
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous direct anodic flow oxidation of aromatic hydrocarbons to benzyl amides†
Mikhail A.
Kabeshov
,
Biagia
Musio
 and
Steven V.
Ley
*
and
Steven V.
Ley
*
Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW, UK. E-mail: svl1000@cam.ac.uk
First published on 2nd November 2017
Abstract
The continuous production of benzyl amides by anodic oxidation in flow was developed. The stability and productivity of the equipment was examined over time and monitored by means of in-line UV analysis. The applicability of the method to twelve substrates was demonstrated.
Methods for the site-selective C–H functionalisation of hydrocarbons, without the need of installing an additional functional group, are becoming more widely used.1 Their exploitation in multistep synthesis allows reducing the number of steps, increases atom efficiency and reduces waste.2 Nevertheless, most of the modern C–H activation methods, although being usually chemo- and regioselective, require expensive and moisture- or air sensitive transition metal catalysts.3
Electrochemical methods for direct oxidation and reduction are increasingly important for modern synthetic chemistry mainly because they employ electric current instead of more obnoxious agents.4 Electrochemistry is therefore becoming more frequently used in the design of new atom-efficient, safe and more sustainable chemical processes.5 Also a range of products can sometimes be obtained from a common starting material by controlling either the electric current or potential.6
Carrying out electrochemical experiments in a continuous manner increases greatly their throughput and general applicability.7 A higher surface-to-volume ratio, characteristic for micro- and mesofluidic conditions, causes better conductivity of the reaction media, resulting in a lower concentration of the ancillary electrolyte required.8 Furthermore, a shorter distance between the anode and cathode enables the processes which involve highly unstable, reactive intermediates to proceed smoothly and with high level of selectivity due to fast subsequent chemical reactions.9 It has previously been shown that replacing batch electrolysis with the continuous methods can reduce the formation of the overoxidation products since the reactor is constantly replenished with the starting materials and the desired products are removed to a benign environment.7d
The discovery reported here relates to the development of an electrochemical method to convert aromatic hydrocarbons into the benzyl amides. It is well known that alcohols can be converted into amides by the Ritter reaction – when reacting with nitriles and water under strong acidic conditions.10 This reaction proceeds through the formation of a carbocation which is sequentially trapped first by nitrile followed by a molecule of water. If instead the carbocation is generated by two-electron oxidation from the hydrocarbon, following the same trapping steps, the amide can be obtained analogously. This reaction has been observed when toluene and a few electron-rich toluene derivatives were oxidized electrochemically at the anode in batch mode using acetonitrile as a solvent and an undivided cell.11 Unfortunately, these amides could only be isolated in low yields mainly due to overoxidation.11a Considering that flow electrochemistry methods are often advantageous over the batch alternatives in terms of synthetic applicability, efficiency and throughput, we focused on the development of a continuous direct anodic oxidation of aromatic hydrocarbons to benzyl amides.
All the experimental studies were performed using an Ammonite® 8 reactor (V = 1 mL)12 equipped with a platinum disk anode, circular stainless steel cathode, and perfluoroelastomer FFKM gasket. This unit, with the carbon polymer anode, was previously applied to the electrochemical methoxylation of N-formylpyrrolidine and p-methoxybenzyl alcohol deprotection allowing high conversion of the substrate in a single run.13
The reaction conditions for the continuous electrolysis at the constant current of p-tert-butyltoluene 1a in acetonitrile (0.1 M) as a model substrate were explored varying electrolyte, electric current and the concentration of water (Table 1).13
|
|
||||
|---|---|---|---|---|
| Entry | [2], M | [H2O], M | Current, A (charge, F) | Conversion 3a,b % |
| a Black triangle represents input solution, white triangle represents output solution. b Determined by 1H NMR using the calibrated external standard (neat TMSCl contained in the inner tube of a coaxial NMR tube). | ||||
| 1 | LiBF4, 0.1 | None | 0.20 (2.5) | 8 |
| 2 | LiBF4, 0.1 | 0.05 | 0.20 (2.5) | 60 |
| 3 | LiBF4, 0.1 | 0.1 | 0.20 (2.5) | 66 |
| 4 | LiBF4, 0.1 | 0.2 | 0.20 (2.5) | 56 |
| 5 | LiBF4, 0.05 | 0.1 | 0.20 (2.5) | 54 |
| 6 | LiBF4, 0.15 | 0.1 | 0.20 (2.5) | 59 |
| 7 | LiBF4, 0.1 | 0.1 | 0.23 (2.9) | 57 |
| 8 | Bu4NPF6, 0.01 | None | 0.20 (2.5) | 7 |
| 9 | Bu4NPF6, 0.01 | 0.1 | 0.20 (2.5) | 48 |
| 10 | Bu4NPF6, 0.01 | 0.12 | 0.20 (2.5) | 46 |
| 11 | Bu4NPF6, 0.01 | 0.2 | 0.20 (2.5) | 43 |
| 12 | Bu4NPF6, 0.01 | 0.1 | 0.23 (2.5) | 46 |
| 13 | Bu4NPF6, 0.01 | 0.1 | 0.26 (3.3) | 45 |

First, the efficiency of LiBF4 as an electrolyte was examined. It was shown that the presence of water was crucial for the reaction, since without water added only small amount of the product was detected (entry 1, Table 1). Stoichiometric amounts of water were found to be optimal as an excess led to a decrease in conversion to 3a (entries 2 and 4, Table 1). Similarly, the use of stoichiometric amount of LiBF4 also provided the highest conversion to 3a (compare entries 5 and 6 with entry 3, Table 1). The lower conversion to 3a was observed at a higher current due to over-oxidation with the p-tert-butylbenzaldehyde formation (determined by 1H NMR in the crude reaction mixture) while some of the starting material 1a was still present (entry 7, Table 1).14
Under the optimum conditions when using LiBF4 as an electrolyte (entry 3, Table 1), it was found that the system was not stable over time and that the conversion towards the amide 3a deteriorated significantly (from 66% to 38% in 3 hours). The formation of an inorganic solid inside the cell channel was observed over the time that caused clogging and voltage instability. This solid, which reacted violently with water with the formation of hydrogen gas, is likely to contain lithium metal in agreement with the previous reports.11c Subsequent loss of the current efficiency was therefore the most probable reason for the decreased conversion over time.
In order to find sustainable alternative conditions, the anodic oxidation of p-tert-butyltoluene 1a was optimised using an electroneutral electrolyte Bu4NPF6 (entries 8–13, Table 1). As in the case of LiBF4, a stoichiometric amount of water was found to be optimal for the experiments with Bu4NPF6 (entries 8–11, Table 1) providing the highest conversion to the desired product 3a (48% conversion; entry 9, Table 1). Applying higher current did not improve the conversion due to the overoxidation side reactions (entries 12 and 13, Table 1). Under these new conditions, the system remained stable and fully homogeneous over time.
One possible reason of the higher efficiency of the system based on the LiBF4 electrolyte during the initial period, when compared to Bu4NPF6, could be the difference in the pH of the two reaction mixtures (Scheme 1).
 | ||
| Scheme 1 Electrochemical processes for the systems using LiBF4 and Bu4NPF6 as electrolytes. | ||
In the case of LiBF4, the lithium cation Li+ is initially readily reduced at the cathode not affecting the pH of the reaction mixture (red rectangle, Scheme 1). On the other hand, the anodic oxidation produces 1 eq. of H+ per 1 F of electric current passed, resulting in an acidic pH of the reaction mixture as confirmed experimentally (pH ∼ 0–1). Conversely, when Bu4NPF6 is used as an electrolyte, as 1 eq. of H+ per 1 F of electric current passed is produced in the same way at the anode, 1 eq. of H+ per 1 F is consumed at the cathode, maintaining the overall neutral pH of the reaction mixture (green rectangle, Scheme 1). Knowing that the reduction potential E(2H+/H2) at the cathode is proportional to ln([H+]) according to the Nernst equation, the reduction of H+ to H2 and the overall electrolysis should proceed easier and at a lower voltage in the presence of a Brønsted acid.
With this information available, the influence of various Brønsted acid additives on the anodic oxidation of p-tert-butyltoluene 1a was explored (Table 2).
| Entry | Acid additive | [Acid], M | Conversion 3a,b % |
|---|---|---|---|
a c(1a) = c(H2O) = 0.1 M, c(Bu4NPF6) = 0.01 M, flow rate 500 mL min−1.
b Determined by 1H NMR using the calibrated external standard (neat TMSCl contained in the inner tube of a coaxial NMR tube); isolated yield in brackets.
c Solvent system: acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dichloromethane = 3:1. :dichloromethane = 3:1.
|
|||
| 1 | TsOH·2H2O | 0.1 | N/A |
| 2c | TsOH·2H2O | 0.1 | 48 |
| 3 | TFA | 0.1 | 68 |
| 4 | MSA | 0.1 | 72 (63) |
| 5 | MSA | 0.05 | 60 |
| 6 | MSA | 0.13 | 70 |
The continuous flow electrolysis could not be performed in pure acetonitrile as a solvent and toluenesulfonic acid (pKa = −2.8) as an additive due to a solid formation (entry 1, Table 2). The addition of dichloromethane as a co-solvent (25% vol) was beneficial to keep the system homogeneous, but the amide 3a was still obtained with low conversion (48% conversion; entry 2, Table 2). When using trifluoroacetic acid (TFA; pKa = 0.23), a higher conversion to 3a was observed (68% conversion; entry 3, Table 2). This result could be further improved by using methanesulfonic acid (MSA; pKa = −1.9; 72% conversion; entry 4, Table 2). Changing the concentration of MSA did not lead to any further improvement in the formation of 3a (entries 5 and 6, Table 2). Under the optimised conditions (entry 4, Table 2) the system remained fully homogeneous over the time.
Next, the stability of the electrolytic system was investigated (Fig. 1). Thus a mixture of the hydrocarbon 1a (0.1 M), H2O (0.1 M), MSA (0.1 M) and Bu4NPF6 (0.01 M) in acetonitrile was continuously passed (500 μL min−1) through the reactor applying constant current of 0.20 A (2.5 F).
 | ||
| Fig. 1 Anodic oxidation of p-tert-butyltoluene 1a monitored by Flow-UV™. | ||
An online steady state monitoring was best achieved by means of a UV/vis detector (Flow-UV™)15 which was installed after the reactor (Fig. 1). The reaction pleasingly proceeded with the constant productivity of the amide 3a equal to 0.38 g h−1 over 9 hours (Fig. 1).
Once the stability and the applicability of the developed procedure to a large scale production were demonstrated, the generality of the methodology over a range of aromatic hydrocarbons was studied (Scheme 2). To simplify the downstreaming process, the efflux of the reactor was mixed with a second stream (500 μL min−1) containing an ammonia solution in methanol (0.7 M). Full neutralisation of the acid was beneficial to reduce decomposition of the amide products 3 under strong acidic conditions. The evaporation of solvent in vacuo was sufficient to obtain the crude products 3 (Scheme 2).
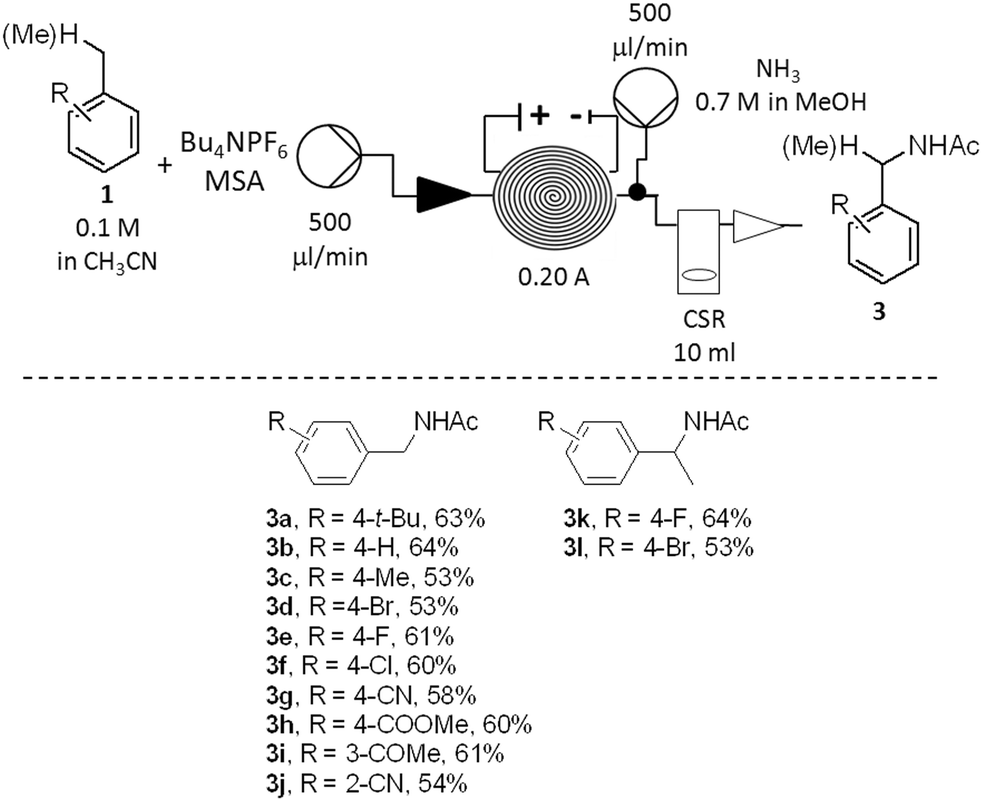 | ||
| Scheme 2 Scope of the anodic oxidation of aromatic hydrocarbons (isolated yields are listed). | ||
The amide derivatives of various para- (3a–3g; 3k–3l), meta- (3i) and ortho-substituted (3j) benzene derivatives were obtained in good yields. The anodic oxidation was successful for the preparation of either moderately electron-rich (3a, 3c), electron-neutral (3b, 3f, 3k) or electron-poor (3d, 3f–3j, 3l) derivatives. A number of functional groups were tolerated by the method: such as halogen (3d–3f, 3k–3l), nitrile (3g, 3j), ketone (3i), ester (3h). The synthesis of both primary (3a–3j) and secondary (3k–3l) benzyl amides was also successfully achieved. In all the cases, the amides 3 were isolated as the major products. Residual starting materials were the second major components of all the reaction mixtures while only traces of other by-products, such as substituted benzaldehydes or benzyl alcohols, were minor components (<2%).
Conclusions
In conclusion, the preparation of benzyl amides from a number of aromatic hydrocarbons was achieved by a stable and continuous flow anodic oxidation. The addition of a Brønsted acid was crucial to maintain the process stability over 9 hours with the productivity of 9.12 g per day. The results described here open up to the application of electrochemical methodologies to large-scale production.Conflicts of interest
There are no conflicts or financial interest to declare.Acknowledgements
The authors are grateful to EPSRC (grants EP/K009494/1 and EP/K039520/1) for financial support. The authors are also grateful to Dr Bashir Harji (Cambridge Reactor Design Ltd) for technical support and assistance.Notes and references
- (a) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016 CrossRef CAS PubMed; (b) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (c) S. M. Paradine, J. R. Griffin, J. Zhao, A. L. Petronico, S. M. Miller and M. C. White, Nat. Chem., 2015, 7, 987 CrossRef CAS PubMed.
- (a) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (b) D. Y.-K. Chen and S. W. Youn, Chem. – Eur. J., 2012, 18, 9452 CrossRef CAS PubMed; (c) C. Leitner and T. Gaich, Chem. Commun., 2017, 53, 7451 RSC.
- (a) K. S. Egorova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 12150 CrossRef CAS PubMed; (b) F. Rodesly, J. Oble and G. Poli, J. Mol. Catal. A: Chem., 2017, 426, 275 CrossRef; (c) N. V. Tzouras, I. K. Stamatopoulos, A. T. Papastavrou, A. A. Liori and G. C. Vougioukalakis, Coord. Chem. Rev., 2017, 343, 25 CrossRef CAS.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2017 DOI:10.1002/anie.201707584 , ASAP.
- (a) K. D. Moeller, Synlett, 2009, 8, 1208 CrossRef; (b) B. R. Rosen, E. W. Werner, A. G. O'Brien and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5571 CrossRef CAS PubMed; (c) J. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2009, 108, 2265 CrossRef PubMed; (d) E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302 CrossRef CAS PubMed; (e) A. G. O'Brien, A. Maruyama, Y. Inokuma, M. Fujita and P. S. Baran, Angew. Chem., Int. Ed., 2014, 53, 11868 CrossRef PubMed; (f) H.-B. Zhao, Z.-J. Liu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 12732 CrossRef CAS PubMed; (g) X.-Y. Qian, S.-Q. Li, J. Song and H.-C. Xu, ACS Catal., 2017, 7, 2730 CrossRef CAS.
- G. Laudadio, N. J. W. Straathof, M. D. Lanting, B. Knoops, V. Hessel and T. Noël, Green Chem., 2017, 19, 4061 RSC.
- (a) D. S. P. Cardoso, B. Šljukić, D. M. F. Santos and C. A. C. Sequeira, Org. Process Res. Dev., 2017, 21, 1213–1226 CrossRef CAS; (b) K. Watts, W. Gattrell and T. Wirth, Beilstein J. Org. Chem., 2011, 7, 1108 CrossRef CAS PubMed; (c) S. Suga, M. Okajima, K. Fujiwara and J.-I. Yoshida, J. Am. Chem. Soc., 2001, 123, 7941 CrossRef CAS PubMed; (d) J. Kuleshova, J. T. Hill-Cousins, P. R. Birkin, R. C. D. Brown, D. Pletcher and T. Underwood, Electrochim. Acta, 2012, 69, 197 CrossRef CAS; (e) R. Hayashi, A. Shimizu, Y. Song, Y. Ashikari, T. Nokami and J.-I. Yoshida, Chem. – Eur. J., 2017, 23, 61 CrossRef CAS PubMed; (f) D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2017 DOI:10.1021/acs.chemrev.7b00360 , ASAP.
- (a) R. A. Green, R. C. D. Brown and D. Pletcher, J. Flow Chem., 2016, 6, 191 CrossRef; (b) M. Atobe, H. Tateno and Y. Matsumura, Chem. Rev., 2017 DOI:10.1021/acs.chemrev.7b0353 , ASAP; (c) Y. Liu and X. Jiang, Lab Chip, 2017 10.1039/c7lc00627f , ASAP.
- M. A. Kabeshov, B. Musio, P. R. D. Murray, D. L. Browne and S. V. Ley, Org. Lett., 2014, 16, 4618 CrossRef CAS PubMed.
- (a) D. Jiang, T. He, L. Ma and Z. Wang, RSC Adv., 2014, 4, 64936 RSC; (b) A. Guérinot, S. Reymond and J. Cossy, Eur. J. Org. Chem., 2012, 19 CrossRef; (c) J. J. Ritter and P. P. Minieri, J. Am. Chem. Soc., 1948, 70, 4045 CrossRef CAS PubMed; (d) R. Sanz, A. Martinez, V. Guilarte, J. M. Alvarez-Gutierrez and F. Rodriguez, Eur. J. Org. Chem., 2007, 4642 CrossRef CAS.
- (a) I. N. Rozhkov and I. Y. Alyev, Tetrahedron, 1975, 31, 977 CrossRef CAS; (b) L. Eberson and B. Olofsson, Acta Chem. Scand., 1969, 23, 2355 CrossRef CAS; (c) E. A. Mayeda and L. L. Miller, Tetrahedron, 1972, 28, 3375 CrossRef CAS; (d) T. Tajima, H. Ishii and T. Fuchigami, Electrochem. Commun., 2002, 4, 589 CrossRef CAS.
- Ammonite by Cambridge reactor Design (CRD): http://www.cambridgereactordesign.com/ammonite/ammonite.html (last visited on 19/09/17) Search PubMed.
- (a) R. A. Green, R. C. D. Brown, D. Pletcher and B. Harji, Electrochem. Commun., 2016, 73, 63 CrossRef CAS; (b) R. A. Green, R. C. D. Brown and D. Pletcher, Org. Process Res. Dev., 2015, 19, 1424 CrossRef CAS; (c) R. A. Green, K. E. Jolley, A. A. M. Al-Hadedi, D. Pletcher, D. C. Harrowven, O. De Frutos, C. Mateos, D. J. Klauber, J. A. Rincón and R. C. D. Brown, Org. Lett., 2017, 19, 2050 CrossRef CAS PubMed.
- Lower flow rates with the current adjusted did not affect conversion to 3a whereas the concentration of the substrate 1a is reported to be optimal for 2e− oxidation processes using the Ammonite 8 set-up .
- Flow-UV by Uniqsis Ltd: http://www.uniqsis.com/paProductsDetail.aspx?ID=Flow-UV (last visited on 19/09/17) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental methods, general procedure for the anodic oxidation, characterization of the synthesized compounds and copies of NMR spectra. See DOI: 10.1039/c7re00164a |
| This journal is © The Royal Society of Chemistry 2017 |