
Photo-induced ring-closure via a looped flow reactor†
Evelien
Baeten
ab,
Maarten
Rubens
a,
Kilian N. R.
Wuest
cd,
Christopher
Barner-Kowollik
 *cd and
Tanja
Junkers
*ab
*cd and
Tanja
Junkers
*ab
aPolymer Reaction Design Group, Institute for Materials Research (IMO), Universiteit Hasselt, Martelarenlaan 42, 3500 Hasselt, Belgium. E-mail: tanja.junkers@uhasselt.be
bIMEC Associated Lab IMOMEC, Wetenschapspark 1, 3590 Diepenbeek, Belgium
cMacromolecular Architectures Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128 Karlsruhe, Germany
dSchool of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia. E-mail: christopher.barnerkowollik@qut.edu.au
First published on 26th October 2017
Abstract
Looped flow processes are an efficient and versatile tool to synthesize complex macromolecular materials. Especially for light-induced ring-closure reactions, which typically require low concentrations, looped flow processes are critical for upscaling. Here, such a reactor was designed to carry out a photo-induced ring-closure reaction via a photo-enol reaction for the synthesis of cyclic polymers, leading to a reduction of required solvent in the synthesis by over a factor of 40, hence giving access to a more economical and fully scalable process.
Continuous flow processes have been increasingly investigated as an alternative to conventional batch chemistry in the last decade. Providing a high level of control over reaction parameters, fast heat exchange and high reaction efficiencies, continuous flow approaches are of particular interest for organic synthesis, yet increasingly so in the field of advanced polymer design.1–9 Contemporary research focuses on the use of specialized reactor set-ups for specific reactions and on the use of reactor cascades (multistep reactions in ‘one flow’) to carry out several chemical transformations in sequence.10–12 Recently, looped flow processes – where the main reactor consists of a closed looped tubular system – have been (re)investigated for the synthesis of complex macromolecular materials.13 In 1990, liquid phase (free radical) polymerizations of olefins were carried out in a ‘loop reactor’ on an industrial scale.14–16 Later, emulsion polymerizations were carried out in a continuous ‘loop reactor’.17,18 Only recently, the first ‘looped flow process’ for controlled radical polymerizations was reported, where well-defined multiblock copolymers were prepared via reversible addition fragmentation chain transfer (RAFT) polymerization.13 Yet, similar processes are also achievable with near identical efficiency via linear continuous flow processes.12 Thus, the main potential of looped flow processes – the ability to use higher reactant concentrations compared to a batch process as well as gaining flexibility in reaction times compared to linear flow systems – has not fully been exploited in the polymer field.
The principle of looped flow processes is based on the use of a recycle loop, whereby the solution circulates in a closed reactor circuit. Looped flow processes are not truly continuous since no outlet flow is generated. Yet, such reactors have the same advantages as other continuous flow processes due to similar reactor characteristics. Thereby, they allow the gradual addition of reagents, as equivalent to the batchwise ‘slow-addition’ method via the use of a dropping funnel or a syringe pump. Generally, such methods can be employed to reduce side reactions or to establish higher product concentrations. Hence, the productivity of the process can be increased significantly when compared to the original batch or linear continuous flow processes. Here, a looped flow reactor was designed to carry out a photo-induced ring-closure reaction generating cyclic polymers (refer to Scheme 1). The combination between a photochemical transformation and a loop reactor is intriguing. Photoreactions allow for localization of the reaction and hence differentiation is possible between the loop and the reservoir with respect to where the reaction is occurring. While the loop can herein provide defined reaction conditions and superior light efficiency in the small diameter tubing, no reaction will occur in the reservoir. This is in sharp contrast to thermally activated reactions, which would occur then in the flow and the batch compartment of the reactor setup. Cyclic polymers are interesting because of their more compact nature compared to their linear analogues and are thus the most simplistic version of a compacted polymer chain.19–21 Generally, cyclic polymers have a smaller hydrodynamic volume compared to their linear counterparts, but also exhibit higher glass transition temperatures, lower intrinsic viscosities and higher critical solution temperatures.22–28 Yet, to synthesize these cyclic polymers in high purity, a highly diluted (<0.1 g L−1) reaction solution is typically required to avoid intermolecular coupling. This seriously hampers scalability when a traditional process is used and requires excessive amounts of solvent even for small product quantities. A first flow process for the synthesis of cyclic polymers has already been described, yet requires 17.3 L of solvent to produce 1 g of cyclic polystyrene.23 While in this study the flow reactor already helped to reach higher reaction efficiencies and made handling of large solvent volumes somewhat easier, the core problem – the sheer use of large volumes of solution – was not solved. Hence, a looped flow process is developed herein as an alternative, in order to reduce the required amount of solvent by increasing the product concentration.
 | ||
| Scheme 1 Light-induced ring-closure reaction between the dithioester and o-methyl-substituted aromatic aldehyde end groups of a linear methacrylate precursor polymer. | ||
First, a flow reactor is constructed to carry out a light-induced reaction in the reactor loop (refer to Scheme 2 and Fig. S1†). The main feature of the looped flow reactor is the loop pump (‘L’), providing a continuous recycle stream from the reservoir (‘R’, solvent + product) through the loop. An injection pump (‘P’) takes care of injecting the α,ω-functionalized linear precursor gradually into the reactor system via the use of a check valve (‘C’). The injected solution is immediately diluted with the recycle stream via the use of a static mixing tee (‘M’), after which it directly undergoes the light-induced ring-closure reaction under the influence of a UV lamp (peak wavelength of 312 nm). Via the difference in flow rate between the loop and the dosing from pump P, no significant volume increase in the loop is required when injecting the precursor from a concentrated solution at a low rate. Since the starting material concentration in this setup is at all times low – while the product can accumulate – it is possible to work in a highly diluted concentration regime without the need for excessive amounts of solvent. More technical details on the setup can be found in the ESI.† The advantages of such a setup are evident. The loop features small optical path lengths that allow the photoreaction to be conducted with very high efficiency and for short residence times. Illumination of a batch reactor at the same concentration regime would result in slower reaction rates. At the same time, the reactor would be readily scaled. By using flow reactors, photoreactions can be easily scaled to several hundreds of grams per reactor run without loss of reaction efficiency. Hence, by simply extending the length of the loop (and adjustment of the flow rate to maintain the same loop residence time), an upscale can be reached. Since herein only a proof of concept of the reactor concept is given, a relatively small loop volume was chosen for efficient use of the reactants.
 | ||
| Scheme 2 Looped flow reactor representation for the light-induced ring-closure of an α,ω-functionalized linear precursor toward cyclic polymers. | ||
For cyclization, the well-known photo-enol ring-closure reaction is used, based on a difunctional RAFT agent (see Scheme 1).29–31 The photo-enol reaction is light-triggered, which provides convenient access to reaction conditions which cause the reaction to take place only in the reaction loop, but not in the syringe. In addition, the reaction of a photo-enol with dithiobenzoates is very fast, allowing for high conversions on a short time scale. Using a flow reactor increases the efficiency of the light reaction even further.32,33 An α,ω-functionalized linear polymethacrylate (PMA) was prepared as a precursor polymer via RAFT polymerization with an α-methylbenzaldehyde functional RAFT agent. To ensure a high end group fidelity, a temperature of 70 °C and a low azobisisobutyronitrile (AIBN) equivalence (0.05 eq. compared to the RAFT agent) were employed, while monomer conversions of less than 100% were targeted. The α,ω-functionalized linear PMA precursor was obtained with 68% conversion, a number average molecular weight of 7000 g mol−1 and a dispersity of 1.3 (refer to Fig. 1 and the ESI† for more details). The slight broadening can be explained by the non-ideal match between the acrylate monomer and the dithiobenzoate RAFT agent. Yet, the presence of light cannot also be completely avoided during sample preparation, leading to the presence of a high molecular weight shoulder related to the intermolecular coupling of several polymer chains. After isolation of the linear polymer, the light-induced coupling of the functional end groups of this precursor polymer was employed to investigate the required reaction conditions. Therefore, a 5 mg mL−1 solution was prepared by dissolving 19 mg precursor polymer in 3.8 mL acetonitrile and purging with argon. As for the reactor set-up, the looped flow reactor was employed, thus injecting the precursor polymer via a syringe pump and diluting it with pure acetonitrile via the loop pump to mimic the recycle stream. However, the reaction mixture was not recirculated back into the reactor but was collected instead for analysis. Initially, the precursor polymer was diluted by a factor of 50, leading to a total precursor polymer concentration of 0.1 g L−1. Despite the low concentration, the light-induced reaction is extremely fast. A complete shift of the polymer distribution to lower apparent molecular weights – caused by the smaller hydrodynamic volume of the cyclic polymer – could be observed (Fig. 1), indicating the quantitative conversion for 15 s and 30 s residence times. No significant differences could be observed between the cyclic polymer formed after 15 s and after 30 s residence times, yet to stay on the side of caution, 30 s residence time was employed for further testing. Still, an increase of the high molecular weight fraction within the distribution is observed for both residence times, indicating the formation of intermolecular coupling products during the cyclization reaction.
 | ||
| Fig. 1 GPC chromatograms of the linear precursor polymer and the obtained cyclic polymers after 15 s and 30 s residence times (with a concentration of 0.1 g L−1). | ||
Thus, to reduce this intermolecular coupling, different precursor polymer concentrations were tested by varying the flow rate of the injection pump. Three initial concentrations were assessed: 0.1 g L−1, 0.05 g L−1 and 0.025 g L−1, the results of which are given in Table 1 and Fig. 2 (it should be noted that the reaction was in total screened in a broader concentration and residence time range; however, only relevant data is discussed herein). The success of the intramolecular coupling is at all times clearly indicated by the shift to lower apparent molecular weights. Critically, a clear decrease of intermolecular coupling is observed when decreasing the concentration of the linear precursor. Although the differences between 0.05 g L−1 and 0.025 g L−1 are minimal, 0.025 g L−1 was employed for further experiments to err on the side of caution and to assure maximum purity of the produced cyclic polymer.
| Type of polymer | Concentration (g L−1) | M appn (g mol−1) | Đ | M appp (g mol−1) |
|---|---|---|---|---|
| Precursor | — | 7100 | 1.25 | 8800 |
| Cyclic | 0.1 | 7200 | 1.26 | 7900 |
| Cyclic | 0.05 | 6500 | 1.29 | 8000 |
| Cyclic | 0.025 | 6400 | 1.26 | 8200 |
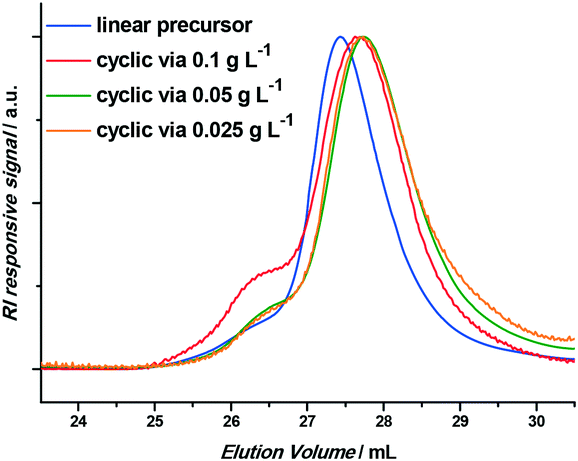 | ||
| Fig. 2 GPC chromatograms of the linear precursor polymer and the obtained cyclic polymers when employing 0.1 g L−1, 0.05 g L−1 and 0.025 g L−1 as precursor polymer concentrations. | ||
Next, the cyclic polymers were prepared via the use of the looped flow reactor by employing the conditions derived above. Hence, a concentration of 0.025 g L−1 of the α,ω-functionalized linear precursor was established by injecting the precursor polymer directly into the loop reactor. Therefore, a 5 mg mL−1 precursor polymer solution was injected into the loop reactor, where it was directly diluted to 0.025 g L−1. The light-induced ring-closure took place in the reactor loop with a residence time of 30 s by injecting the precursor polymer with a flow rate of 10 μL min−1. Simultaneously, the loop pump was used to dilute the precursor polymer with the solvent/product mixture (initial solvent volume of 10 mL) at a flow rate of 2 mL min−1. The reaction was allowed to proceed for 16 h 40 min, until 50 mg precursor polymer was converted into the cyclic polymer in a total volume of 20 mL. The cyclic polymer was collected in a quantitative manner by evaporating the solvent/polymer mixture. Hence, no purification is required. Since the precursor linear polymer and its corresponding RAFT agent are not easily available in larger quantities, no further upscale was carried out. Since the reaction conditions were chosen quite conservatively and because a volume increase in the looped flow reactor could be realized without any problems (refer to the discussion above), production of larger product quantities significantly above the gram scale should proceed without problems.
Compared to previously reported flow procedures, our looped flow process features clear advantages. Zhang and coworkers generated 1 g cyclic polystyrene in 17.3 L, in 3 h by using a reactor with an internal volume of 200 mL. In comparison, our production rate seems rather low (50 mg in 16 h 40 min), yet this is related to the employed reactor with a loop volume of 1 mL rather than the reactor design as noted above. By employing a reactor volume of 20 mL (which can be realized without problems with the same light source by elongating the length of the reactor tubing), equally 1 g would be accessible in the same product quality from our design, whereby the overall volume of solvent would not need to be increased at the same factor. Yet, the major advantage of a looped flow process is clearly the ability to reduce the required solution volumes by establishing a higher product concentration. Consequently, the total required solution volume was decreased by a factor of 43 compared to the reported procedure by Zhang and coworkers.23 Taking into account that a reservoir volume increase is only required to a certain extent to allow for efficient eluent pumping, an improvement of a factor of 400 can be projected for a loop reactor that is scaled to 1 g production.
Conclusions
Looped flow processes provide a continuous alternative to the batchwise ‘slow-addition’ method, by allowing the gradual addition of reagents. Higher product concentrations can be established and thus the productivity of a process can be increased greatly. The looped flow process thus provides a straightforward upscalable procedure to all reactions limited by their concentrations, i.e. the synthesis of cyclic polymers in the present example. To synthesize these cyclic polymers with high purity, a ring-closure coupling reaction must be carried out in highly diluted reaction solutions (<0.1 g L−1). In comparison to previously reported procedures, the required solution volumes could be reduced by a factor of 43.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. B.-K. acknowledges the Queensland University of Technology (QUT) for key support as well as the Australian Research Council (ARC) for support in the form of a Laureate Fellowship. Additional support from the Karlsruhe Institute of Technology (KIT) in the context of the Helmholtz STN program is gratefully acknowledged. E. B., M. R. and T. J. are grateful for funding from the Fonds Wetenschappelijk Onderzoek (FWO). Further, support in the framework of the IAP-PAI P7/05 Functional Supramolecular Systems (BELSPO) is kindly acknowledged.Notes and references
- C. Tonhauser, A. Natalello, H. Löwe and H. Frey, Macromolecules, 2012, 45, 9551–9570 CrossRef CAS.
- D. Wilms, J. Klos and H. Frey, Macromol. Chem. Phys., 2008, 209, 343–356 CrossRef CAS.
- K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406–446 CrossRef PubMed.
- T. Junkers, Macromol. Chem. Phys., 2017, 218, 1600421 CrossRef.
- B. J. Reizman and K. F. Jensen, Acc. Chem. Res., 2016, 49, 1786–1796 CrossRef CAS PubMed.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J.-C. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67 CrossRef CAS PubMed.
- H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noel, Chem. Soc. Rev., 2016, 45, 83–117 RSC.
- I. M. Mándity, S. B. Ötvös and F. Fülöp, ChemistryOpen, 2015, 4, 212–223 CrossRef PubMed.
- I. M. Mándity, S. B. Ötvös, G. Szőlősi and F. Fülöp, Chem. Rec., 2016, 16, 1018–1033 CrossRef PubMed.
- D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680 RSC.
- J.-C. M. Monbaliu, N. Emmanuel and R. Gérardy, Chim. Nouv., 2016, 122, 18–26 Search PubMed.
- E. Baeten, J. J. Haven and T. Junkers, Polym. Chem., 2017, 8, 3815–3824 RSC.
- A. Kuroki, I. Martinez-Botella, C. H. Hornung, L. Martin, E. G. L. Williams, K. E. S. Locock, M. Hartlieb and S. Perrier, Polym. Chem., 2017, 8, 3249–3254 RSC.
- M. A. Ferrero and M. G. Chiovetta, Polym.-Plast. Technol. Eng., 1990, 29, 263–287 CrossRef CAS.
- J. D. Hottovy, F. C. Lawrence, B. W. Lowe and J. S. Fangmeier, Phillips Petroleum Company, Patent CA2044782A1, 1990.
- J. J. Zacca and W. H. Ray, Chem. Eng. Sci., 1993, 48, 3743–3765 CrossRef CAS.
- C. Abad, J. C. De La Cal and J. M. Asua, Chem. Eng. Sci., 1994, 49, 5025–5037 CrossRef CAS.
- D.-Y. Lee, J.-H. Wang and J.-F. Kuo, Polym. Eng. Sci., 1992, 32, 198–205 CAS.
- B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202–2213 RSC.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251–284 CrossRef CAS.
- R. Elupula, J. Oh, F. M. Haque, T. Chang and S. M. Grayson, Macromolecules, 2016, 49, 4369–4372 CrossRef CAS.
- X.-Y. Tu, M.-Z. Liu and H. Wei, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1447–1458 CrossRef CAS.
- P. Sun, J. Liu, Z. Zhang and K. Zhang, Polym. Chem., 2016, 7, 2239–2244 RSC.
- C. D. Roland, H. Li, K. A. Abboud, K. B. Wagener and A. S. Veige, Nat. Chem., 2016, 8, 791–796 CrossRef CAS PubMed.
- J. N. Hoskins and S. M. Grayson, Polym. Chem., 2011, 2, 289–299 RSC.
- Y. Tezuka and H. Oike, Prog. Polym. Sci., 2002, 27, 1069–1122 CrossRef CAS.
- Y. Zhu and N. S. Hosmane, ChemistryOpen, 2015, 4, 408–417 CrossRef CAS PubMed.
- T. Josse, J. De Winter, P. Gerbaux and O. Coulembier, Angew. Chem., Int. Ed., 2016, 55, 13944–13958 CrossRef CAS PubMed.
- K. K. Oehlenschlaeger, J. O. Mueller, N. B. Heine, M. Glassner, N. K. Guimard, G. Delaittre, F. G. Schmidt and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2013, 52, 762–766 CrossRef CAS PubMed.
- T. Gruendling, K. K. Oehlenschlaeger, E. Frick, M. Glassner, C. Schmid and C. Barner-Kowollik, Macromol. Rapid Commun., 2011, 32, 807–812 CrossRef CAS PubMed.
- M. Kaupp, T. Tischer, A. F. Hirschbiel, A. P. Vogt, U. Geckle, V. Trouillet, T. Hofe, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2013, 46, 6858–6872 CrossRef CAS.
- T. Junkers and B. Wenn, React. Chem. Eng., 2016, 1, 60–64 CAS.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00124j |
| This journal is © The Royal Society of Chemistry 2017 |