu3)2 or free PtBu3 were remaining after the reaction.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1985, 83, 1736–1740 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, New York, 1990 Search PubMed.
- A. Kostenko, B. Müller, F.-P. Kaufmann, Y. Apeloig and H.-U. Siehl, Eur. J. Org. Chem., 2012, 1730–1736 CrossRef CAS.
- K. C. Sproul and W. A. Chalifoux, Org. Lett., 2015, 17, 3334–3337 CrossRef CAS PubMed.
- T. Nokami, Y. Yamane, S. Oshitani, J.-k. Kobayashi, S.-i. Matsui, T. Nishihara, H. Uno, S. Hayase and T. Itoh, Org. Lett., 2015, 17, 3182–3185 CrossRef CAS PubMed.
- X. Creary and E. D. Kochly, J. Org. Chem., 2009, 74, 9044–9053 CrossRef CAS PubMed.
- I. Mayer, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS.
- I. Mayer, Chem. Phys. Lett., 2012, 544, 83–86 CrossRef CAS.
- Observed
selectivities for substrate 1b (X = Br, R = Mes), determined by 1H NMR analysis of the crude reaction mixture using 1,3,5-trimethoxybenzene as an internal standard after reaction for 18 h in toluene (0.1 M) employing 5 mol% Pd(QPhos)2: Z/E > 95
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 at 50 °C and 10:90 at 100 °C.
:5 at 50 °C and 10:90 at 100 °C.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, cartesian coordinates of calculated species, experimental procedures, and spectroscopic data are given. See DOI: 10.1039/c6sc05001h |
| This journal is © The Royal Society of Chemistry 2017 |