
Epoxidation of 1-octene under harsh tail-end conditions in a flow reactor I: a comparative study of crystalline vs. amorphous catalysts†
Martina
Aigner
 a,
Nicolás Andrés
Grosso-Giordano
a,
Alexander
Okrut
*a,
Stacey
Zones
*b and
Alexander
Katz
*a
a,
Nicolás Andrés
Grosso-Giordano
a,
Alexander
Okrut
*a,
Stacey
Zones
*b and
Alexander
Katz
*a
aDepartment of Chemical and Biomolecular Engineering, University of California at Berkeley, Berkeley, CA 94720, USA. E-mail: alexander.okrut@berkeley.edu; askatz@berkeley.edu
bChevron Energy Technology Company, Richmond, CA 94801, USA. E-mail: SIZO@chevron.com
First published on 26th July 2017
Abstract
Amorphous silica versus crystalline delaminated-zeolite catalysts consisting of grafted Ti(IV) Lewis-acid active sites were investigated from the perspective of 1-octene olefin epoxidation with ethylbenzene hydroperoxide (EBHP) as oxidant. Reactions were performed at conditions of temperature and concentrations of organic hydroperoxide and inhibitors (epoxide product and alcohol co-product) that mimic the harsh conditions found at the tail-end of the flow reactor for industrial propylene-oxide (PO) synthesis, where there is a current need to improve activity and selectivity, because of deactivation. Catalyst synthesis was performed by grafting a Ti-alkoxide precursor onto framework vacancies (“silanol nests”) of the delaminated zeolite UCB-4, as well as onto amorphous SiO2. Both catalysts were characterized by powder X-ray diffraction (PXRD), nitrogen physisorption at 77 K, and UV-visible spectroscopy before and after catalysis. Experiments at different conversions were performed, and show that crystalline Ti-UCB-4 exhibits a ∼9% higher average selectivity (73% versus 64%) and greater conversion, stability, and robustness upon increasing time on stream relative to amorphous Ti–SiO2. UV-vis spectra are discussed for fresh, spent, and spent/calcined materials and demonstrate that Ti sites in Ti-UCB-4 exist as isolated grafted complexes with four-fold coordination to the zeolite framework, whereas Ti–SiO2 consists of grafted Ti-sites on the silica surface, some of which are isolated but a dominant proportion of which are TiO2 oligomers. The observed increased stability of the crystalline catalyst under tail-end reactor conditions is attributed to the surface pockets of the crystalline material, in which Ti is grafted.
Introduction
Propylene oxide (PO) is an important industrial commodity chemical for polyurethanes, polyols, surfactants, and lubricants. Demand for PO continues to steadily grow, and the current global production capacity is more than 8 × 106 metric tons per year.1 Nearly 50% of worldwide PO production uses an organic hydroperoxide as the required oxidant for transforming the double bond of propylene to the epoxide functional group in PO.1 In this approach, for each generated PO, an organic alcohol co-product is synthesized, sometimes (depending on economics) being as valuable or even more valuable than the PO itself. The most commonly used organic hydroperoxides for PO synthesis are tert-butyl hydroperoxide (TBHP), ethylbenzene hydroperoxide (EBHP), and cumene hydroperoxide. While some PO synthesis processes that rely on TBHP use a soluble molybdenum naphthenate salt as catalyst,1,2 both the cumene and ethylbenzene hydroperoxide routes use a solid epoxidation catalyst for PO production, consisting of supported Ti Lewis-acid sites on amorphous mesoporous silica. It is generally recognized that the active site within such a catalyst consists of a more isolated and coordinatively unsaturated grafted Ti on silica – not coordinatively saturated Ti as found in either anatase or rutile.1 In all PO synthesis processes, for both safety reasons as well as avoidance of by-product formation in the separation train, it is important to convert the vast majority (i.e. over 99%) of the organic hydroperoxide in the tail end of the flow reactor.2 These high conversions create a harsh environment for catalysis, which leads to catalyst active-site inhibition by both the epoxide product as well as the alcohol co-product. When combined with the low organic hydroperoxide concentration present under tail-end conditions, this leads to sluggishness in rate for all currently used catalysts. This sluggishness in turn requires higher temperatures of up to 130 °C at the tail end of the PO synthesis reactor,3 which leads to greater energy consumption, costs, and waste due to reduced catalyst selectivity and deactivation under tail-end conditions.To improve the PO synthesis process when using an organic hydroperoxide as oxidant, there is a great need to reduce catalyst deactivation, while maintaining high selectivity by minimizing synthesis of undesired by-products, specifically under harsh tail-end conditions. These conditions typically represent organic hydroperoxide conversions above 80% within the flow reactor, but because of the catalyst sluggishness and deactivation (vide supra), the amount of catalyst in the tail end typically by far exceeds that which came before it in the flow reactor (i.e. at the entrance to the flow reactor leading up to 80% conversion of organic hydroperoxide). Previously hypothesized mechanisms of solid epoxidation-catalyst deactivation involve the growth of grafted Ti-oxide domains on the silica support surface,4 which are more coordinatively saturated Ti sites and thus less catalytically active.4,5
Recently, we compared the stability of site-isolated grafted Fe(III) sites on both crystalline and amorphous silica supports. After hydrogen-peroxide treatment, the grafted metal sites on the amorphous silica support aggregated, whereas those on the crystalline support did not change.6 Based on this, we hypothesized that when dealing with a grafted Ti(IV) site for olefin epoxidation catalysis, also a hard Lewis-acid cation, a crystalline silica support could result in a more stable, active, and selective catalyst compared with the amorphous silica support, which is the one currently used in industrial catalysts. We further hypothesized that the greatest effect of changing the crystallinity of the silica support could be manifested in the tail-end section of the PO synthesis flow reactor, based on the harshness of the catalysis conditions there (vide supra). Further support for this hypothesis is based on the proven robustness and selectivity, mainly in the form of three-dimensional microporous zeolitic catalysts, which are crystalline and already used in many reactions, such as industrial epoxidation with aqueous hydrogen peroxide, isomerization, dehydration, and alkylation, due to their well-defined active-site environments and high stability.7–11
To test our hypothesis, we compare a model of the currently used solid industrial epoxidation catalyst via organic hydroperoxide routes, which consists of coordinatively unsaturated grafted Ti Lewis-acid sites on an amorphous silica support previously described by Buijink et al.,12 with a crystalline delaminated-zeolite-based catalyst, within the context of tail-end epoxidation flow-reactor performance. Our approach relies on the established procedure of using 1-octene as a relevant terminal-olefin reactant, which is accepted as a reliable model for propylene within the epoxidation literature.13 We investigate and quantify catalyst deactivation and selectivity as a function of time on stream in a flow reactor, using 1-octene with ethylbenzene hydroperoxide as the olefin and organic hydroperoxide, respectively. This olefin and organic hydroperoxide choice have been used universally as model reactants for industrial PO manufacture.13 Previously, we have demonstrated reactant scope in batch-mode epoxidation reactors;14,15 however, the goal here was to compare crystalline versus amorphous catalyst supports within the context of a flow-reactor that operates specifically under tail-end conditions. The reactants chosen allow us to investigate tail-end conditions that consist of higher temperatures of 110 °C. The two expected products of this reaction are 1,2-epoxyoctane and 1-phenylethanol, with the chemical equation given in Scheme 1. A loss in selectivity results from consuming organic hydroperoxide (the limiting reagent) in a manner that does not lead to the synthesis of an epoxide product (e.g., decomposition of organic hydroperoxide to dioxygen and alcohol coproduct). This loss of selectivity results in other, undesirable organic-hydroperoxide decomposition products such as O2 and acetophenone (oxidized alcohol to ketone), and has been already identified as a significant problem for PO synthesis when using organic hydroperoxides.4
 | ||
| Scheme 1 Chemical equation of epoxidation of 1-octene with EBHP. | ||
A schematic depiction of the surface of the amorphous catalyst and its grafting with Ti is shown in Fig. 1. This material exhibits mostly tripodal Ti sites, but also oligomers, whereas a schematic structure of the delaminated-zeolite catalyst used, Ti-UCB-4, also shown in Fig. 1, consists of mostly tetrahedral Ti sites. The delaminated-zeolite catalyst circumvents the typical steric limitations imposed by zeolitic microporous frameworks,16 by increasing available exposed external surface area.17 Our delaminated UCB-4 support material was first described by Ogino et al. in 2013, and is synthesized from the crystalline molecular sieve B-SSZ-70.17,18 Calcination of the latter synthesizes the three-dimensional zeolite B-SSZ-70.18 The crystalline lattice of B-SSZ-70 is well defined with boron atoms located in framework T-sites. As shown in Fig. 1, this ordered crystalline framework structure is delaminated at the precursor stage by treating with a surfactant followed by subsequent breaking of intersheet covalent Si–O–Si and Si–O–B connectivity between layers to affect layer exfoliation, thereby resulting in delaminated zeolite B-UCB-4. The typical increase in the external surface area is approximately a factor of ∼2 greater for the delaminated zeolite over its three-dimensional calcined counterpart (i.e. zeolite without delamination). During synthesis of Ti-UCB-4, the boron atoms occupying framework positions in B-UCB-4 are removed via aqueous acid treatment. The resulting deboronated UCB-4 contains framework defects that are organized into “silanol nests” (Fig. 1), which are subsequently reoccupied by framework titanium atoms, via treatment with titanium n-butoxide. The obtained Ti-UCB-4 therefore represents a crystalline 2D zeolite, with a high external surface area and accessible framework Ti sites. While we expect most of the Ti atoms in Ti-UCB-4 to be located in the framework, where they are coordinated to four framework oxygen atoms, we also expect there to be some Ti still grafted to isolated external-surface silanols, which are not within a silanol nest, and are the main sites on amorphous SiO2. Based on the known structure of related MCM-22 zeolite (which like SSZ-70 also consists of a layered-zeolite precursor consisting of 12-membered-ring cups), these sites are anticipated to be a minority, and the majority should consist of Ti coordinated to silanol nests (see Fig. S1 of ESI†).19 We hypothesize that these nests can offer improvement in the stability of the catalyst, as a result of chelation of multiple framework oxygens to the Ti site, which we recently observed when comparing crystalline delaminated-zeolite and amorphous Fe-containing catalysts.6 In addition, we hypothesize that the nests can also offer improved selectivity due to confinement within the nest site. Such nest confinement effects have been previously invoked to explain increases in catalyst activity and selectivity, and form the basis for commercial liquid-phase ethylbenzene production processes.9,20
 | ||
| Fig. 1 Synthesis of epoxidation catalysts consisting of grafted Ti sites on a siliceous support, comprising either (a) amorphous Ti–SiO2 or (b) crystalline Ti-UCB-4. The procedure for synthesis of the latter involves delamination and deboronation prior to Ti grafting. | ||
Experimental section
Characterization
Liquid samples from catalysis were analyzed using an Agilent gas chromatography (GC) system consisting of a GC 6890A plus with helium as the make-up gas, air as the utility gas and a high-resolution capillary column with 50.0 m length, 320 μm diameter and 1.05 μm in film thickness, equipped with a flame ionization detector (FID). The temperature program runs from 80–180 °C. Powder X-ray diffraction (PXRD) patterns were measured using a Bruker GADDS D-8 diffractometer and Cu-Kα radiation. Data were collected in the 2θ range from 3° to 30° with a step size of 0.02° and a dwell time of 2 s. PXRD peaks at 2θ values of lower than 5° are not discussed due to instrument limitations. Ultraviolet-visible (UV-vis) spectroscopy for solid materials was performed on a Cary 400 spectrophotometer (Varian). Samples were measured with an average time of 0.166 s and a wavelength data interval of 0.5 nm, resulting in a scan rate of 180.7 nm min−1. The results are corrected by measuring a baseline of polytetrafluoroethylene and reflectance data were converted into Kubelka–Munk (F(R)) pseudoabsorbance units.21 The determination of the Ti contents of materials was performed via liquid-phase UV-vis spectroscopy via the following procedure: 20 mg catalyst were mixed with 1 mL pure H2SO4 in a 10 mL volumetric flask and left for 1 h. Then, a few drops of water and 0.11 mL of 30% H2O2 solution were added. The volumetric flask was filled with water to the 10 mL mark. The concentration of titanium in the prepared solution was calculated from the value of absorbance at 408 nm in the UV-vis spectrum using a calibration curve. Micropore volume, external surface area, and total pore volume of solid samples were measured via nitrogen physisorption at 77 K using an ASAP 2020 Accelerated Surface Area and Porosimetry system (Micromeritics). About 100 mg sample were weighed and degassed under vacuum at 250 °C for 4 hours. The analysis gas was nitrogen which was adsorbed at a temperature of 77 K. The resulting data were calculated by the ASAP 2020 software. Scanning electron microscopy (SEM) images (ESI† Fig. S4 and S5) of selected materials were measured by a scanning electron microscope JEOL JSM 6700F.Materials and methods
All reagents used in this work were reagent-grade quality and used as received unless otherwise noted. B-SSZ-70, UCB-4, Ti-UCB-4 and EBHP were synthesized as described previously.14,17Titanium grafting
SiO2 (Selecto silica gel, particles size 32–63 μm) and UCB-4 were calcined at 550 °C for 10 hours. Then, 1 g of each material was dried in a 15.0 mL high-pressure flask at 120 °C for at least 3 hours. Under a stream of argon, 10.0 mL of anhydrous 1-butanol and 1 mL of titanium(IV)-n-butoxide were added. The mixture was stirred at 135 °C for 1 hour. After cooling to room temperature, the white solid products were filtered and washed with 1-butanol. After drying at 120 °C, the white powders were crushed with a pestle and calcined at 550 °C for 10 hours. By UV-vis spectrometry, typical Ti contents of 0.41–0.43 wt% for the Ti-UCB-4 and 1.38–1.67 wt% for Ti–SiO2 were determined.Catalysis in a flow reactor
In a typical experiment, Ti-UCB-4 was pelletized to a particle size of 180–250 μm. Then 18–50 mg of calcined catalyst were packed into a stainless-steel reactor (L = 41 mm, ø = 6 mm) between layers of glass wool. Layers of glass beads before and after the catalyst layer were used to stabilize the catalyst bed in the middle of the reactor and to enable thorough mixing of the reaction solution. A typical stock solution consisted of 1029.0 mmol (115.5 g) of 1-octene, 32.1 mmol (4.4 g) of EBHP, 62.4 mmol (8.7 g) of ethylbenzene, 188.4 mmol (24.2 g) of 1,2-epoxyoctane, 2.7 mmol (1.5 g) of acetophenone, 186.6 mmol (22.8 g) of 1-phenylethanol and 11.9 mmol (1.5 g) of n-nonane as an internal standard. The packed reactor was heated under vacuum at 140 °C for at least 4 hours. After cooling to room temperature, the reactor was flushed with 1-octene and connected to a syringe that contained the reaction solution. The flow rate was controlled using a syringe pump. The reactor was submerged in an oil-bath, which was held at a temperature of 110 °C. Samples were collected for 1 hour over different periods of time during the experiment. In order to allow the system to equilibrate, sample collection started at least 2 h after the experiments began. The samples were analyzed via gas chromatography using n-nonane as internal standard.For practical reasons, two separate experiments were conducted to obtain data for the range of 1–24 hours: first, an experiment over 12 hours was performed and then, an experiment over 24 hours was performed. During the 24 hour experiment, the second range of 12 hours of the experiment were measured. For the experiments using Ti-UCB-4, the data is an average of multiple experimental reproductions. A general observation is the broad scattering of the selectivity values of EBHP to 1,2-epoxyoctane. This is caused by the uncertainty introduced when measuring a relatively small signal change for 1,2-epoxyoctane, due to its presence in large excess under tail-end conditions. Error bars were calculated based on the standard deviation. For the run with the highest EBHP conversion >90%, the flow rate was lowered after 12 hours to increase and set the conversion.
Batch-reactor testing of catalyst recyclability
5.0 mg of calcined catalyst were dried in a vial at 120 °C for at least 4 hours. After cooling to room temperature, 2.5 mL of stock solution were added and the mixture was stirred at 110 °C for a selected time and cooled down to take a sample. After sampling, the mixture was stirred again at 110 °C and after a certain time a second and third sample were taken in the same way as before. The samples were analyzed by gas chromatography using n-nonane as internal standard. A typical stock solution consists of 6.830 mmol (0.765 g) of 1-octene, 0.268 mmol (0.037 g) of EBHP, 12.915 mmol (1.369 g) of ethylbenzene, 1.061 mmol (0.136 g) of 1,2-epoxyoctane, 0.216 mmol (0.026 g) of acetophenone, 1.195 mmol (0.146 g) of 1-phenylethanol and 0.203 mmol (0.026 g) of n-nonane as an internal standard. This experiment was performed with fresh Ti–SiO2 and Ti-UCB-4, and also with spent Ti–SiO2 and Ti-UCB-4, which were already used for at least 50 hours in a flow test and afterwards calcined at 550 °C for 10 h.Results and discussion
Catalyst characterization
Support materials consisting of calcined B-SSZ-70, UCB-4, and amorphous SiO2 were structurally characterized by PXRD (ESI,† Fig. S2), prior to Ti incorporation. Characteristic peaks in the powder pattern of Fig. S2a† at 2θ values of 7.2°, 14.5°, and 26.3° are consistent with previously reported data for calcined SSZ-70,14 and are similar to the powder pattern of calcined UCB-4. This provides support for intact crystallinity of this material following delamination and calcination, since UCB-4 is synthesized from the B-SSZ-70 layered zeolite precursor. Peaks at 7.9° and 10.0° 2θ for UCB-4 are broader and less intense for the delaminated material UCB-4 compared to the starting material B-SSZ-70. Because these peaks correspond to the [011] and [012] axes, this indicates greater disorder with respect to the z-axis orientation after delamination, as expected for zeolitic layers that are randomly oriented along the z-axis.17 This is a desirable outcome as it is consistent with exposing more external surface for Ti insertion, resulting in greater densities of active sites, as only sites on the external surface are expected to be active. The lack of any observed Bragg peaks for amorphous silica in Fig. S2c† is consistent with the lack of long-range order in this material.The same materials were also characterized via N2 physisorption at 77 K, to evaluate porosity and external surface area, which is the relevant surface area for Ti incorporation and catalysis, as a result of the steric bulk of the reagents involved. Fig. S3 (ESI†) shows the N2 adsorption/desorption isotherms as a function of relative pressure for the three materials, and Table 1 summarizes the micropore and mesopore volumes as well as the external surface areas determined by the t-plot method from these data. There are significant differences between all isotherms. The low-pressure uptakes within the isotherms correspond to micropores, and the micropore volume for calcined B-SSZ-70 is 0.17 mL g−1, whereas for UCB-4, it is less – at 0.15 mL g−1. This decrease in micropore volume is consistent with loss of that microporosity that would otherwise reside in between layers.17 The amorphous SiO2 support, however, shows nearly no micropore volume.
There is a clear increase of external surface area for UCB-4 as synthesized from B-SSZ-70, from 74 m2 g−1 in B-SSZ-70 to 113 m2 g−1 in UCB-4. Amorphous SiO2 has a much higher external surface area of 506 m2 g−1 using the same approach – and the similarity of this value to the BET surface area (BET surface area of 402 m2 g−1) suggests that most if not all of the internal mesopores of SiO2 are unconfined and appear like external surface area in t-plot calculations. The value of the total pore volume at a P/P0 of near unity represents the total pore volume, a value that also increases as a result of delamination, when comparing calcined B-SSZ-70 (0.35 mL g−1) and UCB-4 (0.43 mL g−1) materials. Amorphous SiO2 has a higher total pore volume of 0.71 mL g−1 and exhibits hysteresis within the isotherm of Fig. S3c,† as characteristic for a mesoporous material. In summary, delamination of B-SSZ-70 preserves crucial aspects of crystallinity when synthesizing UCB-4 while increasing the external surface area and total pore volume. Notwithstanding, the support material with the highest external surface area and total pore volume is represented by amorphous silica.
Following Ti incorporation, solid-state diffuse-reflectance UV-vis spectroscopy between 200 nm and 500 nm allows investigation of the nature of Ti sites within the materials. The UV-vis spectrum of Ti-UCB-4 is shown in Fig. 2, and consists of one major band spanning 200–328 nm, with a maximum at 210 nm. Ratnasamy et al. assign a band at 210 nm in related crystalline zeolitic Ti–SiO2 catalysts to correspond to isolated Ti(OSi)4 or Ti(OSi)3OH framework sites.22 Based on this, we infer that Ti-UCB-4 comprises isolated Ti framework sites. A slight shoulder around 260 nm indicates presence of Ti sites in non-framework positions, such as those grafted on external-surface isolated silanols.23 In comparison, the UV-vis spectrum of amorphous Ti–SiO2 in Fig. 2 shows a broad band spanning between 200 nm and 350 nm, with a peak maximum at 278 nm and a shoulder at approximately 247 nm. The data show that the vast majority of Ti sites in amorphous Ti–SiO2 are isolated surface-grafted sites, represented by a shoulder at 247 nm.22,24 In addition, the band around 260 nm and higher wavelength in Fig. 2 indicates formation of Ti-oxide oligomers, which may form during calcination. No bulk anatase formation is observed (∼330 nm).22 In summary, the preponderance of framework Ti sites in Ti-UCB-4 versus the grafted surface sites of amorphous Ti–SiO2 suggests both materials to be good candidates for testing the central hypothesis of this manuscript, as it relates to effect of amorphous versus crystalline support environment on Ti-site epoxidation catalysis.
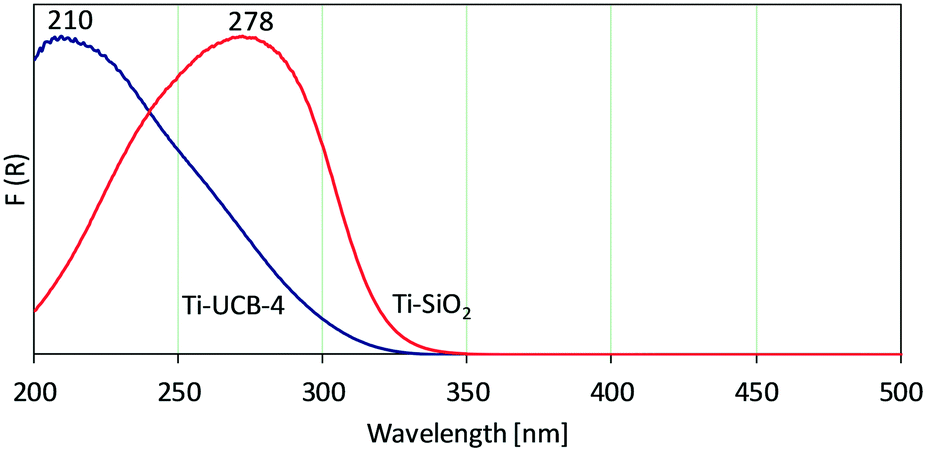 | ||
| Fig. 2 UV-vis spectra of Ti-UCB-4 (blue) and Ti–SiO2 (red). | ||
Tail-end epoxidation catalysis in a flow reactor
We compared crystalline Ti-UCB-4 and amorphous Ti–SiO2 as olefin epoxidation catalysts for the epoxidation of 1-octene with EBHP in a flow reactor, under tail-end conditions. Tail-end conditions correspond to a feed at the entrance of the flow reactor, which represents 80% conversion of a hypothetical entrance-to-reactor feed stream consisting of EBHP and olefin only (i.e. this feed stream consists of negligible epoxide and alcohol coproduct). That is to say, we feed to our flow reactor an amount of epoxide and alcohol that is in large excess relative to EBHP, such that it would appear to correspond to 80% EBHP conversion of a hypothetical entrance-to-reactor feed consisting of EBHP and olefin only. When we state conversion within the discussion below, this refers to a zero-conversion basis at the entrance of our flow reactor (i.e., conversion is defined to be that conversion that is actually achieved within the flow reactor, with the tail-end feed as inlet to the reactor corresponding to 0% conversion within the reactor). We first tested catalysts at a low target EBHP conversion of <35% for a period of 24 hours. The EBHP conversion and selectivity for 1,2-epoxyoctane for the amorphous Ti–SiO2 catalyst are shown in Fig. 3a. This catalyst exhibits an initial EBHP conversion of 67%, which continuously decreases over a 24 hour period, down to a value of 31%. The selectivity for 1,2-epoxyoctane remained relatively constant over this 24 hour period, averaging 63 ± 3%. In comparison, the EBHP conversion for the crystalline Ti-UCB-4 catalyst, shown in Fig. 3b, initially decreases at low time on stream and then remains constant after 15 h until the end of the experiment. The selectivity of EBHP for 1,2-epoxyoctane remains constant for Ti-UCB-4 over the entire 24 hour period and averages 73 ± 4%. | ||
| Fig. 3 Catalysis data for epoxidation of 1-octene with EBHP: lowest conversion (a and b), and highest conversion (c and d) for Ti–SiO2 (red, a, c and e) and Ti-UCB-4 (blue, b, d and e). Conversion of EBHP (•) and selectivity of EBHP to 1,2-epoxyoctane (▲). A summary is given in e. Manually added trend lines to guide the eye show the average selectivity and a decreasing trend for the conversion of EBHP in some ranges. | ||
We next tested our catalysts at high conversion greater than 90%. These data correspond to the highest conversion investigated in this manuscript, and are the most pertinent to analysis of tail-end flow reactor comparisons between the two catalysts, crystalline versus amorphous. Fig. 3c shows data for a target EBHP conversion of greater than 90%, in flow experiments that were run continuously for 72 hours using amorphous Ti–SiO2 as catalyst: EBHP conversion starts at 95%, and continuously drops to 78% during the run. While there is some slight fluctuation observed in the EBHP conversion versus time on stream, the overall trend clearly represents a decrease in the EBHP conversion with time on stream, indicating a deactivating Ti–SiO2 catalyst, with no evidence for a steady state even after 63 h of time on stream for this catalyst in Fig. 3c. The selectivity of the Ti–SiO2 catalyst remains stable around 64 ± 2%. In contrast, Ti-UCB-4 (Fig. 3d) exhibits no clearly observable drop in activity after 37 hours time on stream, demonstrating a clear steady-state performance after 24 h time on stream, without continuing deactivation during the run, at an EBHP conversion of 92%. This EBHP conversion represents only a slight drop in the 99% conversion observed at initial time on stream. The selectivity for the Ti-UCB-4 catalyst also remained stable upon increasing time on stream, and averaged at 73 ± 4%. Altogether, the data above demonstrate that the EBHP conversion decreases much more significantly for Ti–SiO2 relative to Ti-UCB-4 as catalyst, during 1-octene epoxidation catalysis in a flow reactor. In particular, as shown by data in Fig. 3c and d, during a period of 72 hours, the amorphous Ti–SiO2 catalyst deactivates continuously, whereas the crystalline Ti-UCB-4 shows no evidence of deactivation after reaching a steady state operating level after 24 h time on stream.
A hypothesis for the observed initial decrease (especially during the first 15 h time on stream) of the conversion in both catalysts involves the built up of organic-polymer matter, which can block the Ti active sites. This observed stability of Ti-UCB-4 relative to Ti–SiO2 can be rationalized on the basis of Ti sites in the former not being as accessible to such organic-polymer contaminants. This may be a consequence of their location within less accessible (to organic-polymer) framework sites within surface pockets. Further insight into the nature of catalyst deactivation is discussed using UV-vis spectroscopy below (vide infra).
Experiments in two different regions of conversion are summarized in Fig. 3e, to investigate the conversion of EBHP and its correlation to the selectivity for 1,2-epoxyoctane production. Overall, based on data in Fig. 3e, there is nearly no change in the 1,2-epoxyoctane selectivity as a function of the EBHP conversion. The data demonstrate a clear tendency for the delaminated zeolite to be more selective than the amorphous silica catalyst, by approximately 9%. This selectivity difference demonstrates less organic hydroperoxide decomposition to alcohol and molecular oxygen (unproductive decomposition, without oxygen transfer for epoxide synthesis), in the zeolitic Ti-UCB-4 versus amorphous Ti–SiO2 catalyst, at similar organic-hydroperoxide conversions. We infer that this increased selectivity must be due to the location of the Ti sites in the zeolitic catalyst. The majority of the Ti sites in Ti-UCB-4 consist of isolated framework Ti sites that are located near the external surface, in hemispherical cups, which in the fully condensed material form 12-membered ring supercages. Such confined catalytically active sites have been previously described to have a higher selectivity in ethylbenzene synthesis due to an invoked “nest effect”, where two-dimensional steric confinement of reactants bound to active sites in surface pockets leads to higher reaction rates and significantly higher selectivities.20
Rate constant comparisons for Ti-UCB-4 and Ti–SiO2
Pseudo-first order rate constants (assuming an ideal plug-flow reactor) on a catalyst mass and Ti basis were calculated based on the catalysis experiments corresponding to the highest conversion for each catalyst, and results are shown in Table 2. On a mass of catalyst basis, the amorphous silica and crystalline Ti-UCB-4 possess similar rate constants, k′ of 132 mL h−1 gcat−1 for Ti–SiO2 and 103 mL h−1 gcat−1 for Ti-UCB-4. On a per Ti basis, the calculated k for Ti-UCB-4 of 25.0 × 103 mL h−1 g−1 Ti is significantly higher than that for Ti–SiO2 of 9.6 × 103 mL h−1 g−1 Ti. We ascribe this difference to outer-sphere effects that can include a nest effect in the case of the crystalline material, where catalysis occurs at active sites that are located within surface pockets. Previously invoked nest effects account for rate enhancements in ethylbenzene production with MCM-22-based catalysts.9,20| Material | Crystalline | Conversion reaches steady-statea | Average selectivityb | Stable selectivity | Ti-sites in framework | Visual color of spent catalyst | Reaction rate constant k′ mass-based [mL h−1 g−1] | Reaction rate constant k titanium content-based [mL h−1 g−1] |
|---|---|---|---|---|---|---|---|---|
| a Conversion of EBHP. b Selectivity of EBHP for 1,2-epoxyoctane. | ||||||||
| Ti–SiO2 | No | No | 64% (±0%) | Yes | No | Orange | 132 | 9.6 × 103 |
| Ti-UCB-4 | Yes | Yes | 73% (±1%) | Yes | Yes | Light yellow | 103 | 25.0 × 103 |
Ultraviolet-visible spectroscopy of spent catalysts
To characterize catalysts before and after reaction, as well as after calcination of used catalysts following reaction, diffuse-reflectance UV-vis spectroscopy of the crystalline Ti-UCB-4 and amorphous Ti–SiO2 catalysts was performed.UV-vis data for Ti-UCB-4 after catalysis in Fig. 4a exhibits a maximum at 200 nm with two shoulders, one at 210 nm and a second at 230 nm. This can be compared with a maximum of 210 nm for the fresh catalyst, which, as discussed previously, is consistent with isolated Ti framework sites. We interpret the shoulder at 230 nm as representing higher coordinated (6-coordination number) Ti sites, and it may also indicate titanium sites that are not fully condensed to the framework, i.e. containing a titanol.24 Upon calcining this spent catalyst, the spent/calcined Ti-UCB-4 has a maximum absorbance at 221 nm. Because framework sites have been previously attributed to be in the range of 206–220 nm, we infer that sites in spent/calcined Ti-UCB-4 comprise isolated titanium sites in the framework, but with a shift towards titanols (Ti–OH). The visual color of the Ti-UCB-4 material after catalysis is a very pale yellow, and following calcination, it turns white.
 | ||
| Fig. 4 UV-vis spectra of fresh (I), spent (II) and spent and calcined (III) catalyst for a) Ti-UCB-4 and b) Ti–SiO2. | ||
In comparison, Fig. 4b shows the absorption spectrum of the amorphous Ti–SiO2 catalyst, which visually appears dark yellow (nearly orange) after catalysis, in contrast to the white color of the fresh catalyst. The spent catalyst shows a broad band spanning from 200 nm to 400 nm, with a maximum at 260 nm and a clear shoulder at 210 nm. Compared with the fresh Ti–SiO2 catalyst, this band is much broader and has a shoulder at higher energy. Following calcination of the spent catalyst, the material appears visually white, and the breadth of this band narrows considerably in the spent/calcined Ti–SiO2 catalyst, which exhibits a maximum at 260 nm and no shoulder at 210 nm. We infer that the shoulder at 210 nm as well as high wavelength bands in the region above 310 nm in the spent Ti–SiO2 catalyst must be due to organic residue on the catalyst surface, since these bands disappear upon calcination. The disappearance of these bands upon calcination is inconsistent with bands above 310 nm and at 210 nm in the spent Ti–SiO2 catalyst as being due to aggregated Ti and isolated Ti sites, respectively, as suggested previously in the literature.4,12,22,24 In comparison, Ti-UCB-4 lacks these bands and intense coloration following catalysis – suggesting less or no organic residue poisoning sites in this catalyst.
These results further correlate the greater degree of observed stability and selectivity of the crystalline Ti-UCB-4 catalyst relative to the amorphous Ti–SiO2 catalyst with regards to the formation of organic residue during catalysis. We posit that the higher observed selectivity of the crystalline catalyst and lack of organic residue poisoning sites in this catalyst stem from an outer-sphere environment effect on reactivity, which can include a “nest” effect, because of the location of Ti within surface pockets. Such an effect is caused by shape selectivity of the cup in which Ti sites within Ti-UCB-4 reside, which in turn prevents organic residue, which may be polymeric in nature, from plugging up Ti sites. Indeed, similar nest confinement effects have been previously invoked to explain enhanced catalytic rates during ethylbenzene synthesis, also in a liquid-phase reaction as encountered here, by Degnan et al.9,20
In summary, this manuscript compares a crystalline delaminated zeolite Ti-UCB-4 and an amorphous Ti–SiO2 material, for the epoxidation of 1-octene with ethylbenzene hydroperoxide, under tail-end reactor conditions. Table 2 summarizes the properties of each of these catalytic materials. While the rate constant on a mass basis is similar for both, on a Ti basis, the framework sites of the crystalline zeolite significantly outperform those of the silicate. The framework sites comprising Ti-UCB-4 are also more selective and catalytically more robust, in terms of conversion versus time on stream in a flow reactor operating at tail-end conditions, when compared with amorphous Ti–SiO2. Much of this difference in deactivation is correlated with poisoning by an organic residue in Ti–SiO2 and the substantial lack thereof in the former.
Recyclability of catalysts
In order to investigate the possibility of activity loss of the catalysts as a result of possible Ti leaching, a study was conducted on the recyclability of the catalysts. Fresh and spent/calcined amorphous Ti–SiO2 and crystalline Ti-UCB-4 catalysts were tested in a batch reactor system for epoxidation of 1-octene using EBHP at tail-end conditions, using the same mass of catalyst for each run as used in the flow testing. The spent catalysts correspond to a 50 h flow-catalysis experiment in which the conversion was slightly lower than in Fig. 3c and d for the amorphous and crystalline catalysts, respectively. The spent catalysts were calcined in air after catalysis. The results are presented in Fig. 5. There are no significant differences between the amorphous and crystalline catalysts in terms of activity on a mass basis, and the activity does not change when comparing the fresh versus spent/calcined materials in this batch-reactor study. This shows that both amorphous and crystalline catalysts can be recycled. These results are consistent with a lack of Ti leaching in both of these catalysts. We surmise that the observed catalyst deactivation is not the result of either Ti agglomeration or leaching, as both of these would be irreversible processes (i.e. not reversed upon calcination). One interpretation of these results is that deactivation is caused by active-site blockage as a result of organic matter, which may be removed via calcination treatment. Such an interpretation is based on diffuse-reflectance UV/vis spectroscopy of fresh, spent, and spent/calcined catalysts (vide supra). | ||
| Fig. 5 Recyclability of Ti–SiO2 (•) and Ti-UCB-4 (▲) fresh (empty) and spent/calcined (filled). | ||
Conclusions
Grafted Ti Lewis-acid sites synthesized on crystalline UCB-4 and amorphous SiO2 supports. UV-vis data demonstrate mostly isolated framework Ti sites in fresh Ti-UCB-4, which are consistent with fully condensed grafted Ti sites, whereas for Ti–SiO2, this data in consistent with predominantly tripodally grafted Ti sites on the surface, with some surface oligomers. Results of 1-octene epoxidation experiments at tail-end reactor conditions in a flow reactor show a clear benefit of the crystalline framework in the Ti-UCB-4 catalyst, in terms of activity per Ti site (similar activities for Ti–SiO2 and Ti-UCB-4 on a mass basis). The conversion of EBHP reaches a steady state for Ti-UCB-4, with virtually no observed long-term deactivation. In comparison, the conversion for the amorphous Ti–SiO2 never reaches a steady state with time on stream, as a result of observed deactivation. A crystalline environment in Ti-UCB-4 shows 9% higher selectivity than the amorphous environment of Ti–SiO2. UV-vis spectroscopy of both materials after catalysis show organic residues on the surface of Ti–SiO2 but not on Ti-UCB-4. These organic residues can be removed via calcination, to synthesize catalysts that are as active as the fresh materials, according to batch epoxidation experiments. These results demonstrate the benefit of a crystalline versus amorphous framework for the grafting of Ti active sites, for olefin epoxidation catalysis. Because these advantages of the crystalline versus amorphous catalyst become particularly amplified under tail-end reactor conditions, the results suggest a benefit of crystalline grafted Ti catalysts such as Ti-UCB-4, as drop-in replacements to conventional tail-end catalysts used for olefin epoxidation with organic hydroperoxide, in flow reactors.References
- H. Baer, M. Bergamo, A. Forlin, L. H. Pottenger and J. Lindner, Propylene Oxide, Ullmann's Encyclopedia of Industrial Chemistry, 2012 Search PubMed.
- D. Kahlich, U. Wiechern and J. Lindner, Ullmann's Encyclopedia of Industrial Chemistry, 2011 Search PubMed.
- D. L. Trent, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc, 2000 Search PubMed.
- J. K. F. Buijink, J.-P. Lange, A. N. R. Bos, A. D. Horton and F. G. M. Niele, Propylene epoxidation via Shell's SMPO process: 30 years of research and operation, Elsevier, Amsterdam, The Netherlands, 2008 Search PubMed.
- J. M. Notestein, L. R. Andrini, V. I. Kalchenko, F. G. Requejo, A. Katz and E. Iglesia, J. Am. Chem. Soc., 2007, 129(5), 1122 CrossRef CAS PubMed.
- N. A. Grosso-Giordano, A. J. Yeh, A. Okrut, D. J. Xiao, F. Grandjean, G. J. Long, S. I. Zones and A. Katz, Chem. Mater., 2017 DOI:10.1021/acs.chemmater.7b02062.
- A. Corma, Chem. Rev., 1995, 95(3), 559 CrossRef CAS.
- A. Corma, V. Fornés, J. M. Guil, S. Pergher, T. L. M. Maesen and J. G. Buglass, Microporous Mesoporous Mater., 2000, 38(2), 301 CrossRef CAS.
- T. F. Degnan, C. M. Smith and C. R. Venkat, Appl. Catal., A, 2001, 221(1), 283 CrossRef CAS.
- G. Wu, Y. Wang, L. Wang, W. Feng, H. Shi, Y. Lin, T. Zhang, X. Jin, S. Wang and X. Wu, Chem. Eng. J., 2013, 215, 306 CrossRef.
- X. Lu, H. Wu, J. Jiang, M. He and P. Wu, J. Catal., 2016, 342, 173 CrossRef CAS.
- J. K. F. Buijink, J. J. M. van Vlaanderen, M. Crocker and F. G. M. Niele, Catal. Today, 2004, 93–95, 199 CrossRef CAS.
- J. Buijink and V. Van, Titanium catalyst, its preparation and its use in epoxidation reactions (WO2007128666A1), 2007 Search PubMed.
- X. Ouyang, S.-J. Hwang, D. Xie, T. Rea, S. I. Zones and A. Katz, ACS Catal., 2015, 5(5), 3108 CrossRef CAS.
- X. Ouyang, S. I. Zones and A. S. Katz, Highly active, selective, accessible, and robust zeolitic Sn-baeyer-villiger oxidation catalyst (US9687830), 2017 Search PubMed.
- R. C. Runnebaum, X. Ouyang, J. A. Edsinga, T. Rea, I. Arslan, S.-J. Hwang, S. I. Zones and A. Katz, ACS Catal., 2014, 4(7), 2364 CrossRef CAS.
- I. Ogino, E. A. Eilertsen, S.-J. Hwang, T. Rea, D. Xie, X. Ouyang, S. I. Zones and A. Katz, Chem. Mater., 2013, 25(9), 1502 CrossRef CAS.
- S. I. Zones and A. W. Burton, Molecular sieve SSZ-70 composition of matter and synthesis thereof (US7108843B2), 2006 Search PubMed.
- S. Unverricht, M. Hunger, S. Ernst, H. G. Karge and J. Weitkamp, Stud. Surf. Sci. Catal., 1994, 84, 37 CrossRef CAS.
- T. F. Degnan, J. Catal., 2003, 216(1), 32 CrossRef CAS.
- J. H. Nobbs, Color. Technol., 1985, 15(1), 66 Search PubMed.
- P. Ratnasamy, D. Srinivas and H. Knözinger, Adv. Catal., 2004, 48, 1 CAS.
- H. Kochkar and F. Figueras, J. Catal., 1997, 171(2), 420 CrossRef CAS.
- L. Marchese, T. Maschmeyer, E. Gianotti, S. Coluccia and J. M. Thomas, J. Phys. Chem. B, 1997, 101(44), 8836 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00076f |
| This journal is © The Royal Society of Chemistry 2017 |