
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular assembly of platinum-containing polyhedral oligomeric silsesquioxanes: an interplay of intermolecular interactions and a correlation between structural modifications and morphological transformations†
Ho-Leung
Au-Yeung
,
Anthony Yiu-Yan
Tam
,
Sammual Yu-Lut
Leung
and
Vivian Wing-Wah
Yam
*
Institute of Molecular Functional Materials (Areas of Excellence Scheme, University Grants Committee (Hong Kong)) and Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: wwyam@hku.hk
First published on 16th December 2016
Abstract
A series of alkynylplatinum(II) terpyridine complexes functionalized with polyhedral oligomeric silsesquioxane (POSS) moieties has been demonstrated to exhibit drastic color changes and give various distinctive nanostructures with interesting multi-stage morphological transformations from spheres to nanoplates in response to solvent conditions through the interplay of various intermolecular interactions, including hydrophilic–hydrophilic, hydrophobic–hydrophobic, Pt⋯Pt and π–π stacking interactions. These supramolecular architectures can be systematically modified and controlled through the molecular design and the variation of solvent compositions. In particular, drastic changes in color in response to solvent polarity were observed through the incorporation of the charged moieties, representing a new class of potential candidates for functional materials with sensing or imaging capabilities. This class of complexes has been studied by 1H NMR spectroscopy, electron microscopy, UV-vis absorption and emission spectroscopy.
Introduction
Square-planar platinum(II) polypyridyl complexes of d8 electronic configuration have been extensively investigated owing to their interesting photophysical properties.1–10 Their propensity in formation of metal–metal and π–π stacking interactions4–10 has been exploited to give drastic spectroscopic changes and luminescence enhancement in various molecular association processes.5–7 The formation of various distinctive supramolecular architectures8 and metallogels9 has been demonstrated to be driven by Pt⋯Pt and π–π stacking interactions, illustrating the importance of Pt⋯Pt and π–π stacking interactions in governing and contributing to the self-association process. This formation of supramolecular architectures through the formation of Pt⋯Pt and π–π stacking interactions is usually associated with obvious color changes.5,8–10 In particular, Yam and coworkers have reported the morphological transformation of amphiphilic platinum(II) bzimpy complexes5b in response to varying solvent polarity, with associated drastic color changes, illustrating the interplay of Pt⋯Pt interactions and hydrophilic–hydrophilic interactions in governing the aggregation–deaggregation processes. Such drastic color changes upon the formation of aggregates and in response to various external stimuli make them potential candidates in monitoring the self-association process, providing insights into the exploration of supramolecular assembly.Molecular approaches towards nanochemistry have reached a high level of sophistication. The development and design of molecular precursors that are capable of undergoing self-assembly have aroused much attention. They provide access to highly controllable and processable nanoscaled superstructures with various morphologies that can commonly be employed in various research fields.11,12a,b In particular, the exploitation of organosilanes towards the preparation of mesomaterials and porous silica has been extensively explored.12,13 Among them, polyhedral oligomeric silsesquioxane (POSS) is the smallest silica nanoparticle with a rigid cage-like conformation surrounded by multiple organic substituents,14 which determine the overall reactivity, solubility and thermal stabilities.15 These peripheral groups are readily tunable, providing various specific molecular interactions among the POSS molecules, which facilitate the utilization of the rigid POSS core as versatile precursors towards the construction of unique supramolecular architectures.14c,16 In view of these properties, POSSs are potentially ideal candidates for constructing various controlled nanostructures.
Transition metal incorporated organosilanes have therefore attracted growing interest as a new class of functional materials, due to their rich spectroscopic properties arising from the transition metal center and the excellent mechanical properties of the organosilane core. However, the utilization of this class of materials as molecular precursors towards supramolecular chemistry has not been widely explored. Recently, we reported a new class of POSS-functionalized platinum(II) inorganic–organic hybrids,5c demonstrating that the incorporation of this rigid inorganic cage could contribute to interesting transformable nanostructures with notable color and spectroscopic changes in response to various solvent compositions. Such interesting self-association processes were controlled by the interplay of both Pt⋯Pt and hydrophobic–hydrophobic interactions arising from the platinum(II) and POSS moieties respectively. It is envisaged that with the incorporation of additional non-covalent intermolecular interactions, one could further fine-tune and systematically control the aggregation process, leading to the formation of distinct supramolecular architectures. This has prompted us to investigate the interplay of various intermolecular interactions in controlling the self-assembly process. Herein, a series of POSS-functionalized alkynylplatinum(II) terpyridine complexes (Scheme 1) has been synthesized and characterized. They have been systematically modified with the incorporation of various functional moieties to provide a variety of non-covalent interactions for the molecular association process. Their viability in self-association to give nanoscaled superstructures, associated with drastic spectroscopic and luminescence changes in various solvent compositions, has been demonstrated and investigated. The structural correlation in constructing and controlling the formation of these supramolecular architectures has been explored.
 | ||
| Scheme 1 Structures of complexes 1–8. | ||
Results and discussion
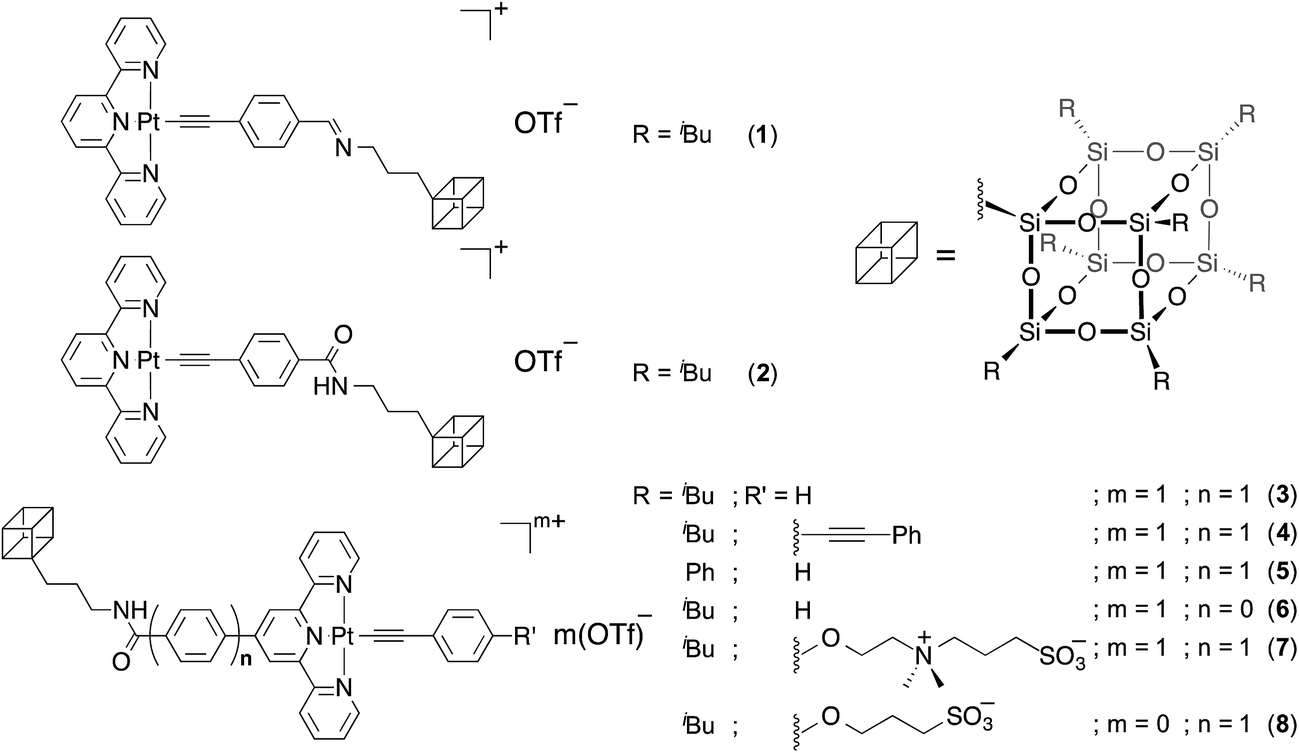
Complex 1, with the platinum(II) terpyridine moiety linked to the heptaisobutyl POSS through an imine linkage, was first prepared by the Cu(I)-catalyzed reaction of the corresponding POSS-functionalized alkyne and the chloroplatinum(II) terpyridine precursor complex. The complex was purified by recrystallization to give dark solids, which gave satisfactory elemental analyses and was well characterized by 1H NMR spectroscopy, IR spectroscopy, and FAB mass spectrometry. Dissolution of 1 in THF gives pale orange solutions, and the corresponding UV-vis absorption spectrum is depicted in Fig. 1. With reference to the previous work on alkynylplatinum(II) terpyridine complexes,4b,c,5a,6a,b the high-energy absorption band at ca. 300–350 nm is assigned as intraligand (IL) [π → π*] transitions of alkynyl and terpyridine ligands, while the low-energy absorption band at ca. 400–500 nm is assigned as a metal-to-ligand charge transfer (MLCT) [dπ(Pt) → π*(tpy)] transition mixed with alkynyl-to-terpyridine ligand-to-ligand change transfer (LLCT) character. It is interesting to note that this complex exhibits strong color changes, from yellow (30% H2O) to red (70% H2O), upon an increase in water content as depicted in Fig. 1a. UV-vis absorption studies reveal the drastic color changes, which are accompanied by the formation of a new absorption shoulder at around 560 nm (Fig. 1b), which is typically assigned as a metal–metal-to-ligand charge transfer (MMLCT) transition.4b,c,5a,6a,b The absorbance of this shoulder is found to grow with increasing water content of the sample solution, accompanied by a drop in intensity of the MLCT absorption band at 442 nm to give well-defined isosbestic points. This newly formed absorption shoulder is believed to be of MMLCT origin and can be rationalized by the ground-state aggregation brought about by the formation of Pt⋯Pt and/or π–π interactions among the platinum(II) terpyridine moieties, assisted by hydrophobic–hydrophobic interactions arising from the presence of the hydrophobically functionalized POSS moiety and the increase in solvent polarity. A plot of the absorbance at ca. 550 nm versus water content reveals that a critical water content of around 62% is required to trigger such an aggregation process. Further evidence for the occurrence of aggregation comes from the solvent-dependent 1H NMR experiments at various THF–water compositions (Fig. S1†), revealing 1 would undergo a self-assembly process in aqueous media. Well-resolved proton signals with chemical shifts consistent with the chemical formulation are initially observed in the 20% water–THF mixture. However, further raising the water content to 60% would lead to a significant upfield shift and broadening of the signals corresponding to the terpyridine moieties, indicating the presence of molecular aggregation at high water compositions, showing consistent results with those from the UV-vis absorption studies.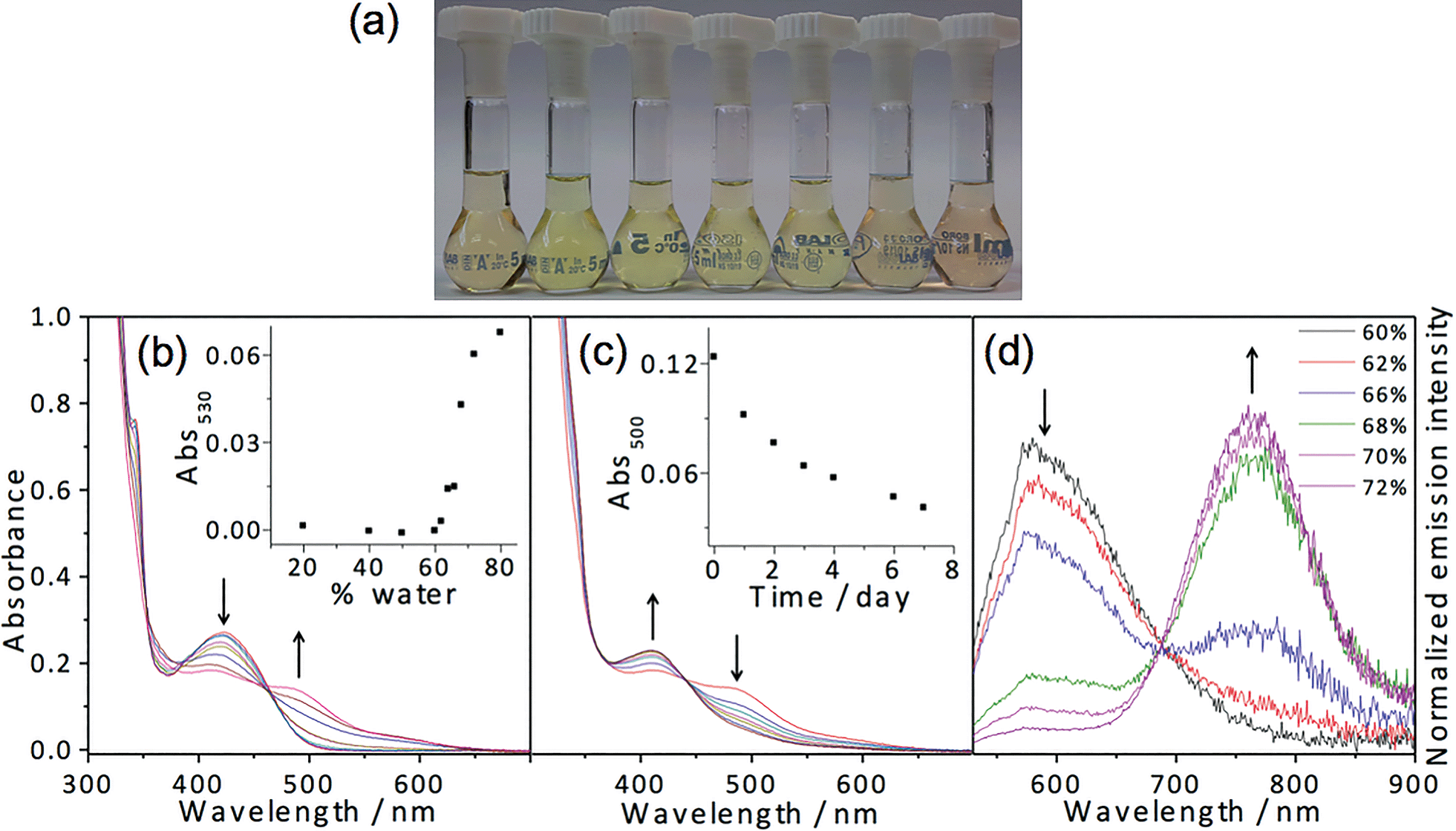 | ||
| Fig. 1 (a) Solutions of 1 in THF–water mixtures (percentage of water in THF from left to right: 0, 20, 40, 50, 60, 70 and 80%). (b) UV-vis absorption spectral changes of 1 in THF with increasing water content from 50 to 80%. (c) Time-dependent UV-vis absorption spectral changes of 1 in 80% water–THF mixture from t = 0 to t = 7 days. (d) The corresponding corrected emission spectral changes in response to the change in water composition from 0 to 90% normalized at 688 nm. | ||
Notably, complex 1 exhibits reversed color changes of the solution (80% H2O), red to yellow, upon leaving the solution under ambient conditions for several days. The corresponding time-dependent UV-vis absorption study (Fig. 1c) reveals the drop in the low-energy MMLCT band over time, indicating the weakening and declining of the Pt⋯Pt and/or π–π interactions. This phenomenon could be rationalized by the hydrolysis of the imine linkages that results in the detachment of hydrophobic POSS moieties, diminishing the driving forces towards ground state aggregation processes. This detachment of the POSS moieties demonstrates its importance towards the self-assembly process, and also illustrates the significance of hydrophobic–hydrophobic interactions in governing the self-assembly process in the system.
In light of this, complexes 2–8 have been designed and prepared with the replacement of the imine linkages with the more robust amide linkages that would not readily hydrolyze under ambient conditions. Complex 2 was prepared as a structural analogue of 1, demonstrating the enhanced stability of the system with the introduction of the amide linker. Another series of complexes, 3–8, was prepared with systematic modifications at the alkyne, at the pincer ligands and at the POSS moieties for rationalizing the underlying factors that govern the self-assembly process. In particular, the effect of π–π stacking has been rationalized and compared through the incorporation of an extended π-conjugated alkynyl ligand in complex 4, while the hydrophobicity of the POSS moieties and the planarity of the pincer ligands towards the aggregation process were illustrated through complexes 5 and 6 respectively. The charged moieties were incorporated in complexes 7 and 8 for investigating the effect of charge and the electrostatic effects towards the supramolecular chemistry. These complexes were prepared similar to that of 1 by the Cu(I)-catalyzed reaction, and were purified and well characterized. Dissolution of 2–6 gives yellow solutions in THF with similar UV-vis absorption patterns and solvent-dependent phenomena (Fig. S2–S6†). Temperature-dependent electronic absorption spectroscopy of 3 with a 65% water–THF mixture has been performed (Fig. S7†), in which a drop in the low-energy absorption tail and a rise in the higher-energy MLCT transition with clear isosbestic points are observed. Such phenomena can be ascribed to the deaggregation process at increasing temperature, causing the weakening of Pt⋯Pt and/or π–π stacking interactions, revealing the low-energy absorption to be associated with aggregation and of MMLCT origin. Further evidence for the presence of aggregate species comes from the solvent-dependent 1H NMR experiment of 3 (Fig. S8†), with upfield shifts and broadening of the proton signals observed at high water content, indicating the presence of significant molecular interactions. Despite all complexes exhibiting similar solvent-dependent phenomena, slight differences in the critical water compositions required to trigger the growth of the MMLCT transitions are observed. Complexes 3, 4 and 6 with heptaisobutyl-substituted POSS could readily self-associate and aggregate at relatively lower water contents (around 56%, 50% and 54% respectively) as revealed in their corresponding solvent-dependent electronic absorption spectra, while the MMLCT transition is triggered at a slightly higher water composition of 58% in the heptaphenyl POSS-functionalized 5.
The study of the effect of solvent composition on the emission properties of 1–6 in THF–water mixtures has also been performed. Upon excitation at the isosbestic wavelength, all complexes show similar emission spectra. Complex 1 in THF exhibits an emission band at about 626 nm (Fig. 1d), which is assigned to an excited state of predominantly 3MLCT character. The drop in emission intensity, together with enhancement of a new NIR luminescence at around 765 nm are observed with increasing water content in the water–THF mixture. With reference to previous alkynylplatinum(II) terpyridine systems,4b,c,5a,6a,b the significantly red-shifted luminescence maxima, together with the self-aggregation revealed from 1H NMR and UV-vis absorption studies, suggest that the emission is of 3MMLCT origin. Similar phenomena have been observed in complexes 2–6 (Fig. S2–S6†), with red-shifted and enhanced luminescence at high water content, which can be attributed to the presence of Pt⋯Pt and π–π stacking interactions as a result of solvent-induced self-assembly. The critical solvent compositions required to trigger the 3MMLCT emission are consistent with those from the UV-vis studies.
This critical solvent composition towards aggregation is known to be thermodynamically related.17a A lower critical solvent composition usually indicates a more favorable aggregation process under similar conditions. The difference in critical solvent composition among 3–6 could probably be attributed to the difference in hydrophobicity of the POSS moieties. It is likely that the highly hydrophobic isobutyl-substituted POSS in 3, 4 and 6 would be able to provide a sufficiently strong hydrophobic–hydrophobic interaction for aggregation to take place at lower solvent polarity, which gives rise to intermolecular Pt⋯Pt and π–π stacking interactions that account for the MMLCT transitions. The lesser extent of aggregation at low water composition in 5, as revealed from the corresponding electronic absorption spectra, could be ascribed to the relatively less hydrophobic phenyl-functionalized POSS, which would result in a weaker driving force and account for the slight increase in critical solvent polarity for the self-assembly processes. A significantly lower critical solvent fraction towards aggregation has been observed in 4 (around 50%), which could be ascribed to the extended π-conjugated alkynyl system that would provide a stronger π–π stacking interaction, resulting in the stabilization of the aggregated state, eventually favoring the aggregation process. The substituents of the POSS moieties as well as additional π–π stacking interactions are shown to play a significant role in governing the occurrence and extent of self-association. The underlying thermodynamics have been studied by fitting the curves of 5 (Fig. S9†) to the nucleation–elongation model17a for solvent-dependent self-assembly, giving a ΔG0 value of around −69 ± 6.1 kJ mol−1. A more negative ΔG0 value has been observed in this system compared to that previously reported in a simple organic system17a and an alkynylgold(I) system17b (ΔG0 = −40 kJ mol−1 and −47 kJ mol−1 respectively). Such differences in behavior could be rationalized by the highly hydrophobic POSS moieties as well as the formation of Pt⋯Pt interactions that contribute to a more thermodynamically favorable aggregation process. Attempts to fit complexes 1–4 and 6 into the model have not been successful due to the limitation of solubility of the complexes in the non-solvent media, resulting in insufficient data points, especially in the plateau region, that could be obtained experimentally, which limits the fitting of the thermodynamic data to the model with sufficient certainty.
Effect of π-conjugated alkynyl ligand towards self-association
In order to study the morphological changes in a variety of solvent compositions, transmission electron microscopy (TEM), scanning electron microscopy (SEM) and atomic force microscopy (AFM) have been employed to establish the identities of the aggregated species at various THF–water compositions. TEM, SEM and AFM images of 4 in THF–water (30% water) solution show nano-rings with internal and external diameters of around 180 nm and 300 nm respectively (Fig. 2a–c). Upon the addition of water, the solution turns red (60% water), accompanied by the interesting morphological transformation into plate-like nanostructures, with lengths of a few μm and thicknesses of about 12 nm, as revealed from the corresponding TEM, SEM and AFM studies (Fig. 2d–f respectively). In contrast to 4, complex 3 with a less extensive conjugated phenyl-substituted alkynyl shows only short helix-like fibers with lengths of a few hundreds of nm at low water content (30% water, Fig. 3a and b), while no well-defined morphology could be observed upon a further increase of the water composition. Such dramatic differences in aggregation behavior in response to the variation of solvent polarities among complexes 3 and 4 could probably be attributed to the differences in the alkynyl moieties. The replacement of phenylacetylene with alkynyl moieties of extended π-conjugation in 4 could enhance the π–π stacking interactions, which provides additional driving forces for ordered molecular association. The π–π stacking interactions among the alkynyl moieties, together with the formation of Pt⋯Pt interactions, may govern the conformational changes of the aggregated species, which favor the one-dimensional stacking of the alkynyl moieties and result in the formation of plate-like nanostructures at 60% water composition. In contrast, less conjugated alkynyl ligands in 3 would result in weaker π–π stacking interactions. Such insufficient stacking among the alkynyl moieties may favor the adoption of a less ordered aggregation process in polar media, which accounts for the absence of well-defined supramolecular architectures at higher water content. | ||
| Fig. 2 Electron micrographs of the superstructures prepared from 4 (2 × 10−4 M). (a) TEM, (b) SEM and (c) AFM images in a 30% water–THF mixture; (d) TEM, (e) SEM and (f) AFM images in a 60% water–THF mixture. Insets: height profile of the nanostructures at the selected cross-section (indicated as red lines). | ||
 | ||
| Fig. 3 (a) TEM and (b) SEM images of the superstructures prepared from 3 (2 × 10−4 M) in a 30% water–THF mixture. (c–f) Electron micrographs of the superstructures prepared from 6 (2 × 10−4 M). (c) TEM and (d) SEM images in a 30% (v/v) water–THF mixture; (e) TEM and (f) SEM images in a 60% (v/v) water–THF composition. | ||
Effect of pincer ligand towards self-association
To further investigate the structural variation of the pincer ligands of the complexes and their π–π stacking abilities towards the aggregation behavior, complex 6 as a structural analogue of 3, without a phenyl linking group on the terpyridyl moiety, was prepared. The phenyl group connected to the terpyridine unit in 3 is well-known to adopt a twisted conformation as a result of the steric hindrance among the peripheral protons.18 This slightly twisted structure between the aromatic rings in 3 and 4 could lead to the perturbation of π–π stacking interactions among the terpyridine moieties and result in different aggregated structures. Compared to complex 3, complex 6 with highly structural similarity shows significantly different morphologies under similar mixed-solvent conditions. Instead of ring-like nanostructures, spherical aggregates with diameters of about 120 nm are observed in 30% water in THF solution (Fig. 3c and d). Morphological transformation into plate-like nanostructures with longer lengths of a few μm and widths of around 90 nm is observed upon further increasing the water composition to 60% (Fig. 3e and f). Such differences could be ascribed to the difference in the terpyridine units, where the extra aromatic group in 3 would contribute to the adoption of a twisted conformation, which would weaken the π–π stacking interactions. In contrast to 3, better stacking among the terpyridine moieties is found in complex 6, accounting for the adoption of plate-like nanostructures with a high water content. Such plate-like aggregates closely resemble that observed in complex 4 under similar conditions, probably suggesting their similarity in packing arrangement in polar media due to the one-dimensional stacking of the terpyridine and alkynyl units contributed from both the Pt⋯Pt and π–π stacking interactions. Both complexes 4 and 6 are structurally similar and are derived from 3 with slight modifications to the alkynyl and terpyridine moieties respectively. The extended π-conjugation in the alkynyl moieties as well as improved stacking among the terpyridine units have resulted in enhanced π–π stacking interactions, which eventually lead to a morphological transformation, with the assistance of Pt⋯Pt interactions, upon raising the solvent polarity, which was not observed in complex 3 due to insufficient π–π stacking interactions. Such phenomena reveal the importance of terpyridine and alkynyl moieties in providing π–π stacking interactions towards the governing of the self-assembly processes.In addition to the terpyridine and alkynyl moieties, modification of the POSS units has also been studied. Complex 5 functionalized with heptaphenyl-substituted POSS shows different aggregated species compared to 3 under similar conditions. Entangled fibril-like nanostructures with lengths of around 500 nm and widths of about 100 nm have been observed in a THF–water (30% water) mixture (Fig. S10†). Such different assembly behavior in a low water content can be attributed to the modification of the POSS moieties. The variation of hydrophobicity as well as enhanced rigidity of the POSS substituents in 5 may contribute to a different packing conformation under similar conditions, which eventually contributes to the differences in morphologies, illustrating the significant role of POSS moieties in governing the self-assembly process. The loss of well-defined architectures has been observed upon further increasing the water composition (60%), which could probably be ascribed to the adoption of a random aggregation process due to the weaker π–π stacking interactions contributed by the alkynyl and terpyridine moieties, similar to the findings in complex 3.
It is worth noting that 1 shows circular aggregates in a 30% water–THF mixture (internal and external diameters of around 90–120 nm and 150–200 nm respectively), which transform into long fibrils of a few μm in length at around 70% water composition (Fig. 4), accompanied with a significant color change from yellow to red. Prolonged standing in an ambient atmosphere results in decolorization of the mixture due to hydrolysis of the imine linkage. The corresponding TEM micrographs reveal a significant drop in the population and size of the circular and fibril aggregates in 30% and 70% mixtures respectively (Fig. S11†). Such a phenomenon can be ascribed to the dissociation of the POSS moieties, leading to the loss of the hydrophobic driving force, which would eventually result in the deaggregation of the complexes. This can rationalize the spectroscopic changes and changes in the electron micrographs, revealing the importance of the POSS moieties in governing the self-assembly process. The corresponding amide analogue 2 was also probed with electron microscopy at a 30% water–THF mixture (Fig. S12†), revealing circular aggregates of internal and external diameters of around 50 nm and 140 nm respectively, demonstrating similar self-assembly behavior compared to that of its imine counterpart. However, further increase of the water content to 70% gradually destabilizes the assemblies and leads to precipitation of the system, limiting the morphological studies at higher water compositions.
 | ||
| Fig. 4 Electron micrographs of the superstructures prepared from 1 (2 × 10−4 M). (a) TEM and (b) SEM images in a 30% (v/v) water–THF mixture; (c) TEM and (d) SEM images in a 70% (v/v) water–THF composition. | ||
The hydrophobic moieties as well as π–π stacking interactions have been demonstrated to govern the self-assembly processes in response to the variation of the solvent composition. In addition to these interactions, it is anticipated that the platinum(II) metal center also plays an important role in governing such processes. In order to further investigate the role of the platinum(II) center in governing the self-association of the complexes, the microscopic images of their pure organic counterparts with solely π–π stacking and hydrophobic interactions have been investigated under similar conditions. TEM images of the precursor terpyridine ligands of complexes 3–6 reveal the absence of well-defined morphologies under similar mixed-solvent conditions. Such an absence of well-ordered aggregated species reveals the importance of the platinum(II) metal center that results in the planarization of the terpyridine moieties through the coordination of metal centers, facilitating better π–π overlap that results in stronger π–π stacking interactions towards the formation of the supramolecular architecture. Such strengthening of the π–π stacking interactions may also bring the platinum atoms into close proximity to induce the formation of Pt⋯Pt interactions. This formation of π–π stacking and/or Pt⋯Pt interactions, together with the formation of hydrophobic–hydrophobic interactions, could probably control the stacking arrangement of the platinum(II) terpyridine moiety. The aggregation among the hydrophobic POSS groups resulting from the reduced solvation brought about by the increase in solvent polarity, together with the stacking among the platinum(II) terpyridine moiety via π–π stacking and/or Pt⋯Pt interactions, would limit the packing arrangement of the aggregate species into a head-to-head manner. Such a presence of both interactions gradually limits the packing arrangement, and probably contributes to the formation of well-defined architectures, which is not commonly achievable in the pure organic counterparts with solely π–π stacking and/or hydrophobic–hydrophobic interactions.
Electrostatic effect towards the self-assembly behavior
In other to further investigate the effect of substituents on the self-assembly processes, charged polar alkynyl moieties have been introduced. Amphiphilic platinum(II) complexes 7 and 8 have been prepared. A charged zwitterionic sulfobetaine alkynyl tail has been incorporated into 7, providing significant hydrophilic driving forces to the system, and anionic sulfonate has been introduced to achieve charge neutrality in 8, reducing electrostatic repulsion among the complexes. Their photophysical properties and aggregation behavior have also been studied. Interestingly, dissolution of sulfobetaine functionalized 7 in THF gives a significantly more red-shifted solution compared to those observed for 1–6. The corresponding UV-vis electronic absorption studies (Fig. 5) reveal the presence of a low-energy absorption band at around 505 nm, which is typically assigned as the MMLCT transition. The corresponding emission spectra also reveal a significantly red-sifted structureless emission maximum at ca. 740 nm, which is assigned to be of 3MMLCT origin, suggesting that complex 7 exhibits significant ground state aggregation in THF. Notably, this complex exhibits dramatic color changes from red (THF) to yellow (50% water) and back to red (70% water) in response to various water compositions, as depicted in Fig. 5. An initial increase in the water content results in drops in intensity of the MMLCT band in both the UV-vis absorption and emission spectra, suggesting the weakening of the Pt⋯Pt and/or π–π interactions. Upon a further increase in the water composition, the low-energy MMLCT absorption band reappears, indicating the formation of Pt⋯Pt and/or π–π interactions at higher water content, while the emission is quenched in the increased water composition. Such unique color changes could probably be ascribed to an aggregation–partial deaggregation–aggregation process upon changing the composition of the water–THF mixture. The initial aggregated state in THF could be attributed to the domination of hydrophilic–hydrophilic interactions among the charged sulfobetaine moieties, while the gradual increase in the water composition would lead to improved solvation of the moieties, resulting in a less aggregated state of the molecules. Further increasing the water composition would lead to the reduced solvation of the POSS moieties, which eventually leads to an increase in the hydrophobic–hydrophobic interactions, restoring the aggregated state. A more detailed mechanism for the process will be discussed in the following paragraphs.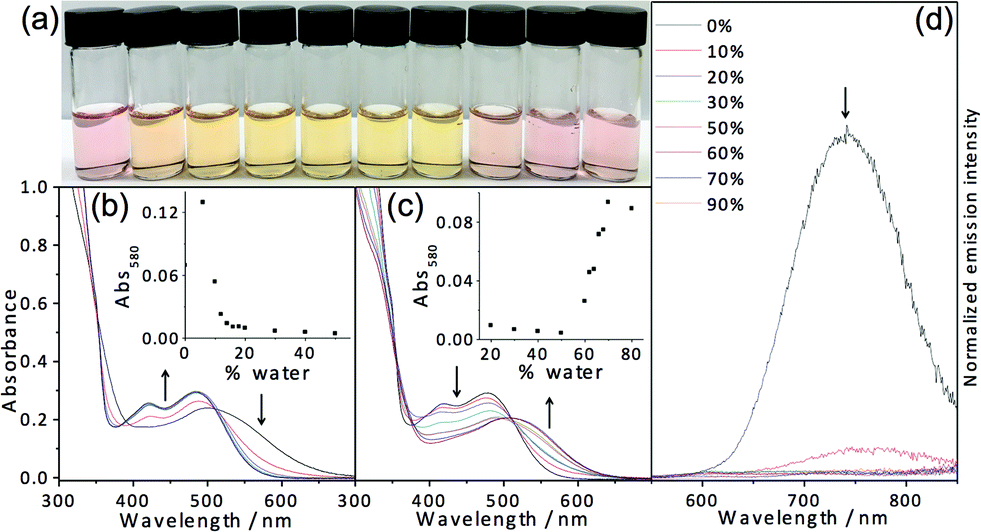 | ||
| Fig. 5 (a) Solutions of 7 in THF–water mixtures (percentage of water in THF from left to right: 0, 10, 20, 30, 40, 50, 60, 70, 80 and 90%). UV-vis absorption spectra of 7 in THF with increasing water content from (b) 2 to 40%; (c) 40 to 80%. (d) The corresponding corrected emission spectral changes in response to the change in water composition from 0 to 90%. | ||
To further investigate the aggregation process, TEM and SEM are employed to probe the identities of the aggregated species at various water–THF compositions. Interestingly, this complex exhibits three stages of morphological transformation in response to the solvent media. Short fibrils with lengths of a few μm are initially observed in the 10% water–THF mixture (Fig. 6a and b). A slight increase in water composition to 30% would result in spherical aggregates with diameters of around 150–200 nm (Fig. 6c and d). Further increasing the water composition to 50% would lead to the appearance of circular aggregates with internal and external diameters of around 100 nm and 250 nm respectively (Fig. 6e and f). A continual increase in the water composition to 70% would trigger the third stage of the morphological transformation into plate-like species with widths of ca. 200 nm and lengths of a few μm (Fig. 6g). TEM images at a 55% water content reveal intermediate species showing the evolution of fibrils from the ring-like aggregates (Fig. 6h), suggesting that the plate-like aggregates at high water content are potentially developed from the circular aggregates.
 | ||
| Fig. 6 Electron micrographs of the superstructures prepared from 7 (2 × 10−4 M). (a) TEM and (b) SEM images in a 10% (v/v) water–THF mixture; (c) TEM and (d) SEM images in a 30% (v/v) water–THF mixture; (e) TEM and (f) SEM images in a 50% (v/v) water–THF mixture; (g) TEM image in a 70% (v/v) water–THF mixture. (h) TEM image of 7 in a 55% (v/v) water–THF mixture showing the morphological transformation from nano-rings to fibrils through an intermediate state with the fibrils evolving from the ring-like aggregates. | ||
By consideration of the amphiphilicity of the complex, together with the electron micrographs as well as other spectroscopic studies, a possible mechanism for the aggregation–partial deaggregation–aggregation process is proposed, as depicted in Fig. 7. The zwitterionic sulfobetaine moieties are considered as having higher affinity towards aqueous media, while the POSS-functionalized terpyridine is generally more hydrophobic. In THF, it is likely that the hydrophobic POSSs would remain dispersed and solvated, while the charged hydrophilic tails would aggregate in a manner to minimize the contact area with the less polar media. Such aggregation is believed to be driven by the domination of the hydrophilic–hydrophilic interactions among the sulfobetaine moieties, which then bring the platinum(II) moieties into close proximity and giving rise to the formation of Pt⋯Pt and/or π–π interactions, contributing to the low-energy transitions in the spectroscopic studies. Such formation of hydrophilic–hydrophilic interactions together with Pt⋯Pt and/or π–π interactions would limit and restrict the alignment of the aggregates, resulting in linear growth and formation of fibrils in the low water composition (10%, Fig. 7a). A further increase in water composition would result in better solvation of the hydrophilic sulfobetaine tails, and thus the aggregates are no longer tightly held, resulting in a partial deaggregation state with the weakening of the Pt⋯Pt and/or π–π interactions, accounting for the drops in intensity of the MMLCT absorption band. Multilayer micelle-like spherical aggregates are then formed, as revealed in the corresponding electron micrographs, with the POSS moieties located on the surface, projected outwards to the non-polar media, while the charged zwitterionic groups are located mainly in the core of the aggregates. Such a self-assembly process is mainly governed by hydrophilic–hydrophilic interactions (Fig. 7b). Upon the addition of water in THF to an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) ratio, a further increase in the solvation of the sulfobetaine moieties is observed. Such an increase in solvation further weakens the hydrophilic–hydrophilic interactions, leading to the formation of less tightly-packed, expanded circular aggregates. Further addition of water results in the reduced solvation of the hydrophobic POSS moieties, causing them to aggregate through the hydrophobic–hydrophobic interactions that bring the platinum(II) terpyridine moieties into close proximity, resulting in the formation of Pt⋯Pt and/or π–π interactions, providing additional forces for further regulating the aggregation processes. With this alignment of the Pt⋯Pt interactions of the alkynylplatinum(II) terpyridine complexes, the POSS organosilane hybrids would gradually transform into linear plates from the circular species upon an increase in the solvent polarity (Fig. 7c). The proposed transformation is supported by the corresponding TEM images at a 55% water content, revealing the evolution of ring-like aggregates into fibrils.
:1 (v/v) ratio, a further increase in the solvation of the sulfobetaine moieties is observed. Such an increase in solvation further weakens the hydrophilic–hydrophilic interactions, leading to the formation of less tightly-packed, expanded circular aggregates. Further addition of water results in the reduced solvation of the hydrophobic POSS moieties, causing them to aggregate through the hydrophobic–hydrophobic interactions that bring the platinum(II) terpyridine moieties into close proximity, resulting in the formation of Pt⋯Pt and/or π–π interactions, providing additional forces for further regulating the aggregation processes. With this alignment of the Pt⋯Pt interactions of the alkynylplatinum(II) terpyridine complexes, the POSS organosilane hybrids would gradually transform into linear plates from the circular species upon an increase in the solvent polarity (Fig. 7c). The proposed transformation is supported by the corresponding TEM images at a 55% water content, revealing the evolution of ring-like aggregates into fibrils.
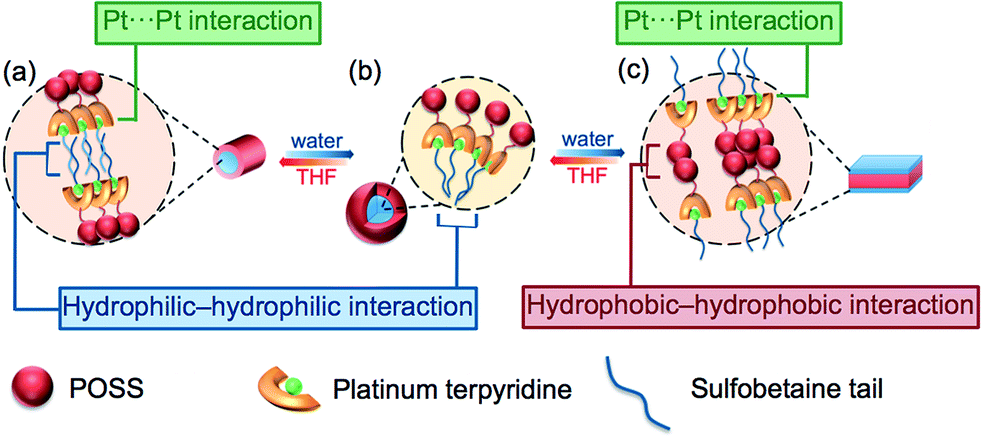 | ||
| Fig. 7 Schematic drawing of the morphological transformation of 7 in THF with varying solvent compositions. (a) 10% water–THF mixture, formation of fibrils due to predominantly hydrophilic–hydrophilic and Pt⋯Pt interactions; (b) 30% water–THF mixture, formation of spherical aggregates mainly due to hydrophilic–hydrophilic interactions with water molecules trapped inside; (c) 70% water–THF mixture, formation of plate-like aggregates due to the domination of hydrophobic–hydrophobic and Pt⋯Pt interactions. | ||
The three stages of morphological transformation are generally due to the interplay of hydrophilic–hydrophilic, hydrophobic–hydrophobic, Pt⋯Pt and π–π stacking interactions. With different molecular interactions being dominated at various solvent compositions, such a presence of various interactions gradually controls the packing arrangement, contributing to the well-defined architectures, which may not be achievable without the interplay of these various molecular interactions.
In light of the fascinating spectroscopic and microscopic properties brought about by the incorporation of charged moieties in 7, another amphiphilic complex 8 with sulfonate alkynyl has been prepared. It dissolves in THF to give a yellow solution, which is found to be relatively sensitive to water addition. Dramatic changes in apparent color from yellow (THF) to pink are observed upon the addition of around 0.5% water as depicted in Fig. 8. Corresponding electronic absorption studies reveal the development of a low-energy MMLCT absorption band at this low water composition (0.5%), suggesting that this complex exhibits aggregation behavior at an exceptionally low water content. The corresponding electron micrographs reveal the presence of rod-like aggregates, indicating the presence of aggregated species at a low water composition (Fig. S13†). Such sensitivity to water addition could probably be attributed to the charge neutrality of the complex, which minimizes the electrostatic repulsion and thus favors the aggregation process. In the absence of water, complex 8 remains solvated in the media; however, upon the addition of a minimal amount of water, the hydrophobic POSS becomes less solvated. This leads to the domination of hydrophobic–hydrophobic interactions among the POSS moieties, which facilitate the aggregation of the complexes, resulting in the formation of Pt⋯Pt and/or π–π stacking interactions that contribute to the color changes. However, it is worth noting that further increasing the water composition beyond 2% (v/v, water–THF) would result in slight drops in the intensity of the low-energy MMLCT absorption band. This phenomenon could be ascribed to the negative solvatochromism due to the destabilization of the excited state energy upon an increase in solvent polarity as well as the partial deaggregation process where the increase in solvation of the hydrophilic sulfonate moieties in the media would result in the weakening of the Pt⋯Pt and/or π–π stacking interactions. All in all, this complex is shown to aggregate at very low water composition and gives rise to drastic color and spectroscopic changes, which makes this class of complexes a potential candidate for sensing purposes.
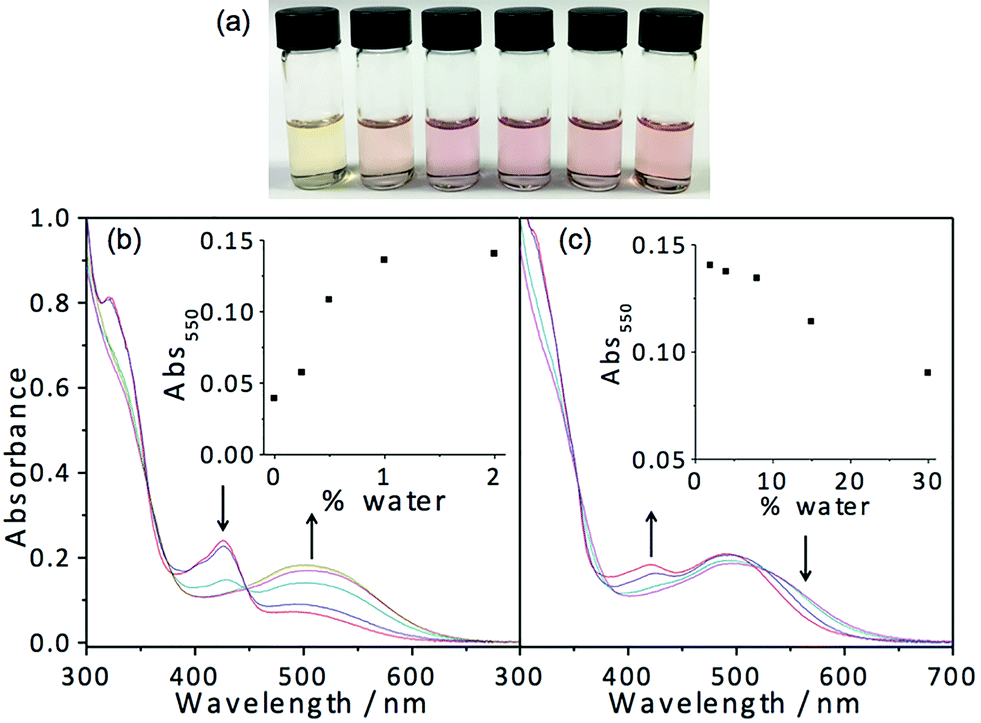 | ||
| Fig. 8 (a) Solutions of 8 in THF–water mixtures (percentage of water in THF from left to right: 0, 0.5, 1, 2, 4 and 8%). UV-vis absorption spectra of 8 in THF with increasing water content from (b) 0 to 2%; (c) 2 to 30%. | ||
Conclusions
To conclude, a series of alkynylplatinum(II) terpyridine complexes functionalized with POSS moieties have been designed and synthesized, which demonstrated the capability to form various distinct morphologies that can be systematically controlled by structural modification and variation of the solvent polarity. Such a self-association process is found to be governed by the interplay of various molecular interactions, including hydrophobic, hydrophilic, Pt⋯Pt and π–π stacking interactions. These interesting multiple stages of morphological transformations in various solvent media, which are associated with spectroscopic changes, have been fully studied by UV-vis absorption and emission spectroscopy, and electron microscopy. The present study illustrates structural modifications that manipulate intermolecular forces to achieve the gradual modification of the supramolecular architectures. Multiple stages of the morphological transformation have been demonstrated by the introduction of hydrophilic groups. These kinds of amphiphilic platinum(II) complexes have been demonstrated to show drastic solvatochromism through an aggregation and deaggregation cycle, and could serve as potential candidates for a new class of functional materials with sensing or imaging capabilities.Acknowledgements
V. W.-W. Y. acknowledges support from The University of Hong Kong under the University Research Committee (URC) Strategic Research Theme on New Materials. This work has been supported by the University Grants Committee Areas of Excellence (AoE) Scheme (AoE/P-03/08), a General Research Fund (GRF) grant from the Research Grants Council of Hong Kong Special Administrative Region, P. R. China (HKU 17304715), and the National Basic Research Program of China (973 Program; 2013CB834701). H.-L. A.-Y. acknowledges the receipt of a Postgraduate Studentship, and S. Y.-L. L. acknowledges the receipt of a University Postdoctoral Fellowship, both administered by The University of Hong Kong.References
- (a) J. W. Schindler, R. C. Fukuda and A. W. Adamson, J. Am. Chem. Soc., 1982, 104, 3596 CrossRef CAS; (b) D. M. Roundhill, H. B. Gray and C.-M. Che, Acc. Chem. Res., 1989, 22, 55 CrossRef CAS; (c) V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1989, 28, 1529 CrossRef CAS; (d) H. Kunkely and A. Vogler, J. Am. Chem. Soc., 1990, 112, 5625 CrossRef CAS; (e) V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1991, 30, 4446 CrossRef CAS; (f) V. M. Miskowski, V. H. Houlding, C.-M. Che and Y. Wang, Inorg. Chem., 1993, 32, 2518 CrossRef CAS; (g) R. H. Herber, M. Croft, M. J. Coyer, B. Bilash and A. Sahiner, Inorg. Chem., 1994, 33, 2422 CrossRef CAS; (h) W. B. Connick, L. M. Henling, R. E. Marsh and H. B. Gray, Inorg. Chem., 1996, 35, 6262 Search PubMed.
- (a) K. W. Jennette, J. T. Gill, J. A. Sadownick and S. J. Lippard, J. Am. Chem. Soc., 1976, 98, 6159 CrossRef CAS; (b) T. K. Aldrige, E. M. Stacy and D. R. McMillin, Inorg. Chem., 1994, 33, 722 CrossRef; (c) J. J. Novoa, G. Aullóh, P. Alemany and S. Alvarez, J. Am. Chem. Soc., 1995, 117, 7169 CrossRef CAS; (d) M. G. Hill, J. A. Bailey, V. M. Miskowski and H. B. Gray, Inorg. Chem., 1996, 35, 4585 CrossRef CAS; (e) W. B. Connick, R. E. Marsh, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1997, 36, 913 CrossRef CAS; (f) B. Ma, J. Li, P. I. Djurovich, M. Yousufuddin, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2005, 127, 28 CrossRef CAS PubMed.
- (a) H.-K. Yip, L.-K. Cheng, K.-K. Cheung and C.-M. Che, J. Chem. Soc., Dalton Trans., 1993, 2933 RSC; (b) J. A. Bailey, M. G. Hill, R. E. Marsh, V. M. Miskowski, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1995, 34, 4591 CrossRef CAS; (c) G. Arena, G. Calogero, S. Campagna, L. M. Scolaro, V. Ricevuto and R. Romeo, Inorg. Chem., 1998, 37, 2763 CrossRef CAS PubMed; (d) R. Büchner, C. T. Cunningham, J. S. Field, R. J. Haines, D. R. McMillin and G. C. Summerton, J. Chem. Soc., Dalton Trans., 1999, 711 RSC.
- (a) W.-S. Tang, X.-X. Lu, K. M.-C. Wong and V. W.-W. Yam, J. Mater. Chem., 2005, 15, 2714 RSC; (b) V. W.-W. Yam, K. H.-Y. Chan, K. M.-C. Wong and B. W.-K. Chu, Angew. Chem., Int. Ed., 2006, 45, 6169 CrossRef CAS PubMed; (c) K. M.-C. Wong, W.-S. Tang, B. W.-K. Chu, N. Zhu and V. W.-W. Yam, Organometallics, 2004, 23, 3459 CrossRef CAS; (d) K. M.-C. Wong, N. Zhu and V. W.-W. Yam, Chem. Commun., 2006, 3441 RSC.
- (a) V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506 CrossRef CAS PubMed; (b) C. Po, A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12136 CrossRef CAS PubMed; (c) H.-L. Au-Yeung, S. Y.-L. Leung, A. Y.-Y. Tam and V. W.-W. Yam, J. Am. Chem. Soc., 2014, 136, 17910 CrossRef CAS PubMed; (d) K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424 CrossRef CAS PubMed.
- (a) C. Yu, K. H.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19652 CrossRef CAS PubMed; (b) C. Yu, K. H.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Chem.–Eur. J., 2008, 14, 4577 CrossRef CAS PubMed; (c) M. C.-L. Yeung, K. M.-C. Wong, Y. K.-T. Tsang and V. W.-W. Yam, Chem. Commun., 2010, 46, 7709 RSC.
- (a) C. Yu, K. M.-C. Wong, K. H.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2005, 44, 791 CrossRef CAS PubMed; (b) K. M.-C. Wong and V. W.-W. Yam, Coord. Chem. Rev., 2007, 251, 2477 CrossRef CAS; (c) C. Y.-S. Chung and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 18775 CrossRef CAS PubMed.
- (a) W. Lu, S. S.-Y. Chui, K.-M. Ng and C.-M. Che, Angew. Chem., 2008, 120, 4644 CrossRef; (b) M.-Y. Yuen, V. A. L. Roy, W. Lu, S. C. F. Kui, G. S. M. Tong, M.-H. So, S. S.-Y. Chui, M. Muccini, J. Q. Ning, S. J. Xu and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 9895 CrossRef CAS PubMed; (c) Y. Chen, K. Li, W. Lu, S. S.-Y. Chui, C.-W. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 9909 CrossRef CAS PubMed; (d) B. Jiang, J. Zhang, W. Zheng, L.-J. Chen, G.-Q. Yin, Y.-X. Wang, B. Sun, X. Li and H.-B. Yang, Chem.–Eur. J., 2016, 22, 14664 CrossRef CAS PubMed; (e) Y. Mao, K. Liu, L. Meng, L. Chen, L. Chen and T. Yi, Soft Matter, 2014, 10, 7615 RSC; (f) Y. Chen, C.-M. Che and W. Lu, Chem. Commun., 2015, 51, 5371 RSC; (g) S. Y.-L. Leung and V. W.-W. Yam, Chem. Sci., 2013, 4, 4228 RSC; (h) C. Po and V. W.-W. Yam, Chem. Sci., 2014, 5, 4868 RSC; (i) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589 CrossRef CAS PubMed; (j) F. C.-M. Leung, S. Y.-L. Leung, C. Y.-S. Chung and V. W.-W. Yam, J. Am. Chem. Soc., 2016, 138, 2989 CrossRef CAS PubMed; (k) S. Y.-L. Leung, K. M.-C. Wong and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2845 CrossRef CAS PubMed.
- (a) A. Y.-Y. Tam, K. M.-C. Wong, G. Wang and V. W.-W. Yam, Chem. Commun., 2007, 2028 RSC; (b) A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 6253 CrossRef CAS PubMed; (c) T. Tu, W. Fang, X. Bao, X. Li and K. H. Dötz, Angew. Chem., Int. Ed., 2011, 50, 6601 CrossRef CAS PubMed; (d) K.-C. Chang, J. L. Lin, Y. T. Shen, C.-Y. Hung, C.-Y. Chen and S.-S. Sun, Chem.–Eur. J., 2012, 18, 1312 CrossRef CAS PubMed; (e) X.-S. Xiao, W. Lu and C.-M. Che, Chem. Sci., 2014, 5, 2482 RSC; (f) J. L.-L. Tsai, T. Zou, J. Liu, T. Chen, A. O.-Y. Chan, C. Yang, C.-N. Lok and C.-M. Che, Chem. Sci., 2015, 6, 3823 RSC; (g) C. A. Strassert, C.-H. Chien, M. D. G. Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946 CrossRef CAS PubMed.
- (a) V. N. Kozhevnikov, B. Donnio and D. W. Bruce, Angew. Chem., Int. Ed., 2008, 47, 6286 CrossRef CAS PubMed; (b) M. Krikorian, S. Liu and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 2952 CrossRef CAS PubMed; (c) M. E. Robinson, D. J. Lunn, A. Nazemi, G. R. Whittell, L. De Cola and I. Manners, Chem. Commun., 2015, 51, 15921 RSC; (d) A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2016, 8, 10 CrossRef CAS PubMed.
- (a) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853 CrossRef CAS PubMed; (b) T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401 CrossRef CAS PubMed.
- (a) C. Sanchez, B. Julian, P. Belleville and M. Popall, J. Mater. Chem., 2005, 15, 3559 RSC; (b) C. Sanchez, P. Belleville, M. Popall and L. Nicole, Chem. Soc. Rev., 2011, 40, 696 RSC; (c) B. G. Trewyn, I. I. Slowing, S. Giri, H. T. Chen and V. S. Y. Lin, Acc. Chem. Res., 2007, 40, 846 CrossRef CAS PubMed; (d) N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789 RSC.
- (a) C. Sanchez, G. J. A. A. Soler-Illia, F. Ribot, T. Lalot, C. R. Mayer and V. Cabuil, Chem. Mater., 2001, 13, 3061 CrossRef CAS; (b) V. Alfredsson and H. Wennerström, Acc. Chem. Res., 2015, 48, 1891 CrossRef CAS PubMed; (c) N. Song and Y.-W. Yang, Chem. Soc. Rev., 2015, 44, 3474 RSC.
- (a) D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081 CrossRef CAS PubMed; (b) J. Wu and P. T. Mather, Polym. Rev., 2009, 49, 25 CrossRef CAS; (c) W.-B. Zhang, X. Yu, C.-L. Wang, H.-J. Sun, I.-F. Hsieh, Y. Li, X.-H. Dong, K. Yue, R. V. Horn and S. Z. D. Cheng, Macromolecules, 2014, 47, 1221 CrossRef CAS.
- (a) C. M. Leu, M. Reddy, K. H. Wei and C. F. Shu, Chem. Mater., 2003, 15, 2261 CrossRef CAS; (b) F. Mammeri, E. L. Bourhis, L. Rozes and C. Sanchez, J. Mater. Chem., 2005, 15, 3787 RSC; (c) B. M. Moore, S. M. Ramirez, G. R. Yandek, T. S. Haddad and J. M. Mabry, J. Organomet. Chem., 2011, 696, 2676 CrossRef CAS; (d) B. Seurer and B. Coughlin, Macromol. Chem. Phys., 2008, 209, 2040 CrossRef CAS; (e) D. B. Drazkowshi, A. Lee, T. S. Haddad and D. J. Cookson, Macromolecules, 2006, 39, 1854 CrossRef.
- (a) B. Jiang, W. Tao, X. Lu, Y. Liu, H. Jin, Y. Pang, X. Sun, D. Yan and Y. Zhou, Macromol. Rapid Commun., 2012, 33, 762 Search PubMed; (b) W. Zhang, J. Yuan, S. Weiss, X. Ye, C. Li and A. H. E. Müller, Macromolecules, 2011, 44, 6891 CrossRef CAS; (c) H. Hussain, B. H. Tan, G. L. Seah, Y. Liu, C. B. He and T. P. Davis, Langmuir, 2010, 26, 11763 CrossRef CAS PubMed; (d) S. W. Kuo, H. F. Lee, W. J. Huang, K. U. Jeong and F. C. Chang, Macromolecules, 2009, 42, 1619 CrossRef CAS; (e) W. H. Hu, K. W. Huang, C. W. Chiou and S. W. Kuo, Macromolecules, 2012, 45, 9020 CrossRef CAS; (f) Y. Xue, H. Wang, D. Yu, L. Feng, L. Dai, X. Wang and T. Lin, Chem. Commun., 2009, 6418 RSC; (g) X.-H. Dong, B. Ni, M. Huang, C.-H. Hsu, R. Bai, W.-B. Zhang, A.-C. Shi and S. Z. D. Cheng, Angew. Chem., Int. Ed., 2016, 55, 2459 CrossRef CAS PubMed; (h) J. Miao, L. Cui, H. P. Lau, P. T. Mather and L. Zhu, Macromolecules, 2007, 40, 5460 CrossRef CAS.
- (a) P. A. Korevaar, C. Schaefer, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482 CrossRef CAS PubMed; (b) E. Y.-H. Hong, H.-L. Wong and V. W.-W. Yam, Chem. Commun., 2014, 50, 13272 RSC.
- (a) A. Göller and U. W. Grummt, Chem. Phys. Lett., 2000, 321, 399 CrossRef; (b) C. P. Brock and R. P. Minton, J. Am. Chem. Soc., 1989, 111, 4586 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc04169h |
| This journal is © The Royal Society of Chemistry 2017 |