
Epoxidation of 1-octene under harsh tail-end conditions in a flow reactor II: impact of delaminated-zeolite catalyst surface area and structural integrity on catalytic performance†
Martina
Aigner
 a,
Nicolás Andrés
Grosso-Giordano
a,
Christian
Schöttle
a,
Alexander
Okrut
*a,
Stacey
Zones
*b and
Alexander
Katz
*a
a,
Nicolás Andrés
Grosso-Giordano
a,
Christian
Schöttle
a,
Alexander
Okrut
*a,
Stacey
Zones
*b and
Alexander
Katz
*a
aDepartment of Chemical and Biomolecular Engineering, University of California at Berkeley, Berkeley, CA 94720, USA. E-mail: alexander.okrut@berkeley.edu; askatz@berkeley.edu
bChevron Energy Technology Company, Richmond, CA 94801, USA. E-mail: SIZO@chevron.com
First published on 12th October 2017
Abstract
Building on our previous study of delaminated-zeolite catalysts for harsh tail-end conditions in an epoxidation flow reactor employing organic hydroperoxide as oxidant, this manuscript compares approaches for delamination in catalyst prepration. In one, a mild method of delamination is used for synthesis of Ti-UCB-4, in which fluoride in organic solvent is used as a mineralizing agent to affect delamination, while in another, catalyst Ti-SSZ-70-DEL-HIGHPH is synthesized by delamination under high-pH conditions and results in the highest external surface area, similar to that previously reported for ITQ-2. We also compare both materials to the calcined 3-D zeolite consisting of Ti-SSZ-70, a control which underwent no delamination treatment. Results of long-term flow reactor catalytic testing demonstrate a distinct 2.5-fold increase in reaction-rate constant k′ on a mass basis for Ti-UCB-4 relative to 3-D Ti-SSZ-70, and a lack of long-term deactivation for both catalysts. In contrast, for Ti-SSZ-70-DEL-HIGHPH, due to deactivation, no steady-state behavior is observed for either conversion or selectivity with increasing times on stream. A combination of data from powder X-ray diffraction (PXRD), nitrogen physisorption at 77 K, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), and UV-visible (UV-vis) spectroscopy demonstrate Ti-SSZ-70-DEL-HIGHPH to be comprised of a combination of crystalline and amorphous phases. Control experiments demonstrate a negative synergy on catalysis when both phases are combined in a single catalyst, which leads to decreased conversion, at levels below values predicted based on the linear combination of the two phases present.
Introduction
The oxidation of propylene to propylene oxide (PO) is a growing (5% per year growth rate) industrial commodity chemical process, which supplies PO for polyurethanes, polyols, surfactants, and lubricants, among other chemical products.1–3 Organic hydroperoxides such as ethylbenzene hydroperoxide (EBHP), tert-butyl hydroperoxide (TBHP), and cumene hydroperoxide are used as oxidants for propylene epoxidation to PO, and account for nearly 50% of the worldwide production of PO.4 A common feature of these co-product routes is the need to react nearly all of the organic hydroperoxide oxidant (above 99.5% organic hydroperoxide conversion is typical) within the tail end of the PO synthesis reactor, before the separation train. This feature is difficult to accomplish in practice, requiring long sections of reactor, due to both the low forward kinetic driving force for reaction within the tail end, as a result of the low organic hydroperoxide concentration there, as well as the inhibiting effect of high concentrations of accumulated epoxide product and alcohol co-product. Typically, to facilitate the high organic hydroperoxide conversion as required in the tail end of the PO synthesis reactor, high temperatures of up to 130 °C are used,3 which can lead to selectivity losses (in terms of fraction of organic hydroperoxide converted that leads to PO synthesis) as well as impurities that also inhibit catalytic sites.To address this, we have previously proposed the use of a crystalline zeolitic catalyst, based on delaminated zeolite Ti-UCB-4, for the tail-end section of the PO synthesis reactor, which is in contrast to either the soluble (Mo naphthenate) or amorphous (Ti Lewis-acid sites grafted on amorphous silica) nature of conventional PO synthesis catalysts.1,3–5 Delaminated zeolite UCB-4 was used as a support because of its high external surface area, which enables accessibility of catalytic sites to bulky reagents, such as organic hydroperoxides. Previously, we compared Ti-UCB-4 with the conventional solid catalyst based on grafted Ti sites on an amorphous silica support, in the epoxidation of 1-octene with EBHP, which was used as a probe reaction that is directly relevant to PO synthesis, under tail-end conditions in a continuous flow microreactor. Reviews of such microreactors are available elsewhere.6 Our relevant reaction is shown in Scheme 1.7 Tail-end conditions consisted of high temperature (110 °C) and high concentrations of 1,2-epoxyoctane and 1-phenylethanol (corresponding to greater than 80% organic hydroperoxide conversions, based on prior reported concentrations2,5). Our desire to use 1-octene as well as the choice of this temperature is related to it being a reliable surrogate for propylene in testing epoxidation catalysts,2 and our current flow system's limitation to work around atmospheric pressure (i.e. below the boiling point of 1-octene). Our results demonstrated a lack of deactivation,2 displaying true steady state behaviour with time on stream, which was not observed for the amorphous-silica catalyst, and 9% higher selectivity of organic oxidant being converted to epoxide for the crystalline Ti-UCB-4 catalyst. Our data also indicated a lack of dependence of selectivity on organic hydroperoxide conversion for this reaction under our conditions.7
 | ||
| Scheme 1 Chemical equation of the epoxidation of 1-octene with EBHP. | ||
Here, in this manuscript, we were compelled to investigate an alternative approach for delamination, which consists of high pH conditions and is known to result in delaminated materials with much higher external surface areas, compared with those that we have investigated previously. This high-pH approach is inspired by the elegant work of Corma et al., who reported the synthesis of ITQ-2,8,9 and, in particular, Ti-ITQ-2 as an active olefin epoxidation catalyst in batch-reactor mode.10,11 In particular, an appealing feature of the ITQ-2-based materials is their significantly higher external surface area, which is reported to be in excess of 700 m2 g−1, more than 8-fold higher compared to the 3-D zeolite without delamination. This higher external surface area is in contrast to the 1.5-fold enhancements that are typically observed when delaminating SSZ-70 using fluoride anion as mineralizing agent in organic solvent, for UCB-4-based materials, which typically have a surface area of 125 m2 g−1.12,13
Yet, while UCB-4-based materials lack amorphous silica as an impurity phase (no Q2 resonance in 29Si CP/MAS NMR spectrum of material),14 some amorphous silica phase has been previously reported to result as an impurity phase during ITQ-2 synthesis.9,15 Our comparison of high pH versus organic fluoride mineralizing agents for affecting SSZ-70 layered zeolite precursor delamination thus reduces to a question of the possible importance of crystalline-phase purity and high external surface area, when using grafted Ti catalyst sites for olefin epoxidation. In addition, to shed further light on the question of crystalline-phase purity, we investigate a catalyst consisting of grafted Ti sites on a mechanical equal-mass mixture of amorphous silica (506 m2 g−1)7 and crystalline UCB-4 as catalyst support materials. This latter mechanical mixture serves as a control, where the length scale separating amorphous and crystalline phases within the catalyst spans microns (particle size of silica is on the order of 32–63 μm), compared with possible nanoscale phase separation during the high pH hydroxide synthesis. Separately, we also analyze the catalytic performance of a catalyst consisting of the three-dimensional SSZ-70 (no delamination) as a support, in order to directly assess the benefits of delamination in olefin epoxidation catalysis. This comparison is shown in Fig. 1. In all cases, Ti grafting to zeolite surfaces can occur in organized “silanol nests” (shown in Fig. 1), which result from deboronation.
 | ||
| Fig. 1 Schematic description of the synthesis of Ti-SSZ-70, Ti-UCB-4 and Ti-SSZ-70-DEL-HIGHPH. Based on the known structure of related MCM-22 zeolite (which like SSZ-70 also consists of a layered-zeolite precursor consisting of 12-membered-ring cups),13 the vast majority of grafted Ti atoms in Ti-UCB-4 and Ti-SSZ-70 are located in the framework as shown in Fig. 1, where they are coordinated to four framework oxygen atoms, as opposed to Ti grafted on isolated external-surface silanols, which are not within a silanol nest. Ti-SSZ-70-DEL-HIGHPH shows besides this framework mesoporous silica, which we assign to be formed due to the harsh conditions (high-pH) during synthesis. | ||
Previously, in Brønsted-acid catalyzed toluene alkylation with propylene, we demonstrated that the benefits of SSZ-70 delamination included increased surface area as well a shorter diffusions paths to internal active sites. Here, due to the steric bulk of the reagents, we do not expect to have catalytic contributions from internal active sites. From this perspective alone, we hypothesize the benefit of delamination to be limited to the moderate enhancement in external surface area as a result of delamination. The latter implicitly assumes the rates per Ti site to be the same in catalysts that have been delaminated compared with those that have not been, consisting of the same SSZ-70 framework topology. However, due to the complex relationship between Lewis-acid site structure and catalytic function in general,16 it may be that there are other benefits to delamination for epoxidation catalysis, which are above and beyond those predicted by external surface area only, and may involve a different nature in the types of active sites that are formed, as a result of the delamination procedure. This possibility also forms a motivating factor in the current study. Because heterogeneous catalysts comprise nearly 80% of all industrial catalysts,18 the learnings here can be relevant to other systems as well, in which crystalline catalyst frameworks replace those based on amorphous ones.
Experimental
Characterization
Micropore volume, external surface area, and total pore volume of solid samples were measured via Nitrogen physisorption at 77 K using an ASAP 2020 Accelerated Surface Area and Porosimetry system (Micromeritics). About 100 mg sample were weighed and degassed under vacuum at 250 °C for 4 hours prior to analysis. The equilibration interval was 45 seconds. The resulting data were analyzed by the ASAP 2020 software using a Harkins and Jura t-plot.Powder X-ray diffraction (PXRD) patterns were measured using a Bruker GADDS D-8 diffractometer and Cu-Kα radiation. Data were collected in the 2θ range from 3° to 30° with a step size of 0.02° and a dwell time of 2 s. PXRD peaks at 2θ values of lower than 5° are not discussed due to instrument limitations.
High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was conducted with a FEI Tecnai F20 operated at 200 keV. Samples suitable for HAADF-STEM were prepared on amorphous Lacey-carbon films deposited on copper grids (Ted Pella, Inc.).
Diffuse-reflectance Ultraviolet-visible (UV-vis) spectroscopy for solid materials was performed on a Cary 400 spectrophotometer (Varian) fitted with a Praying-Mantis diffuse reflectance attachment from Harrick Scientific Instruments. Samples were measured with an average time of 0.166 s and a wavelength data interval of 0.5 nm, resulting in a scan rate of 180.7 nm min−1. The results are corrected by measuring a baseline of polytetrafluoroethylene and reflectance data were converted into Kubelka–Munk pseudoabsorbance units (F(R)).17 The determination of the Ti contents of all catalysts was performed by Galbraith Laboratories Inc. (Knoxville, TN). Liquid samples from catalysis were analyzed using an Agilent gas chromatography (GC) system consisting of a GC 6890A plus with helium as the make-up gas, air as the utility gas, and a high-resolution capillary column with 50.0 m length, 320 μm diameter, and 1.05 μm in film thickness. The gas chromatograph is equipped with a flame ionization detector (FID). The temperature program runs from 80–180 °C.
Materials and methods
All reagents used in this work were reagent-grade quality and used as received unless otherwise noted. B-SSZ-70, UCB-4, Ti-UCB-4, Ti–SiO2, and EBHP were synthesized as described previously.7,12,14,19SSZ-70-DEL-HIGHPH
SSZ-70-DEL-HIGHPH was synthesized similar to ITQ-2 by Corma et al.20 1.0 g of B-SSZ-70 and 4.0 g of distilled water were combined with a mixture of 5.6 g of CTAB and 13.8 g distilled water. After adding 6.0 g of 40 wt% tetrapropylammonium hydroxide, the slurry was stirred for 1 day at 80 °C. After cooling down to room temperature, the slurry was sonicated (1 second pulse, 0.1 second break) in an ice bath for 1 hour. The pH was subsequently changed from 13 to 2 by adding drops of 6 M aqueous HCl solution. The solid was separated by centrifugation and dried in an oven at 120 °C. After grinding with a mortar and pestle, the obtained material was calcined at 550 °C for 10 hours.8,20Titanium Grafting
SSZ-70-DEL-HIGHPH and SSZ-70 were calcined at 550 °C for 10 hours and dried in 15.0 mL high-pressure flasks at 120 °C for at least 3 hours. Under a stream of argon, 10.0 mL of anhydrous 1-butanol and 1 mL of titanium(IV)-n-butoxide were added to each material. The mixtures were stirred at 135 °C for 1 hour. After cooling to room temperature, the white solid products were filtered and washed with 1-butanol. After drying at 120 °C, the white powders were crushed and calcined at 550 °C for 10 hours. Typical Ti-contents are shown in Table 2. 25.0 mg of calcined Ti-UCB-4 and calcined Ti–SiO2 were mixed and ground together.Catalysis in a flow reactor
In a typical experiment, Ti-UCB-4, Ti-SSZ-70-DEL-HIGHPH, Ti-SSZ-70, and Ti–Ti–SiO2/Ti-UCB-4-MIX were pelletized and ground to a particle size of 180–250 μm. Then 18–55 mg of calcined catalyst were packed into a stainless-steel reactor (L = 41 mm, ø = 6 mm) between layers of glass wool. Layers of glass beads before and after the catalyst layer were used to stabilize the catalyst bed in the middle of the reactor and to enable thorough mixing of the reaction solution. A typical stock solution consisted of 1029.0 mmol (115.5 g) of 1-octene, 32.1 mmol (4.4 g) of EBHP, 62.4 mmol (8.7 g) of ethylbenzene, 188.4 mmol (24.2 g) of 1,2-epoxyoctane, 2.7 mmol (1.5 g) of acetophenone, 186.6 mmol (22.8 g) of 1-phenylethanol and 11.9 mmol (1.5 g) of n-nonane as an internal standard. The packed reactor was heated under vacuum at 140 °C for at least 4 hours. After cooling to room temperature, the reactor was flushed with 1-octene and connected to a syringe that contained the reaction solution. The flow rate was controlled using a syringe pump. The reactor was submerged in an oil-bath, which was held at a temperature of 110 °C. Samples were collected for 1 hour over different periods of time during the experiment. In order to allow the system to equilibrate, sample collection started at least 12 hours after the experiments began. The samples were analyzed via gas chromatography using n-nonane as internal standard.A general observation is the broad scattering of the selectivity values of EBHP to 1,2 epoxyoctane. This is caused by the deviation when measuring the relatively small signal change during 1,2-epoxyoctane formation, which is divided by the relatively large signal change during EBHP consumption. Uncertainties were calculated based on the standard deviation. For all the runs, the flow rate was lowered after 10–12 hours to increase and set the conversion and to avoid situations in which the EBHP conversion approached 100% within uncertainty, at the beginning of catalysis.
Results and discussion
Catalyst characterization
 | ||
| Fig. 2 PXRD results of B-SSZ-70, UCB-4 and SSZ-70-DEL-HIGHPH. | ||
 | ||
| Fig. 3 N2 adsorption/desorption isotherms as a function of relative pressure of B-SSZ-70, UCB-4 and SSZ-70-DEL-HIGHPH. | ||
| Material | Micropore volume [ml g−1] | External surface area [m2 g−1] | Total pore volume [ml g−1] |
|---|---|---|---|
| B-SSZ-70 | 0.17 | 69 | 0.31 |
| UCB-4 | 0.11 | 101 | 0.40 |
| Ti-SSZ-70-DEL-HIGHPH | 0.21 | 579 | 0.93 |
Microporosity in these materials is evident by their high uptakes at low relative pressures, and is quantified to be 0.17 mL g−1 for calcined B-SSZ-70, decreasing to 0.15 mL g−1 in delaminated zeolite UCB-4, consistent with previously reported results.7,12,21,22 We have previously described this decrease in micropore volume between the calcined three-dimensional versus delaminated material as being due to the removal of some microporosity in the delaminated material, which would otherwise be formed in between layers, during condensation of precursor layers when synthesizing the three-dimensional zeolite.7,12,21,22 In contrast, the SSZ-70-DEL-HIGHPH is measured to have a higher micropore volume of 0.21 mL g−1 when compared to calcined B-SSZ-70. This increase in micropore volume cannot be explained by delamination alone and could be a result of the formation of amorphous microporous silica, which could be synthesized under delamination conditions. Specifically, during the acidification step, in principle it is possible to precipitate microporous amorphous silicates from dissolved silicate ions, since acid-catalyzed condensations can yield microporous materials.23 If this occurs during synthesis of SSZ-70-DEL-HIGHPH, then the amorphous phase could also be partially responsible for the increased external surface area of this material.
The aforementioned changes to the exterior surface area and micropore volume motivate two crucial questions in this manuscript: (i) is the 1.5-fold difference in surface area obtained between B-SSZ-70 and UCB-4 significant, when using these materials as supports for catalysis? and (ii) is the much greater external surface area of SSZ-70-DEL-HIGHPH beneficial, when using this material as a support for epoxidation catalysis? We address these questions below, with the understanding that their answers may also be tied in with the type of grafted Ti site that is synthesized on each of the three materials, which are summarized in Fig. 1.
| Material | Ti-SSZ-70 | Ti-UCB-4 | Ti-SSZ-70-DEL-HIGHPH | Ti–SiO2/Ti-UCB-4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
|---|---|---|---|---|
| Ti-content [wt%] | 0.22 | 0.41 | 0.77 | 0.97 |
| Ti per external surface area [μmol m−2] | 0.67 | 0.85 | 0.28 | 0.67 |
High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM)
Both calcined delaminated zeolites UCB-4 and SSZ-70-DEL-HIGHPH were characterized by HAADF-STEM and compared to previously reported calcined B-SSZ-70 in the literature, which reports rectilinear sheets comprised of stacked layers.12,24Fig. 4 shows a typical image of UCB-4, which consists of a less dense arrangement of fragmented zeolite layers and thinner sheets, when compared to the three-dimensional zeolite, consistent with prior reports.12 In contrast, the HAADF-STEM data of calcined SSZ-70-DEL-HIGHPH in Fig. 4 show the presence of zeolite layers similar to UCB-4, except that they are interspersed with what appears to be a poorly structured amorphous phase, which we assign to be amorphous silica. This assignment is consistent with the PXRD results of Fig. 2, and previous studies little to no crystallinity was observed for MCM-22-based zeolites that were delaminated at high pH.9 | ||
| Fig. 4 HAADF-STEM images shows UCB-4 and SSZ-70-DEL-HIGHPH. | ||
Diffuse-reflectance UV-vis spectroscopy prior to catalysis
Ti sites of fresh (prior to reaction) catalysts Ti-SSZ-70, Ti-UCB-4, Ti-SSZ-70-DEL-HIGHPH, and the mechanical mixture consisting of equal masses of Ti–SiO2 and Ti-UCB-4 were characterized by diffuse-reflectance UV-vis spectroscopy. Ti-SSZ-70 and Ti-UCB-4 (Fig. 5) show a similar major band spanning 200–300 nm, with a maximum at 209 nm, consistent with Ti(SiO)4 framework sites.7,25 Therefore, we infer that Ti-SSZ-70 and Ti-UCB-4 comprise similar isolated Ti framework sites. Fig. 5 also shows the spectrum of Ti-SSZ-70-DEL-HIGHPH, with a band maximum at 223 nm and a weak shoulder around 260 nm. Consistent with prior reports, we assign this to sites having connectivity as represented by Ti(OSi)3OH.25 In order to assess the remaining Ti sites, we compare the spectrum of Ti-SSZ-70-DEL-HIGHPH with a spectrum of an equal-mass mechanical mixture consisting of amorphous Ti–SiO2 and crystalline Ti-UCB-4, shown in Fig. 5 and compared to the individual materials comprising the mechanical mixture in ESI† Fig. S1. Similar to Ti-SSZ-70-DEL-HIGHPH, the spectrum of the mechanical mixture also exhibits a plateau from 213–222 nm, which we assign to sites having connectivity Ti(SiO)4 and Ti(OSi)3OH,7,25 and a shoulder around 270 nm. The shoulder around 270 nm coincides with the band corresponding to sites in amorphous catalyst Ti–SiO2, which exhibits a maximum at 278 nm. This shoulder band is assigned to Ti sites that are on an amorphous silica phase – not in a framework site. We thus surmise that delaminated zeolite Ti-SSZ-70-DEL-HIGHPH possesses grafted Ti sites in an amorphous-silica phase as well as framework positions of a crystalline phase, and that the former are similar to sites of Ti–SiO2 according to their UV-Vis characteristics.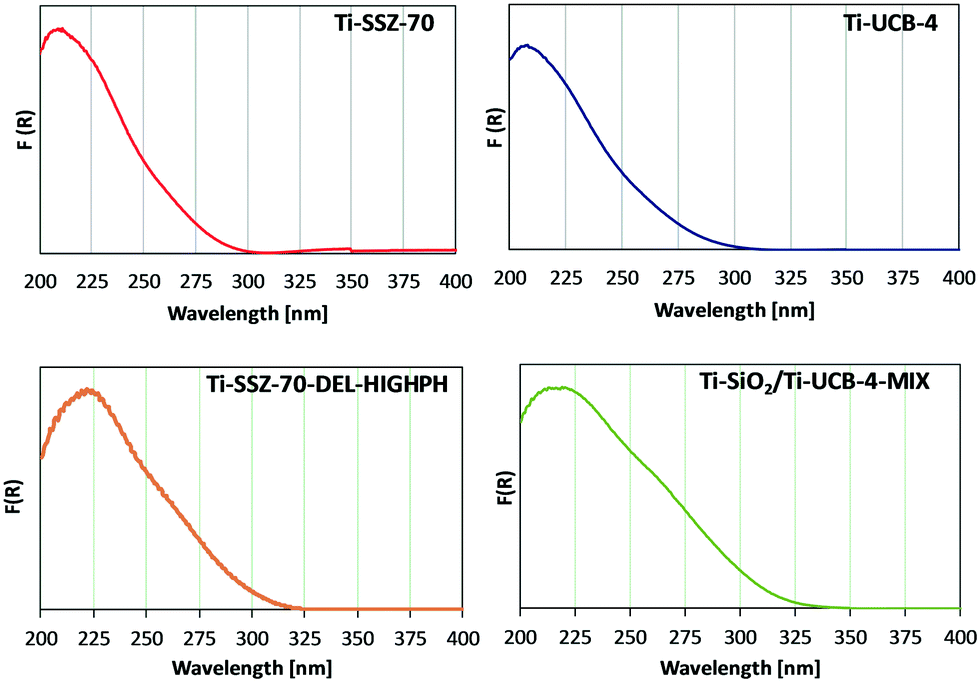 | ||
| Fig. 5 Diffuse-reflectance UV-vis spectra for Ti-SSZ-70, Ti-UCB-4, Ti-SSZ-70-DEL-HIGHPH, and Ti– SiO2/Ti-UCB-4-MIX. | ||
Tail-end epoxidation catalysis in a flow reactor
Our test reaction consists of the epoxidation of 1-octene with EBHP at tail-end conditions in a flow reactor, which we have used previously.7 Compared to catalytic-run duration in our previous study, involving other catalysts,7 we now investigate significantly longer times on stream of nearly a week. We feed to our flow reactor an amount of epoxide and alcohol that is in large excess relative to EBHP, such that it corresponds to 80% EBHP conversion of a hypothetical entrance-to-reactor feed consisting of a higher concentration of EBHP and olefin only (i.e. negligible epoxide and alcohol coproduct). When we refer to conversion within the discussion below, this refers to a zero-conversion basis at the entrance of our flow reactor (i.e., conversion is 0% at the reactor entrance). Pseudo-first order reaction-rate constants (assuming an ideal plug-flow reactor) were calculated on a catalyst mass basis k′ and on a Ti basis k, based on an averaged EBHP conversion between 80 and 100 hours of time on stream (zeolite-based catalysts have reached a steady state by this time on stream). Conversion, selectivity, and reaction-rate constants for Ti-SSZ-70 and Ti-UCB-4 are shown in Fig. 6 and Table 3. The EBHP conversion for Ti-SSZ-70 is observed to slightly drop from an initial value of 63% to 51% after the first 89 hours of time on stream, and the conversion appears to have reached a steady state value after this time, with no signs of further deactivation. | ||
| Fig. 6 Catalysis data for Ti-SSZ-70, Ti-UCB-4, Ti-SSZ-70-DEL-HIGHPH, and Ti-UCB-4/Ti–SiO2-MIX under tail-end conditions. Conversion of EBHP (•), selectivity of 1,2-epoxyoctane (△). | ||
| Material | Crystalline | Conversion reaches steady state | Average selectivity | Selectivity reaches steady state | Ti-sites in framework | Visual color of spent catalyst | Reaction-rate constant k′ mass-based [mL h−1 g−1] | Reaction-rate constant k titanium content-based [mL h−1 g−1] |
|---|---|---|---|---|---|---|---|---|
| Ti-SSZ-70 | Yes | Yes | 74% (±5%) | Yes | Yes | Light yellow | 29 | 13 × 103 |
| Ti-UCB-4 | Yes | Yes | 73% (±2%) | Yes | Yes | Light yellow | 72 | 17 × 103 |
| Ti-SSZ-70-DEL-HIGHPH | Partially | No | 60% (±4%) | No | Partially | Yellow | 20 | 2 × 103 |
| Ti–SiO2/Ti-UCB-4-MIX | Partially | No | 67% (±4%) | No | Partially | Yellow | 29 | 3 × 103 |
| Ti–SiO2 | No | No | 63% (±4%) | No | No | Yellow | 97 | 6 × 103 |
In comparison, when using the delaminated zeolite Ti-UCB-4 as catalyst, the EBHP conversion drops slightly, from an 89% initial conversion to 84% after the first 72 hours of time on stream, as shown Fig. 6. When compared to Ti-SSZ-70, the steady state appears to be reached earlier and the initial drop in EBHP conversion is lower (5% vs. 12%) for Ti-UCB-4. Moreover, the reaction-rate constant on a mass basis k′ for Ti-SSZ-70 of 29 mL h−1 g−1 is lower than k′ for Ti-UCB-4 consisting of 72 mL h−1 g−1. This enhancement in k′ for delaminated Ti-UCB-4 relative to 3-D Ti-SSZ-70 can be partially elucidated on the basis of the 1.5-fold higher external surface area for the former, which we described above leads to the same 1.5-fold higher grafted Ti content for the former (i.e. directly connected to the trend in external surface area). Yet the observed 2.5-fold difference in k′ between these two materials suggests that factors beyond external surface area may also be at play. To further investigate this, we calculated k values on a per Ti basis. The value of k for 3-D Ti-SSZ-70 of 13 × 103 mL h−1 g−1 is significantly lower than the value observed for delaminated Ti-UCB-4 of 17 × 103 mL h−1 g−1. At this point, we do not know the origin of this per-site enhancement in the delaminated material, but acknowledge that it could be a combination of effects consisting of initial deactivation under the harsh tail-end conditions of epoxidation catalysis in the flow reactor here, as well as the possibility of any intrinsic differences within the grafted Ti site as a result of the delamination procedure. The latter we deem to be less likely based on the similarity of the diffuse-reflectance UV-Vis data in Fig. 5, which characterizes the grafted Ti site in both materials. Previously, the reaction rate constants were calculated by using an averaged EBHP conversion over the entire measured time-on-stream period.7 As a result of these different approaches for calculating the reaction rate constants, k and k′ now appear slightly lower for Ti–SiO2 and Ti-UCB-4 than in these former reports.7 The selectivity is defined as the fraction of moles 1,2-epoxyoctane formed per mol EBHP consumed.
Regarding the selectivityfor 1,2-epoxyoctane shown in Fig. 6, which is defined as the fraction of moles 1,2-epoxyoctane formed per mol EBHP consumed, it is stable for Ti-SSZ-70 and averages at 74% (±5%), which is similar to the observed selectivity for Ti-UCB-4, which averages at 73% (±2%) and is also stable. The similarity of the selectivity shown by both materials is consistent with delamination not appreciably changing the nature of the grafted Ti site within the SSZ-70-topology framework. In summary, we observe an increased activity for 2-D Ti-UCB-4 relative to 3-D Ti-SSZ-70 of 2.5-fold on a mass basis, and attribute this to a combination of higher surface area and slightly higher per-Ti-site activity, while selectivity for the two catalysts is comparable.
Catalysis data and reaction-rate constants of Ti-SSZ-70-DEL-HIGHPH are also shown in Fig. 6. The EBHP conversion is observed to decrease from 77% to 51% during the first 162 h of time on stream, without any evidence of this catalyst reaching a steady state in either the conversion or the selectivity. Due to our evidence of an amorphous-silica phase in Ti-SSZ-70-DEL-HIGHPH (vide supra), we chose to compare the catalytic performance of Ti-SSZ-70-DEL-HIGHPH with a catalyst that consists of an equal-mass mechanical mixture of amorphous Ti–SiO2 and crystalline Ti-UCB-4. This latter catalyst exhibits an EBHP conversion that initially drops from 64% to 49% in 183 h, and, like Ti-SSZ-70-DEL-HIGHPH, it continues deactivating without reaching a steady state. A comparison of the activities for Ti-SSZ-70-DEL-HIGHPH and the Ti–SiO2/Ti-UCB-4-MIX indicates that the former exhibits a lower reaction-rate constant per mass k′ of 20 mL h−1 g−1, compared to 29 mL h−1 g−1 for the latter. On a per Ti basis, the calculated k values for both catalysts were similar at 2 × 103 mL h−1 g−1 and 3 × 103 mL h−1 g−1, respectively. These data are consistent with a similar ensemble of grafted Ti sites, on both amorphous and crystalline phases, in both Ti-SSZ-70-DEL-HIGHPH and Ti-UCB-4/Ti–SiO2-MIX catalysts. It suggests this ensemble does not change whether the mixing between the amorphous and crystalline phases is on the nanoscale (as observed in Fig. 6) for Ti-SSZ-70-DEL-HIGHPH versus on the micron particle-size scale of the mechanical mixture in Ti-UCB-4/Ti–SiO2-MIX. This is quite profound given the nearly 1000-fold difference in length scale of phase mixing (relating to mixing of amorphous versus crystalline phases) between these two catalysts. The average selectivities for Ti-SSZ-70-DEL-HIGHPH and Ti-UCB-4/Ti–SiO2-MIX catalysts are 60% (±4%) and 67% (±4%), respectively, and decrease with time on stream. The lack of an observed steady state in the selectivity differentiates these partially amorphous catalysts from the pure crystalline catalysts consisting of Ti-UCB-4 and Ti-SSZ-70, which achieved a stable selectivity of around 74%.
We also compared these two partially amorphous catalysts to a pure amorphous catalyst, Ti–SiO2, over the long-term flow reactor conditions used here in this manuscript. The catalysis data and reaction-rate constants are shown in Fig. 6 and Table 3 for Ti–SiO2. This catalyst is observed to deactivate from an initial EBHP conversion of 88% to 68% after 187 hours of time on stream, without reaching a steady state. The lack of an observed steady state even after long times on stream for Ti–SiO2 is similar to what is observed for Ti-SSZ-70-DEL-HIGHPH and Ti-UCB-4/Ti–SiO2-MIX. The calculated k′ value of 97 mL h−1 g−1 for Ti–SiO2 is significantly higher than for Ti-SSZ-70-DEL-HIGHPH and Ti-UCB-4/Ti–SiO2-MIX, and the calculated k for Ti–SiO2 of 6 × 103 mL h−1 g−1 is 2-to-3-fold higher on a per Ti basis when compared to both partially amorphous catalysts. We surmise that the observed increase in catalyst activity for the fully amorphous Ti–SiO2 when compared with the partially amorphous Ti-SSZ-70-DEL-HIGHPH and Ti-UCB-4/Ti–SiO2-MIX catalysts is due to higher grafted Ti site densities, as described in Table 2, as well as higher per-Ti-site activity in Ti–SiO2. We do not yet understand the origin of the latter, but it points to a negative synergistic effect under tail-end reaction conditions, when a catalyst possesses both amorphous and crystalline phases, in that the catalyst activity is worse on a per-Ti basis than the linear combination of the two phases separately.
UV-Vis spectroscopy after catalysis
UV-vis spectroscopy was also used to characterize Ti sites in the spent catalysts (i.e. after the catalytic runs of Fig. S1†) as well as after spent-catalyst calcination (i.e. spent/calcined materials). The spectra are shown in ESI† Fig. S1. We discussed above data for Ti-SSZ-70 and Ti-UCB-4, which indicate isolated framework Ti(IV) sites with a band at 209 nm, as well as data for Ti-SSZ-70-DEL-HIGHPH, which exhibits a band at 220 nm and a weak shoulder around 260 nm, indicating isolated framework Ti(IV) sites with some Ti-oxide oligomers on the surface. After using these materials for catalysis, each material shows a mix of two different Ti sites and absorbance at higher wavelength (>300 nm). After calcining the spent materials, we observe a return of the bands to lower wavelengths, toward where the bands originally were prior to catalysis. These results are supported by visual observations, as these materials change in color following calcination to a white material for all catalysts. We surmise that the origin of the higher wavelength band following catalysis is the result of organic residue on the catalyst surface, rather than any type of irreversible deactivation as a result of Ti-site aggregation into more extended Ti-oxide domains, since in the case of the latter, calcination would not have caused a change in the bands. We observe the two delaminated materials to show more complete regeneration following calcination, compared to Ti-SSZ-70. Comparing Ti-SSZ-70-DEL-HIGHPH to Ti–SiO2/Ti-UCB-4-MIX, while both catalysts exhibit similar bands for the fresh and spent catalysts, the latter does not regenerate as well as the former following calcination. Such lack of regeneration of catalytic active sites supported on amorphous supports vs. full regeneration for sites on crystalline delaminated zeolite supports has also been recently observed when testing Fe(III) sites under harsh catalytic conditions, and demonstrates the advantage of using crystalline catalyst frameworks to replace traditional amorphous ones.26Conclusion
Table 3 summarizes a comparison of the delaminated and 3-D catalysts investigated in this study. The data demonstrate a definitive benefit to delamination in Ti-UCB-4, over the non-delaminated 3-D material Ti-SSZ-70, in terms of both higher grafted Ti site coverages (on a catalyst mass basis) as well as higher per-Ti-site activities, though both catalysts exhibit steady-state conversions that do not deactivate over increasing times on stream. In contrast, use of aqueous high-pH conditions for affecting delamination in Ti-SSZ-70-DEL-HIGHPH leads to a catalyst that does not exhibit a steady state conversion over any of the investigated times on stream, and exhibits lower selectivity than Ti-UCB-4, which was delaminated under mild conditions using fluoride as mineralizing agent, in organic-solvent media. The Ti-SSZ-70-DEL-HIGHPH catalyst behaves similar to Ti-UCB-4/Ti–SiO2-MIX, which represents an equal-mass mechanical mixture of crystalline and amorphous phases. We deem both Ti-SSZ-70-DEL-HIGHPH and Ti-UCB-4/Ti–SiO2-MIX catalysts to be partially amorphous on the basis of PXRD, N2 physisorption, and HAADF-STEM data. When compared to a fully amorphous Ti–SiO2 catalyst, we observe the per-Ti site activities to be 2.3-fold lower in both partially amorphous catalysts. This represents a heretofore unrecognized negative synergy, when both amorphous and crystalline phases coexist within the same catalyst (whether mixing between the two phases is on the nanoscale or micron scale). Altogether, our results demonstrate that delamination of a layered zeolite precursor can benefit Lewis-acid olefin epoxidation catalysis, when using grafted Ti sites, but that this benefit does not necessarily scale or even relate to the enhancement in accessible surface area, and critically depends on the method of delamination. These results emphasize the importance of the zeolite delamination method on catalytic properties, and specifically highlight that preserving the structural integrity of the crystalline zeolite framework during delamination can more than supersede the importance of high external surface area. Ti-UCB-4, with high-surface area and crystallinity, combines both aspects advantageously.Conflicts of interest
The funding for this research partially came from Chevron Energy Technology Co. and S. I. Z. is an employee of this company and, more generally, is also a stockholder in Chevron Corp.Acknowledgements
Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Funding from the National Science Foundation is gratefully acknowledged (PFI:AIR-TT 1542974). The authors are grateful to the Management and Transfer of Hydrogen via Catalysis Program funded by Chevron Corporation, as well as Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-FG02-05ER15696 for financial support of this work. C.S. acknowledges Deutsche Forschungsgemeinschaft (DFG) for a research fellowship.References
- S. T. Oyama, Mechanisms in homogeneous and heterogeneous epoxidation catalysis, Elsevier, 2011 Search PubMed.
- J. K. F. Buijink, J.-P. Lange, A. N. R. Bos, A. D. Horton and F. G. M. Niele, Propylene epoxidation via Shell's SMPO process: 30 years of research and operation, Elsevier, Amsterdam, The Netherlands, 2008 Search PubMed.
- D. L. Trent, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc, 2000 Search PubMed.
- H. Baer, M. Bergamo, A. Forlin, L. H. Pottenger and J. Lindner, Propylene Oxide, Ullmann's Encyclopedia of Industrial Chemistry, 2012 Search PubMed.
- J. K. F. Buijink, J. J. M. van Vlaanderen, M. Crocker and F. G. M. Niele, Catal. Today, 2004, 199, 93–95 Search PubMed.
- A. Tanimu, S. Jaenicke and K. Alhooshani, Chem. Eng. J., 2017, 327, 792 CrossRef CAS.
- M. Aigner, N. A. Grosso-Giordano, A. Okrut, S. Zones and A. Katz, React. Chem. Eng., 2017 10.1039/C7RE00076F.
- A. Corma, V. Fornes, S. B. Pergher, T. L. M. Maesen and J. G. Buglass, Nature, 1998, 396(6709), 353 CrossRef CAS.
- S. Maheshwari, E. Jordan, S. Kumar, F. S. Bates, R. L. Penn, D. F. Shantz and M. Tsapatsis, J. Am. Chem. Soc., 2008, 130(4), 1507 CrossRef CAS PubMed.
- L. A. Baumes, A. Blansché, P. Serna, A. Tchougang, N. Lachiche, P. Collet and A. Corma, Mater. Manuf. Processes, 2009, 24(3), 282 CrossRef CAS.
- A. Corma, U. Diaz, V. Fornes, J. L. Jorda, M. Domine and F. Rey, Chem. Commun., 1999, 779 RSC.
- I. Ogino, E. A. Eilertsen, S.-J. Hwang, T. Rea, D. Xie, X. Ouyang, S. I. Zones and A. Katz, Chem. Mater., 2013, 25(9), 1502 CrossRef CAS.
- R. C. Runnebaum, X. Ouyang, J. A. Edsinga, T. Rea, I. Arslan, S.-J. Hwang, S. I. Zones and A. Katz, ACS Catal., 2014, 4(7), 2364 CrossRef CAS.
- X. Ouyang, S.-J. Hwang, D. Xie, T. Rea, S. I. Zones and A. Katz, ACS Catal., 2015, 5(5), 3108 CrossRef CAS.
- R. Schenkel, J. O. Barth, J. Kornatowski and J. A. Lercher, Stud. Surf. Sci. Catal., 2002, 142, 69 CrossRef.
- P. Nandi, W. Tang, A. Okrut, X. Kong, S.-J. Hwang, M. Neurock and A. Katz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(7), 2484 CrossRef CAS PubMed.
- (a) J. H. Nobbs, Color. Technol., 1985, 15(1), 66 CrossRef.
- J. J. Bravo-Suárez, R. V. Chaudhari and B. Subramaniam, In Novel Materials for Catalysis and Fuels Processing, ACS, 2013 Search PubMed.
- R. Millini, G. Perego, W. O. Parker, G. Bellussi and L. Carluccio, Microporous Mater., 1995, 4(2), 221 CrossRef CAS.
- A. Corma, V. Fornés, J. M. Guil, S. Pergher, T. L. M. Maesen and J. G. Buglass, Microporous Mesoporous Mater., 2000, 38(2), 301 CrossRef CAS.
- S. I. Zones and A. W. Burton, Molecular sieve SSZ-70 composition of matter and synthesis thereof (US7108843B2), 2006 Search PubMed.
- R. Millini, G. Perego, W. O. Parker Jr., G. Bellussi and L. Carluccio, Microporous Mater., 1995, 4, 221 CrossRef CAS.
- A. Katz and M. E. Davis, Nature, 2000, 403(6767), 286 CrossRef CAS PubMed.
- X. Ouyang, Y.-J. Wanglee, S.-J. Hwang, D. Xie, T. Rea, S. I. Zones and A. Katz, Dalton Trans., 2014, 43(27), 10417 RSC.
- P. Ratnasamy, D. Srinivas and H. Knözinger, Adv. Catal., 2004, 48, 1 CAS.
- N. A. Grosso-Giordano, A. J. Yeh, A. Okrut, D. J. Xiao, F. Grandjean, G. J. Long, S. I. Zones and A. Katz, Chem. Mater., 2017, 29(15), 6480–6492 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00138j |
| This journal is © The Royal Society of Chemistry 2017 |