
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed enantioselective Heck alkenylation of trisubstituted allylic alkenols: a redox-relay strategy to construct vicinal stereocenters†
Chun
Zhang
a,
Brandon
Tutkowski
b,
Ryan J.
DeLuca
a,
Leo A.
Joyce
c,
Olaf
Wiest
 b and
Matthew S.
Sigman
*a
b and
Matthew S.
Sigman
*a
aDepartment of Chemistry, University of Utah, 315 South 1400 East, Salt Lake City, Utah 84112, USA
bDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556-5670, USA
cProcess Research & Development, Merck Research Laboratories, Rahway, New Jersey 07065, USA
First published on 9th December 2016
Abstract
An enantioselective, redox-relay Heck alkenylation of trisubstituted allylic alkenol substrates has been developed. This process enables the construction of vicinal stereocenters in high diastereo- and enantioselectivity and allows the formation of enolizable α-carbonyl methyl-substituted stereocenters with no observed epimerization under the reported reaction conditions.
Introduction
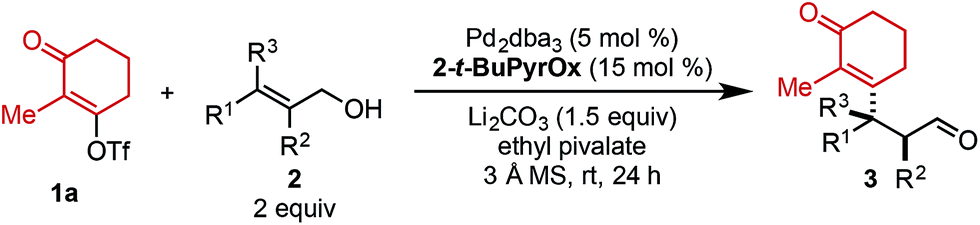
In a Heck reaction using multi-substituted alkenes, the initial syn-carbopalladation sets two vicinal stereocenters by virtue of the migratory insertion process.1 Unfortunately, the resultant Pd–alkyl undergoes facile β-hydride elimination, which generally eliminates the stereochemistry imparted by migratory insertion. Recently, we have reported a modern variant of the Heck reaction, wherein the directionality and stereochemical fidelity of β-hydride elimination can be controlled and, thus, the initial stereochemical consequence of migratory insertion is not lost. Termed redox-relay Heck reactions, the unsaturation of the alkene is conserved as it is transferred to a different position on the alkyl chain, most commonly by oxidation of an alcohol to a carbonyl.2 These reactions have been rendered enantioselective and are effective on both disubstituted alkenes to form tertiary stereocenters and trisubstituted alkenes to form quaternary stereocenters (Scheme 1A).2 However, the potential power of the syn migratory insertion has not been realized as only a single stereocenter has been set.2–5 Therefore, we set out to investigate if vicinal centers can be forged through the use of trisubstituted alkenols of type 2 in enantioselective redox-relay Heck reactions (Scheme 1B). In this case, such stereocenters6 could be strategically constructed if we took advantage of the propensity for the alkenyl electrophile (1) to add at the alkene carbon distal to the alcohol functionality, producing a new Csp2–Csp3 bond and set an adjacent stereocenter in a single migratory insertion event.7 In order to render this transformation enantioselective, the chiral Pd–ligand complex must differentiate the two similar prochiral faces of sterically encumbered trisubstituted alkenol 2, which can be challenging on the basis of past reports.8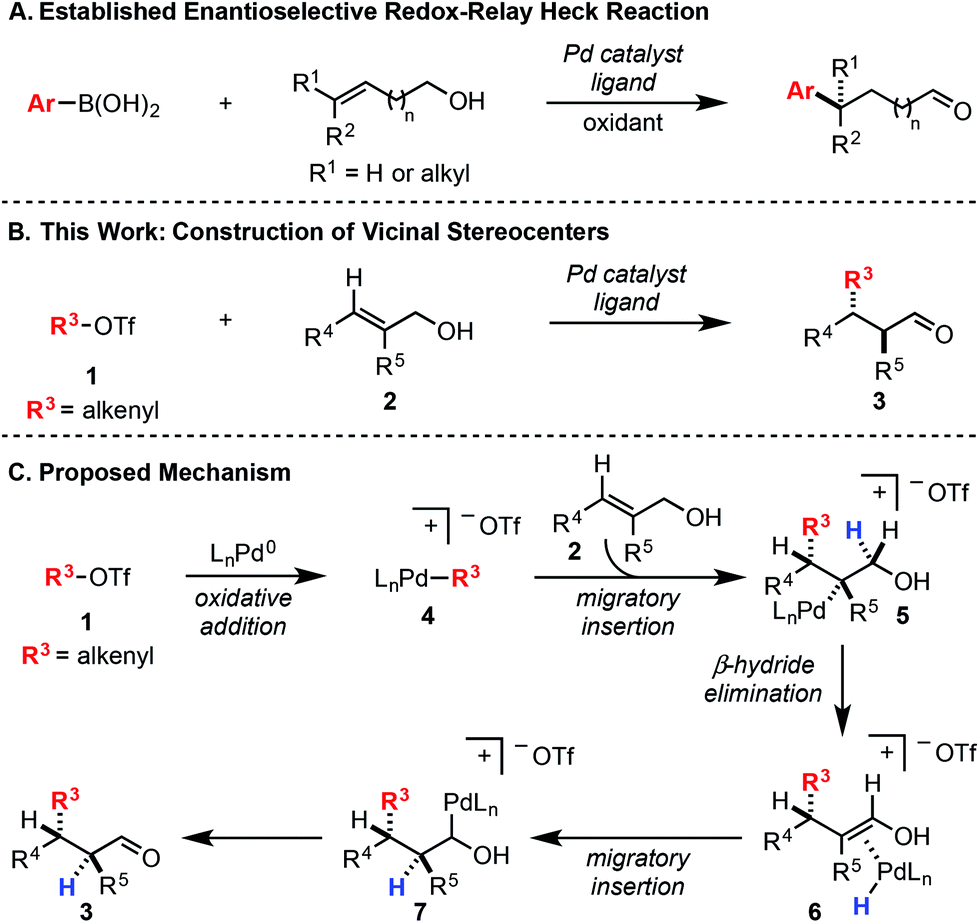 | ||
| Scheme 1 (A) Previous work with di- and trisubstituted alkenes to form tertiary and quaternary stereocenters. (B) Proposed synthesis of vicinal stereocenters using a redox-relay Heck strategy. (C) Mechanistic rationale for the construction of vicinal stereocenters. | ||
Mechanistically, we propose the reaction initiates with oxidative addition of alkenyl triflate 1 with Pd(0) to produce cationic Pd–alkenyl intermediate 4 (Scheme 1C).2c Alkenol 2 can undergo migratory insertion into the Pd–alkenyl bond to furnish Pd–alkyl intermediate 5. The cationic Pd–alkyl species 5 undergoes β-hydride elimination to deliver Pd–enol intermediate 6, which subsequently leads to reinsertion to yield intermediate 7. This type of Pd–alkyl intermediate is unique since the alcohol and Pd-catalyst are bonded to the same carbon atom. As a result, alcohol oxidation occurs (either through β-hydride elimination or an E2-type elimination9), reminiscent of the final step in the Wacker process.10 This yields final product 3 and Pd(0), closing the catalytic cycle.
Results and discussion
Preliminary investigations to develop this process were conducted with trisubstituted alkenol 2a using reaction conditions previously shown to effectively promote such processes. Unfortunately, commonly employed amide solvents, such as dimethylacetamide (DMA) and dimethylformamide (DMF), produced the desired redox-relay Heck product (3a) in low yield and enantioselectivity (entries 1 and 2, Table 1). A variety of solvents were then evaluated and only ester-derived solvents resulted in significant improvements. For example, ethyl acetate (EtOAc) enhanced the yield of product 3a to 45% and enantioselectivity to 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 (entry 3). Isopropyl acetate (i-PrOAc) and ethyl isobutyrate also resulted in similar yield and selectivity (entries 4 and 5). However, changing the solvent to ethyl pivalate delivered product 3a in 62% yield and 95:5 er. It should be noted that the enantioselectivity can be correlated to the natural bond orbital (NBO) charge of the oxygen or nitrogen atom of the ester or amide functional group of the solvent (see ESI for details†).11 Next, an assortment of bases was screened, as an equivalent of triflic acid is formed with each catalytic turnover. The addition of 1.5 equivalents of NEt3 slowed the desired reaction and resulted in trace product formation (entry 7). In contrast, adding 1.5 equivalents of Li2CO3 improved the yield of product 3a to 74% with no erosion in selectivity (entry 8). Exchanging the cation of the carbonate base from lithium to sodium or potassium resulted in slightly lower yields (entries 9 and 10).
:6 (entry 3). Isopropyl acetate (i-PrOAc) and ethyl isobutyrate also resulted in similar yield and selectivity (entries 4 and 5). However, changing the solvent to ethyl pivalate delivered product 3a in 62% yield and 95:5 er. It should be noted that the enantioselectivity can be correlated to the natural bond orbital (NBO) charge of the oxygen or nitrogen atom of the ester or amide functional group of the solvent (see ESI for details†).11 Next, an assortment of bases was screened, as an equivalent of triflic acid is formed with each catalytic turnover. The addition of 1.5 equivalents of NEt3 slowed the desired reaction and resulted in trace product formation (entry 7). In contrast, adding 1.5 equivalents of Li2CO3 improved the yield of product 3a to 74% with no erosion in selectivity (entry 8). Exchanging the cation of the carbonate base from lithium to sodium or potassium resulted in slightly lower yields (entries 9 and 10).
|
|
|||||
|---|---|---|---|---|---|
| entry | solvent | base | % yield | dr | er |
| a Each entry represents the isolated yield on 0.25 mmol scale. er values were determined by SFC or HPLC. Entries 7–10 used 1.5 equiv of base. | |||||
| 1 | DMA | — | 14 | >20:1 |
71:29 |
| 2 | DMF | — | 15 | >20:1 |
75:25 |
| 3 | EtOAc | — | 45 | >20:1 |
94:6 |
| 4 | i-PrOAc | — | 39 | >20:1 |
94:6 |
| 5 | Ethyl isobutyrate | — | 49 | >20:1 |
95:5 |
| 6 | Ethyl pivalate | — | 62 | >20:1 |
95:5 |
| 7 | Ethyl pivalate | NEt3 | trace | — | — |
| 8 | Ethyl pivalate | Li2CO3 | 74 | >20:1 |
95:5 |
| 9 | Ethyl pivalate | Na2CO3 | 70 | >20:1 |
95:5 |
| 10 | Ethyl pivalate | K2CO3 | 61 | >20:1 |
95:5 |
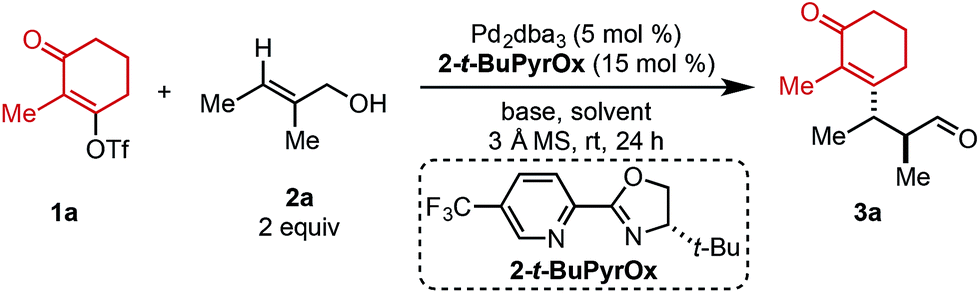
Using ethyl pivalate as solvent and Li2CO3 as base, the substrate scope of trisubstituted allylic alkenols was investigated (Table 2). Simple alkyl groups, such as methyl (3a) and ethyl (3b), at the R1 position on the alkene resulted in 74% and 84% yields, respectively. Introduction of a phenethyl moiety (3c) at the R1 position gave the desired product in 41% yield and 95:5 er. The inclusion of methyl (3d) and ethyl (3e) at the R3 position provided the corresponding ketone products in 60% and 54% yields, with increased enantioselectivity (97:3 and 98:2 er, respectively). An n-octyl alkyl group was examined giving 51% yield of 3f with 98:2 er. Evaluation of phenethyl (3g), benzyl (3h), and phenyl (3i) at R3 indicated that, as the phenyl group was positioned closer to the resulting carbonyl moiety, slightly lower yields and enantioselectivities were observed. In addition, the reaction could tolerate an isopropyl (i-Pr) group at the R3 position furnishing product 3j in 45% yield and 98:2 er. A trimethylsilyl (TMS) group could also be positioned α to the resultant carbonyl in high enantioselectivity, albeit in 19% yield (3k). The absolute configuration of product 3d was determined to be (2S,3S) using electronic circular dichroism (see ESI for details†).12 The other products were assigned by analogy to product 3d.
|
|
|---|
| a Each entry represents the isolated yield on 0.25 mmol scale. er values were determined by SFC or HPLC. |
|

When cyclopropyl-containing substrate 2l was subjected to the reaction conditions, the ring opening product (3l) was isolated in 40% yield and 97:3 er (Scheme 2). This α,β-unsaturated product could arise through Pd-mediated ring opening of β-cyclopropyl Pd–alkyl intermediate 8 to yield enol 9 that could tautomerize to ketone 10. Primary Pd–alkyl intermediate 10 can then undergo β-hydride elimination to produce a terminal alkene that isomerizes to the internal position to produce α,β-unsaturated product 3l.13 Ultimately, this confirms that the Pd-center migrates to the carbon attached to the alcohol.
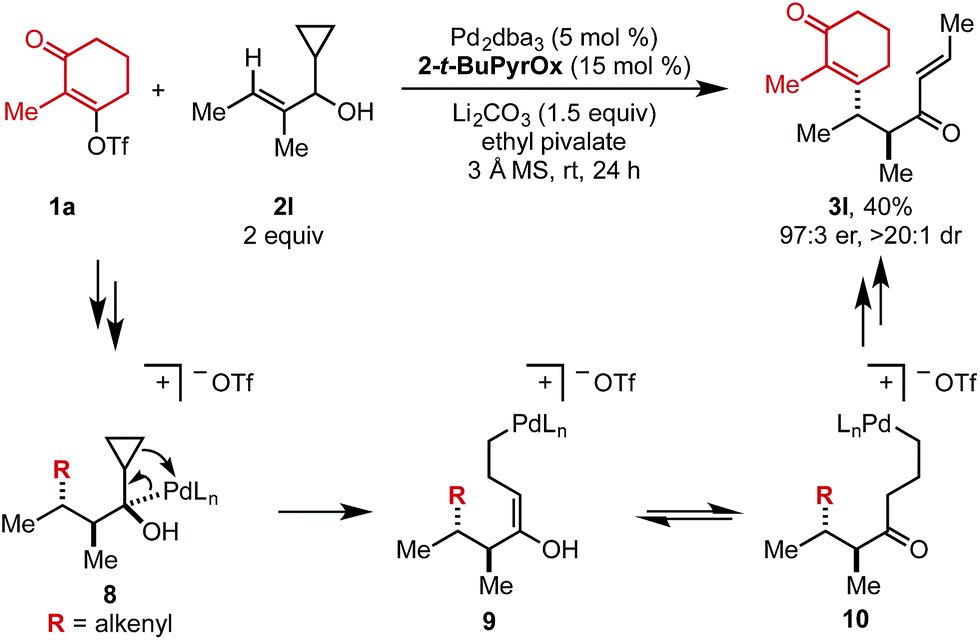 | ||
| Scheme 2 Putative mechanism for the Heck/cyclopropyl ring opening cascade. | ||
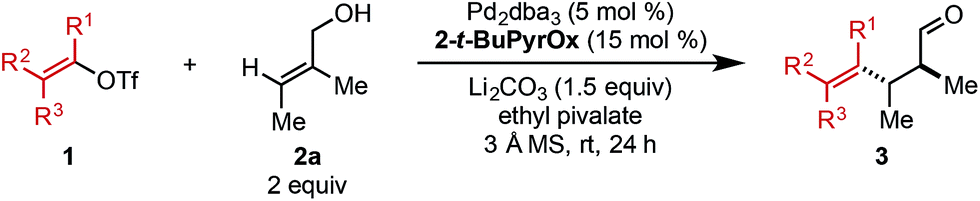
Next, the scope of alkenyl triflates was explored including a variety of tri-and tetrasubstituted alkenyl triflates (1, Table 3). Starting with 2-substituted cyclohexenone triflates, an 85% yield was isolated for the ethyl-substituted product (3m). As the apparent steric impact of the aliphatic group was increased to Bn (3n) and i-Pr (3o), lower product yields were observed. In the case of a 2-bromo-substituted triflate, only 22% yield was isolated (3p). Enol triflates containing a methyl-substituted cyclopentenone (1q) and 5-membered lactone (1r) delivered the corresponding products in excellent yield and good selectivity. Reaction with β-keto ester derived (Z)-enol triflate yielded the (Z)-alkene product in 72% yield (3s). In contrast, reaction with the (E)-enol triflate gave a near equal mixture of (E)- and (Z)-tetrasubstituted alkene products in 56% yield (3t). Interestingly, the (E)-alkene product isomer has a 97:3 er, while the (Z)-alkene isomer product has a 91:9 er, the same er as observed when (Z)-enol triflate 1s was used. This result can be explained through isomerization of (E)-enol triflate 1t producing a mixture of (E)- and (Z)-enol triflates. Reaction with Pd(0) would produce distinct Pd–alkenyl species and ultimately deliver alkene isomeric products with different enantioselectivities (97:3 and 91:9 er). In addition, a TMS-containing enol triflate furnished product 3u in 86% yield and 90:10 er. An enol triflate containing a phthalimide provided product 3v in 58% yield and 91:9 er. Furthermore, (+)-nootkatone derivative 3w was produced in 31% yield and 8:1 dr. Estrone derivative 3x was synthesized in 79% yield and >20:1 dr. Lastly, the enol triflate derived from cholesterol delivered product 3y in 26% yield and 8:1 dr. During our investigation of chiral triflate reagents (1w–1y), we found that the (R)-t-BuPyrOx ligand gave superior diastereoselectivities (see ESI for additional details†).
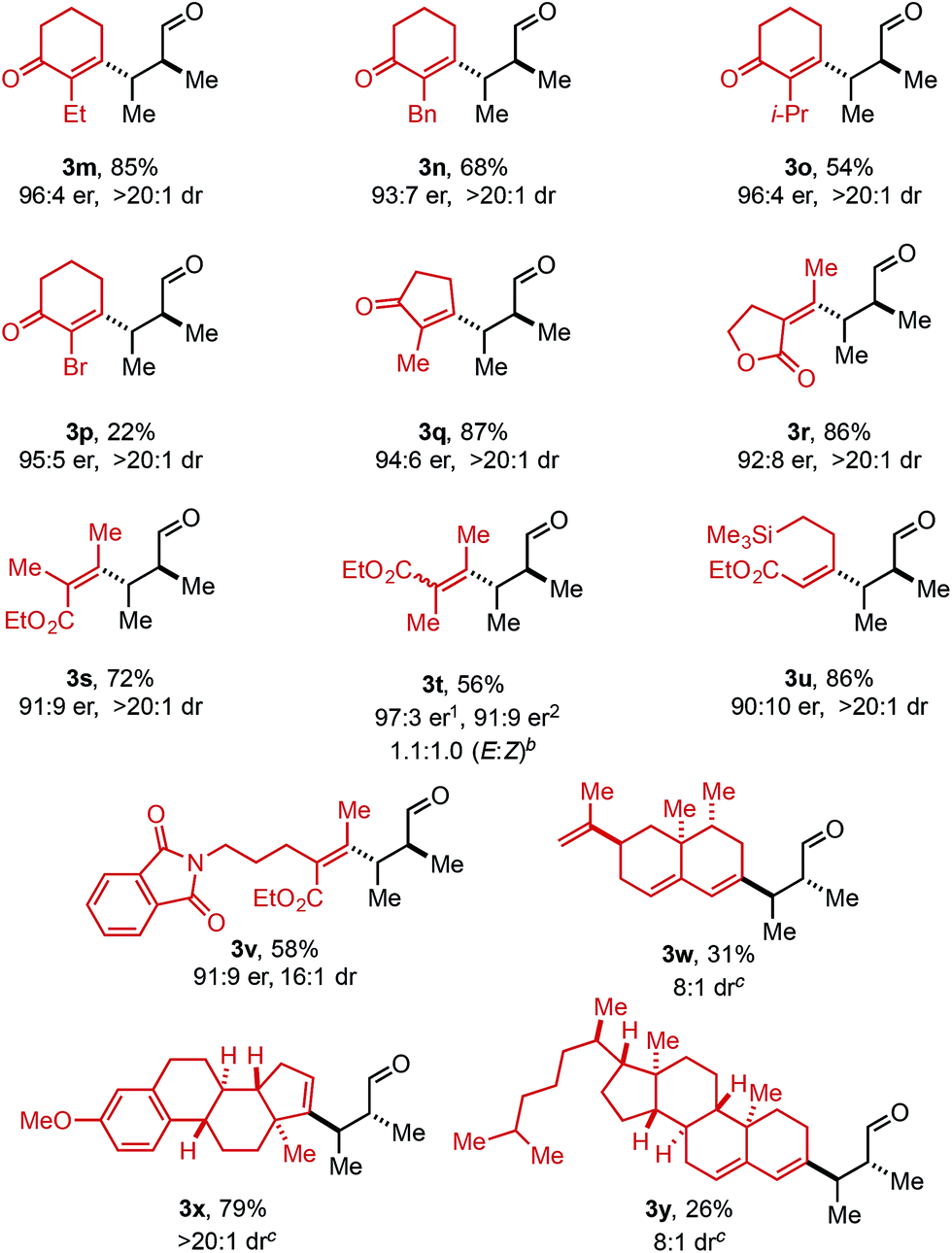
In an effort to expand this redox-relay strategy beyond allylic alkenols, homoallylic alkenol 2z was subjected to the optimized reaction conditions and gave product 3z in 31% yield and 89:11 er. This result, albeit promising, suggests the current system is optimized for allylic substrates.
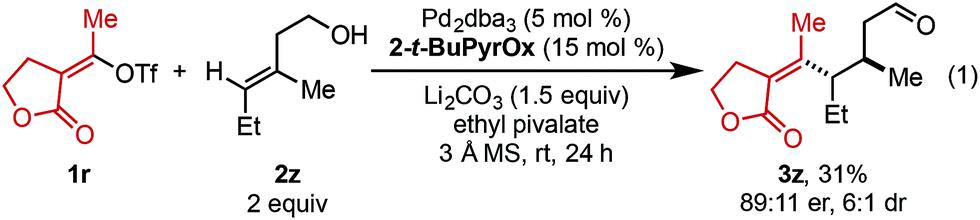
As this alkene class is distinct from others previously evaluated, we were interested in exploring how related substructures performed under these reaction conditions. Accordingly, several alkenes with unique substitution patterns were sdudied (Table 4). The model substrate, (E)-alkenol 2a, yielded the desired redox-relay product in 95:5 er. In contrast, (Z)-alkenol 2aa delivered the product in a significantly reduced enantioselectivity (57:43 er), underscoring the importance of the substitution pattern on the trisubstituted alkene for face selection. For comparison, disubstituted alkene 2ab provided the corresponding product in 89:11 er, while 1,1-disubstituted alkene 2ac gave product in 79:21 er.
|
|
|---|
| a The isolated yields of products 3a–3ac are shown. Each entry represents the isolated yield on 0.25 mmol scale. er values were determined by SFC or HPLC. |
|
The structural origin of the difference in enantioselectivity between the (E)- and the (Z)-alkenes was elucidated computationally in analogy to our previous work.9 The different conformations and configurations at the metal center in the stereodetermining alkene migratory insertion step for (E)-alkenol 2a and (Z)-alkenol 2aa with electrophile 1a mediated by a Pd–PyrOx catalyst were calculated using the M06/6-31+G*/LANL2DZ level of theory in G0914 (for details see ESI†). The lowest energy transition structures leading to the (S)- and (R)-products starting from 2a are shown in Fig. 1A. TSAR is 1.5 kcal mol−1 higher in free energy than TSAS, in good agreement with the experimentally determined selectivity (95:5 er, 1.74 kcal mol−1). This free energy difference is due to a through-space steric repulsion between a hydrogen on the carbon α to the hydroxyl group on the (E)-alkenol substrate and the t-Bu group on the ligand with an H–H distance of 2.03 Å in TSAR. This unfavorable interaction is not present in TSAS. Surprisingly, TSAS has the alkenol positioned proximally to the pyridine ring and the alkenyl group next to the t-Bu oxazoline portion of the ligand. As a result, a potentially stabilizing interaction between a hydrogen on the electron deficient pyridine ring and the hydroxyl group of the alkenol substrate with an H–O distance of 2.11 Å and a C–H–O angle of 144.9° is observed.
 | ||
| Fig. 1 Optimized geometries of the isomers of the transition states for substrates 2a and 2aa. Bond distances (in Å) and activation free energies are shown. Data and coordinates for these structures and all structures calculated can be found in the ESI.† | ||
Fig. 1B shows the lowest energy transition structures TSBS and TSBR leading to the (S)- and (R)-products for (Z)-alkenol 2aa. TSBR is only 0.7 kcal mol−1 higher in free energy than TSBS. This difference matches, within the expected error of the calculations, the experimentally determined selectivity (57:43 er). The free energy difference is smaller for 2aa due to a combination of several unfavorable interactions present in both transition structures. In TSBR, there are two close contacts between the t-Bu group on the ligand and the alkenyl substrate (H–H = 2.16 Å and H–H = 2.18 Å). In TSBS, there is a close contact between a hydrogen on an alkenol methyl group and a hydrogen on the pyridine ring of the ligand (H–H = 1.96 Å). The same potentially stabilizing interaction between a hydrogen on the electron deficient pyridine ring of the ligand and the hydroxyl group of the alkenol substrate seen in TSAS was also observed in both structures. The similarity of the interactions in TSBR and TSBS leads to the low selectivity observed experimentally for this substrate.
Conclusions
In summary, we have developed an enantioselective redox-relay Heck reaction of trisubstituted allylic alkenols to deliver vicinal stereocenters in generally high enantio- and diastereoselectivity. This reaction is conducted under mild reaction conditions that allows the construction of enolizable α-carbonyl methyl-substituted stereocenters with no or little observed epimerization. Systematic evaluation of the substitution pattern on the alkenol substrate has revealed the importance of the double bond geometry to achieve high enantioselectivity. Computational studies were used to explore this finding and showcase a significantly different network of interactions responsible for high enantioselectivity. This information will be utilized in the development of new variants of this reaction with an eye towards expansion to homoallylic alcohols and related substrate classes.Acknowledgements
The synthetic aspects were supported by National Institute of Health (NIGMS R01GM063540) and the modeling was supported by NSF (CHE-1361296 and CHE-1565669). We gratefully acknowledge the Center for High Performance Computing (CHPC) at the University of Utah and the Center for Research Computing (CRC) at the University of Notre Dame. We acknowledge former group members, Dr Vaneet Saini and Dr Cristiane S. Schwalm, for partial substrate syntheses.Notes and references
- (a) M. S. Sigman and E. W. Werner, Acc. Chem. Res., 2012, 45, 874 CrossRef CAS PubMed; (b) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed; (c) G. T. Crisp, Chem. Soc. Rev., 1998, 27, 427 RSC; (d) R. F. Heck, Acc. Chem. Res., 1979, 12, 146 CrossRef CAS.
- (a) C. Zhang, C. B. Santiago, J. M. Crawford and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 15668 CrossRef CAS PubMed; (b) C. Zhang, C. B. Santiago, L. Kou and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 7290 CrossRef CAS PubMed; (c) H. H. Patel and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 3462 CrossRef CAS PubMed; (d) T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340 CrossRef CAS PubMed; (e) T.-S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830 CrossRef CAS PubMed; (f) E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455 CrossRef CAS PubMed.
- For selected reviews on carbometallation, see: (a) M. Shibasaki, E. M. Vogl and T. Ohshima, Adv. Synth. Catal., 2004, 346, 1533 CrossRef CAS; (b) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed. For early examples of intermolecular asymmetric Heck reactions with acyclic olefins, see: (c) K. Yonehara, K. Mori, T. Hashizume, K.-G. Chung, K. Ohe and S. J. Uemura, J. Organomet. Chem., 2000, 603, 40 CrossRef CAS; (d) Y. Koga, M. Sodeoka and M. Shibasaki, Tetrahedron Lett., 1994, 35, 1227 CrossRef CAS. For recent examples of intermolecular asymmetric Heck reactions of acyclic electronically biased olefins, see: (e) K. S. Yoo, J. O'Neil, S. Sakaguchi, R. Giles, J. H. Lee and K. W. Jung, J. Org. Chem., 2010, 75, 95 CrossRef CAS PubMed; (f) K. S. Yoo, C. P. Park, C. H. Yoon, S. Sakaguchi, J. O'Neil and K. W. Jung, Org. Lett., 2007, 9, 3933 CrossRef CAS PubMed.
- For selected examples using a redox-relay strategy, see: (a) H. Renata, Q. Zhou and P. S. Baran, Science, 2013, 339, 59 CrossRef CAS PubMed; (b) S. Aspin, A.-S. Goutierre, P. Larini, R. Jazzar and O. Baudoin, Angew. Chem., Int. Ed., 2012, 51, 10808 CrossRef CAS PubMed; (c) B. J. Stokes, S. M. Opra and M. S. Sigman, J. Am. Chem. Soc., 2012, 134, 11408 CrossRef CAS PubMed; (d) N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854 CrossRef CAS PubMed.
- For examples of isomerization/migration reactions, see: (a) A. Vasseur, J. Bruffaerts and I. Marek, Nat. Chem., 2016, 8, 209 CrossRef CAS PubMed; (b) T. Kochi, T. Hamasaki, Y. Aoyama, J. Kawasaki and F. Kakiuchi, J. Am. Chem. Soc., 2012, 134, 16544 CrossRef CAS PubMed; (c) M. L. Crawley, K. M. Phipps, I. Goljer, J. F. Mehlmann, J. T. Lundquist, J. W. Ullrich, C. Yang and P. E. Mahaney, Org. Lett., 2009, 11, 1183 CrossRef CAS PubMed; (d) F. Berthiol, H. Doucet and M. Santelli, Tetrahedron, 2006, 62, 4372 CrossRef CAS; (e) Y. Wang, X. Dong and R. C. Larock, J. Org. Chem., 2003, 68, 3090 CrossRef CAS PubMed; (f) S. Bouquillon, B. Ganchegui, B. Estrine, F. Hénin and J. Muzart, J. Organomet. Chem., 2001, 634, 153 CrossRef CAS; (g) R. C. Larock, W.-Y. Leung and S. Stolz-Dunn, Tetrahedron Lett., 1989, 30, 6629 CrossRef CAS.
- For selected reviews on organocatalytic methods to form chiral aldehyde products, see: (a) M. J. Gaunt, C. C. C. Johansson, A. McNally and N. T. Vo, Drug Discovery Today, 2007, 12, 8 CrossRef CAS PubMed; (b) A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 CrossRef PubMed; (c) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed.
- For references about the significance of alkenyl motifs, see: (a) A. Saklani and S. K. Kutty, Drug Discovery Today, 2008, 13, 161 CrossRef CAS PubMed; (b) K. K.-C. Liu and L. S. Sakya, Mini-Rev. Med. Chem., 2004, 4, 1105 CrossRef CAS PubMed; (c) K. C. Nicolaou and S. A. Snyder, Classics in Total Synthesis II. More Targets, Strategies, Methods, Wiley-VCH, Weinheim, 2003 Search PubMed; (d) K. C. Nicolaou and E. J. Sorensen, Classics in Total Synthesis, VCH, Weinheim, 1996 Search PubMed.
- For reviews on the asymmetric Heck reaction, see: (a) D. M. Cartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122 RSC. For a review on the synthesis of vicinal stereocenters see: (b) E. A. Ilardi, C. E. Stivala and A. Zakarian, Chem. Soc. Rev., 2009, 38, 3133 RSC; (c) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363 CrossRef CAS PubMed; (d) H. Ito and T. Taguchi, Chem. Soc. Rev., 1999, 28, 43 RSC. For selected examples about the synthesis of vicinal stereocenters, see: (e) W.-B. Liu, C. M. Reeves, S. C. Virgil and B. M. Stoltz, J. Am. Chem. Soc., 2013, 135, 10626 CrossRef CAS PubMed; (f) B. M. Trost, N. Cramer and S. M. Silverman, J. Am. Chem. Soc., 2007, 129, 12396 CrossRef CAS PubMed; (g) B. M. Trost and Y. Zhang, J. Am. Chem. Soc., 2007, 129, 14548 CrossRef CAS PubMed.
- (a) M. J. Hilton, L.-P. Xu, P.-O. Norrby, Y.-D. Wu, O. Wiest and M. S. Sigman, J. Org. Chem., 2014, 79, 11841 CrossRef CAS PubMed; (b) L. Xu, M. J. Hilton, X. Zhang, P.-O. Norrby, Y.-D. Wu, M. S. Sigman and O. Wiest, J. Am. Chem. Soc., 2014, 136, 1960 CrossRef CAS PubMed.
- B. W. Michel, L. D. Steffens and M. S. Sigman, in Organic Reactions, John Wiley & Sons, Inc., 2014, vol. 84, pp. 75–414 Search PubMed.
- For examples demonstrating the relationship between selectivity and solvent, see: (a) D. A. MacManus and E. N. Vulfson, Enzyme Microb. Technol., 1997, 20, 225 CrossRef CAS; (b) C. Reichardt, Angew. Chem., Int. Ed. Engl., 1979, 18, 98 CrossRef.
- (a) G. Pescitelli and T. Bruhn, Chirality, 2016, 28, 466 CrossRef CAS PubMed; (b) E. C. Sherer, J. R. Cheeseman and R. T. Williamson, Org. Biomol. Chem., 2015, 13, 4169 RSC; (c) E. C. Sherer, C. H. Lee, J. Shpungin, J. F. Cuff, C. X. Da, R. Ball, R. Bach, A. Crespo, X. Y. Gong and C. J. Welch, J. Med. Chem., 2014, 57, 477 CrossRef CAS PubMed.
- (a) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051 RSC; (b) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (c) I. Marek, S. Simaan and A. Masarwa, Angew. Chem., Int. Ed., 2007, 46, 7364 CrossRef CAS PubMed; (d) A. de Meijere, Chem. Rev., 2003, 103, 931 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc04585e |
| This journal is © The Royal Society of Chemistry 2017 |