DOI:
10.1039/C7RE00082K
(Review Article)
React. Chem. Eng., 2017,
2, 611-627
Liquid–liquid microflow reaction engineering
Received
6th June 2017
, Accepted 18th August 2017
First published on 18th August 2017
Abstract
Microflow reaction is important for the miniaturization of chemical devices and systems. From an engineering perspective, excellent characteristics, including narrow residence time distribution, rapid reactant mixing, fast mass and heat transfer, and effective droplet morphology control, of segmented liquid–liquid flow in microspace are summarized in this review based on recent research progress in the areas of microfluidics, flow chemistry, micro total analysis, and microreaction engineering. Via engineering, important roles of segmented liquid–liquid flow, such as improvements in production yield, inline purification of intermediates and products, creative droplet reactors, and preparation of particle materials with various structures, are introduced. The scale-up of micro devices and large-scale production of chemicals through microflow technology are also discussed, showing good prospects in research and industrial application.
Introduction
Chemical synthesis using microflow chips or microreactors has rapidly developed over the past two decades. Similar to the electronics industry, miniaturization of devices and systems has brought significant improvement in the efficiency and safety of chemical processes,1 which has led this field from being doubted to completely accepted by academia and industry.2,3 Nowadays, research and development of microreactors,4 microfluidics,5 flash and flow chemistry6,7 and micro total analysis (μ-TAS)8 proposed based on fields of engineering, physics, chemistry, and biology have been highly integrated, leading to many interdisciplinary research topics. Advanced microflow systems with compact equipment have resulted in a change from batch operation to continuous operation in lab, which builds a bridge between chemistry and chemical engineering and allows much faster commercialization of new chemical processes.9,10
To implement microflow chemical processes in lab, basic platforms are easily assembled using a few commercial units including metering pumps, micromixers, microchannel chips, delay loops of microtubes, temperature controllers, and back-pressure regulators. Using this kind of microflow platforms, many chemical reactions have been continuously carried out with improved yield, efficiency, and safety as compared to those in conventional batch reactors.7 Several microflow devices can also be integrated for implementing multistep reactions and separations.11 High-performance liquid chromatographs (HPLC), flow infrared spectrometers (IR), or inline mini mass spectrometers can be connected to a microflow platform to in-line monitor chemical components in time.12,13 This build-up model allows microflow platforms to work like mini factories in lab,14 realizing chemical production in refrigerator-sized reaction systems.15
Owing to the advantages in device build-up, operation, and good performance in running chemical processes, microflow chemical systems have been widely applied in the synthesis of pharmaceutical chemicals,16 fine chemicals,17 particles,18 and polymer materials.19 To extend the application, chemical, mechanical, and material engineers have been continually improving these mini plants, with advances in mixing, heat exchange, safety, and automation,20 allowing chemists to develop new products or novel synthesis routes without worrying about engineering problems.21 Unlike chemists, who are more focused on chemical routes, reaction mechanism, and chemical products, chemical engineers in this area are more interested in reaction engineering issues such as phase behavior, reaction kinetics, transport mechanism, and reaction time control.22,23 In different types of microflow systems, homogeneous processes are more reported in the literatures,24,25 probably because they are much easier to implement in small devices. Homogeneous systems where chemicals are fully mixed also show chemical rules clearly, since they are less influenced by engineering problems, such as mass transfer controlled reaction kinetics.11,25,26 Although running a heterogeneous system is relatively difficult in microspace, it is still gaining attention because of the unique advantages of multiphase processes. For example, low concentrations of chemicals are often used in homogeneous reactions to overcome the limitation of component solubility. However, it is difficult to employ low production capacity in an industrial process. Liquid–liquid or gas–liquid two-phase reacting systems may allow high concentration of reactant in the liquid.27 A multiphase system is also important for purification.9 In many cases, excess reactant or adverse byproduct should be removed quickly from the reaction system.28 Liquid–liquid extraction is one of the best methods for realizing separation in microflow processes. In addition, multiphase microflows have numerous flow patterns, which are effective templates for preparing functional materials using microreactions.29
Considering the necessity of understanding the basic laws and advantages of multiphase microflows for carrying out chemical processes, this review highlights typical multiphase microflow studies in recent years from a reaction engineering perspective, which are less summarized in the literature. For greater focus on our topic, only liquid–liquid reaction processes employing microflow systems are introduced and additional phases other than liquids are not involved in mass transfer or reaction. Therefore, some relevant processes like liquid–liquid–solid catalyst systems are not engaged. Nevertheless, some relevant knowledge can be found in this review to help understand those processes.30–32 In the following sections, the engineering characteristics of liquid–liquid microflows are summarized first. Then, the features and advances of liquid–liquid microflow reactions are discussed using many typical examples and some potential industrial applications. Finally, an outlook for the development on microreaction processes is suggested, which might be helpful for future chemical processes and technologies.
Engineering characteristics of liquid–liquid microflows
Multiphase flows in microspace have been studied in detail for the past two decades, especially in the area of microfluidics.33 Many research papers have explored typical flow patterns of liquid–liquid flows in microchannels or microtubes and control mechanisms of generation, break-up and coalescence of small droplets have been well indicated.34–37 To focus on reaction engineering topics, this paper does not cover all details of fluid dynamics of liquid–liquid phases in microchannels. Interested readers can find relative reports in review papers of ref. 28 and 38–41. In different kinds of liquid–liquid microflows, flow patterns with segmented or tiny droplets are mostly used in chemical syntheses. We can call them slug flows as the droplet length (Ldroplet) is not less than the channel with (w) or the tube diameter (d), Ldroplet ≥ w or D, and bubbly flows as the droplet diameter (ddroplet) is much smaller than w and d, ddroplet ≪ w or d. In the process of material preparation, double emulsions and systems containing Janus droplets, which contain oil–water–oil or water–oil–water phases, are sometime used. For these liquid–liquid microflow processes, we will show their reaction engineering characteristics in this section, including residence time distribution, mixing and transport properties, and material morphology control, which are fundamentals for developing microflow reaction process.
Narrow residence time distribution
According to the basic principles of reaction engineering, residence time distribution (RTD), which shows reaction time difference between different fluid elements in a reactor, is very important for reactant conversion and product yield.42 Batch reactors do not have RTD, but when running a continuous stirred-tank reactor (CSTR), the residence time function gives a long tail, which means the flow-out fluid has increased reaction time. Using a tubular reactor can narrow RTD. But because of radial velocity distribution and axial diffusion, RTD is still different from that of an ideal plug flow reactor (PFR) without considering assisting effects, such as secondary flow induced by curved flow path.43,44 However, this non-ideal RTD in a microflow reactor can be improved by using liquid–liquid slug flows.
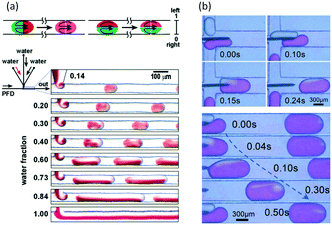
In microspace, flow is mainly dominated by capillary force with low Ca number and We number,45 which represent the ratios of viscous and inert force to capillary force, respectively (Ca = μu/γ, We = ρu2d/γ, where μ is viscosity, u is velocity, γ is interfacial tension, ρ is density of fluid, and d represents tube diameter or a characteristic scale of microchannel, such as w). Since non-wetting dispersed fluids tend to fully fill the cross-section of the flow path under the effect of capillary pressure, the fluids are well segmented by their interfaces at low Ca and We. Regulated by the closed interface on the edge of droplet, the parabolic distribution of velocity in single phase becomes circular flows in each phase, making the dispersed droplets work as mini batch reactors,46 as shown in Fig. 1(a).47 As the interfaces around the droplets are closed, RTD curves for the tracer in droplets are close to the Dirichlet function if liquid slugs between droplets do not become of non-uniform volume (sometimes slugs will be non-uniform in the downstream of droplet generation point because of uncontrolled leakage flow in the gaps between droplets and device wall). For the continuous phase, which involves wetting on the channel wall and connection by corners and liquid films between droplet and device, the tracer can move from a concentrated part to a dilute part, and this axial diffusion gives a distribution of the residence time. However, according to the results in Fig. 1(b),48,49 RTDs are still narrow, which could provide a controllable reaction time. A good method for obtaining sharp RTDs for both the continuous and dispersed phase is to change the wetting property of the flow path to a contact angle of nearly 90°, which makes both phases discontinuous, as shown in Fig. 1(c).50
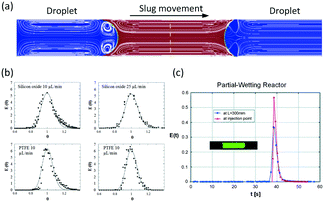 |
| | Fig. 1 Flow simulation of slug flow and RTD tests. (a) Characteristic circulatory patterns in segmented droplets and liquid slugs (adapted from ref. 47 with permission of Elsevier). (b) RTDs of continuous fluids in different microchannel reactors (adapted from ref. 49 with permission of the American Chemical Society). (c) Narrow RTDs in a partial wetting microchannel reactor (adapted from ref. 50 with permission of the Royal Society of Chemistry). | |
The average residence time (τi) of phase i can be calculated from reactor volume (V), phase hold-up (ϕi), and flow rate (Qi) or by dividing reactor length (L) by average velocity of the phase (ui), as shown in eqn (1).
| | 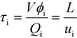 | (1) |
For a tubular reactor with a round cross-section, the hold-up of dispersed liquid plugs can be evaluated using eqn (2) if Ca number of droplet (Cadroplet = μcudroplet/γ) < 0.01.28
| | 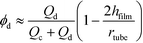 | (2) |
where
μc is viscosity of the continuous phase,
udroplet is relative velocity of droplets to the tube,
Qc and
Qd are flow rates of the continuous and dispersed phase, respectively,
rtube is tube radius, and
hfilm is thickness of lubricated film between the droplet and tube wall, which is a ratio of Ca
droplet2/3 at Ca
droplet < 0.01.
51 In a flow path with a square cross-section, experiments have exhibited that the velocity of droplets is larger than average speed of the surrounding continuous phase.
52 The relative velocity of the two phases has a linear relation with Ca
droplet1/3 at Ca
droplet < 1. According to this relationship, the relative velocity of the two phases is low at low Ca
droplet, and therefore, RTD of the continuous phase is narrow at low Ca as the substance interchange is weak between liquid slugs. Since it is difficult to evaluate the continuous phase flow rate through the droplets, it is better to use an experiment to determine RTD function exactly.
Intensified mixing and transport processes
As discussed in the previous section, flow behaviors of all phases are important for reaction time in liquid–liquid microflow reactors. In addition, they are also important for mixing and mass/heat transport in microreaction processes. Turning a homogeneous mixing process to a heterogeneous process with immiscible carrier fluids is sometimes a good method for mixing enhancement.53 The differences between homogeneous and slug flows during mixing processes have been revealed in an early study.54 As shown in Fig. 2(a), homogeneous mixing is dependent on diffusion in microspace. Without assistance from secondary flow55 or external fields,56 this diffusion is rather slow at a low Reynolds number. However, in slug flow systems, circulations in segmented liquids can accelerate reactant mixing if the reactants are correctly placed, as shown in Fig. 2(a).57,58 This mixing can be further enhanced in winding paths by breaking axisymmetric circulation flow and redistributing to-be-mixed substances.59,60 Recent experiments have shown that segmented slug flow not only intensifies mixing but also allows precise matching of microliter amounts of reagents based on coalescence of droplets, as shown in Fig. 2(b).61,62 This precise feeding of reactants is much more ingenious than conventional batch processes using injections.
 |
| | Fig. 2 Mixing in droplets. (a) Effect of initial fluid position on mixing in droplet and red dye distribution in flowing droplets (adapted from ref. 54 with permission of the American Chemical Society). (b) Microlancet-induced droplet coalescence and mixing in a droplet with time (adapted from ref. 62 with permission of the Royal Society of Chemistry). | |
In addition to fast mixing in droplets, mass transfer of substance passing through interface is also rapid according to several experiments.63 Volumetric mass transfer coefficients in microspace are known to be 2 or 3 orders of magnitude larger than those in many conventional devices.64,65Fig. 3(a) shows a 2D simulation of mass transfer between segmented liquids.66 Although this 2D process is not exactly the same as the experimental one in 3D space, we find that convection flows around droplet caps help transport of acetic acid. In this situation, acetic acid moves quickly from the interface to the liquid interior.67Fig. 3(a) does not consider mass transfer from the side of the dispersed phase. This is only acceptable for flows at low Ca (Cadroplet < 10−2) in round tubes or capillaries with negligible liquid films of continuous fluids beside the droplets. Considering that mass transfer through all parts of the droplets to have similar rates, eqn (3) reported by Susanti et al.68 can be used to evaluate volumetric mass transfer coefficient in tubular microflow devices.
| | 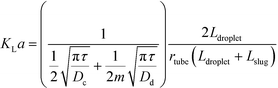 | (3) |
where
KLa is volumetric mass transfer coefficient, which shows as transfer rate per unit mass transfer time and per unit concentration difference;
τ is average residence time of two phases,
τ =
Vtube/(
Qc +
Qd);
Vtube is tube volume;
Dc and
Dd are diffusion coefficients in the continuous and dispersed phase, respectively; and
Ldroplet and
Lslug are lengths of droplets and liquid slugs, respectively, as shown in
Fig. 3(b). In more complicated situations, mass transfer from the side of the droplet is slower than that from the caps because of the low flow rate in liquid films.
69Fig. 3(a) and (b) also show that circulation is only fast at the middle inline and the edges of the droplets and slugs. Different from turbulent flow in stirred reactors, which has eddy break-up to help mixing, laminar flow in droplets only transports substance along the streamline. Therefore, this circulation flow only assists substance movement in the axial direction. However, in the center of the whirlpool, fluid is relatively stationary and mass transfer in this part mainly relies on diffusion. When the mass transfer substance forms circles in a droplet, as shown in
Fig. 3(c), convection mass transfer of substance is useless and the following mixing between the red and blue parts based on diffusion has a reduced rate, as shown by the long logarithmic coordinate time in the figure.
70 Moreover, if a droplet is small and away from the wall, the velocity difference beside it along the radial direction of the tube will be small, making the droplet more rigid and the mass transfer inside more dependent on diffusion.
71 Further, if the droplets have large mass transfer areas, the mass transfer rate will be still acceptable.
72 These complicated phenomena show that prediction of mass transfer coefficient is very difficult for the various controlling steps. Although some semi-empirical formulae for the mass transfer coefficient have been reported,
73 there is an urgent need for more theoretical analysis and simulation on mass transfer mechanisms in microspace helpful for design and operation of liquid–liquid microflow reactors.
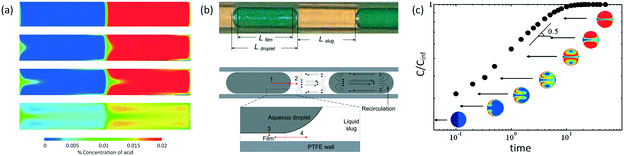 |
| | Fig. 3 Mass transfer in liquid–liquid slug flow. (a) Concentration maps showing distribution of acetic acid as it diffuses from the organic to aqueous phase (adapted from ref. 66 with permission of Elsevier). (b) Mass transfer details in tubular microdevices for physical extractions (adapted from ref. 68 with permission of the American Chemical Society). (c) Variation of normalized concentration inside a droplet as a function of time. The colors in the droplets show concentration distributions from simulation. The microchannel, whose width is close to droplet diameter, as well as the continuous phase are not shown in the figure (adapted from ref. 70 with permission of the American Chemical Society). | |
To further enhance mass transfer, winding flow paths are workable owing to enhancement of convective mass transfer between phases.74 In addition, it is important to increase mass transfer area. Constrictive structures75 and packed bed microreactors,17,76 as shown in Fig. 4(a)–(c), are good approaches for realizing mass transfer enhancement. Owing to droplet split and Rayleigh–Plateau instability break-up,65,77 the two phases can be mixed as emulsions, as shown in Fig. 4(d), achieving a surface-to-volume ratio >230![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2 m−3.75 Adding an inert gas to the liquid–liquid system has also been proved to be a promising method. The gas phase on one hand changes the circulation that aids convective mass transfer and droplet surface renewal,78 and on the other hand, a chaotic gas can further rupture the dispersed liquid to tiny droplets with large mass transfer area.79,80 An optimal mass transfer enhancing effect has been observed at a gas volume ratio of 0.09.81 Above this ratio, mass transfer coefficient reduces, probably due to long bubbles leading to large distances between successive liquids. Inputting mechanical energy into a liquid–liquid flow system using oscillatory equipment or an ultrasonicator will increase mass transfer greatly,82,83 but narrowness of RTD may be affected. Additional heat from fluid friction should also be carefully considered in this situation.
000 m2 m−3.75 Adding an inert gas to the liquid–liquid system has also been proved to be a promising method. The gas phase on one hand changes the circulation that aids convective mass transfer and droplet surface renewal,78 and on the other hand, a chaotic gas can further rupture the dispersed liquid to tiny droplets with large mass transfer area.79,80 An optimal mass transfer enhancing effect has been observed at a gas volume ratio of 0.09.81 Above this ratio, mass transfer coefficient reduces, probably due to long bubbles leading to large distances between successive liquids. Inputting mechanical energy into a liquid–liquid flow system using oscillatory equipment or an ultrasonicator will increase mass transfer greatly,82,83 but narrowness of RTD may be affected. Additional heat from fluid friction should also be carefully considered in this situation.
 |
| | Fig. 4 Mass transfer enhancement by tiny droplets in bubbly flow. (a) Constrictively tubular reactor (adapted from ref. 75 with permission of John Wiley and Sons). (b) Schematic and (c) image of a packed bed microreactor (adapted from ref. 17 and 76 with permission of Elsevier, respectively). (d) Preservation of emulsified liquid–liquid flow in a constrictively tubular reactor (adapted from ref. 75 with permission of John Wiley and Sons). | |
With respect to heat transfer performance of microflows, microreactors have been confirmed to achieve excellent heat exchange between the fluid and microchannel wall,3 which is very important for realizing accurate temperature control of chemical reactions. Usually, in tubular microflow reactors, inside fluids are heat-transfer resistant, and heat transfer coefficients can be evaluated by eqn (4).84
| | 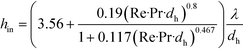 | (4) |
where
hin is heat transfer coefficient, showing heat transfer rate per unit temperature difference, unit time, and unit heat transfer area; Pr is Prandtl number of the fluid (Pr =
μCp/
λ);
μ is viscosity;
Cp is heat capacity;
λ is thermal conductivity; and
dh is hydraulic diameter of the flow path. Considering the similarity between mass and heat transfer, heat exchange between two liquids should also be fast; therefore, the tube wall is not the only path for removing heat. Excess solvents or additional inert components are also able to accept a part of reaction heat.
85 However, very few studies have focused specifically on heat transfer between liquids in microspace. Considering heat conduction rate is actually faster than the diffusion rate in most liquids (heat diffusion coefficient is usually about 10-fold larger than mass diffusion coefficient in most liquids),
86 heat transfer between phases should also be rapid. Using a microdispersion device with an inline phase separator, volumetric heat transfer coefficients for bubbly flows are found to be from 5 to 25 MW m
−3 K
−1.
87 Owing to this good heat transfer performance, temperature difference between reacting phase and heat removal phase (or solvent phase) may be neglected for strong exothermal reactions, as indicated by simulation in
Fig. 5.
88 This phenomenon is important for developing a new cooling method for reactions in microreactors, especially for those in which it is much more difficult to remove reaction heat from the microchannel or tube walls.
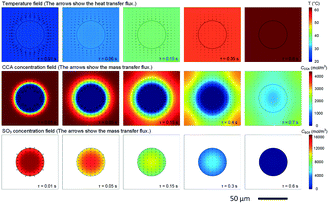 |
| | Fig. 5 Temperature and concentration distributions around a tiny droplet. Variations in temperature and concentration in the reaction between cyclohexanecarboxylic acid (CCA) solution (the continuous phase) and oleum (droplet) with reaction time (adapted from ref. 88 with permission of Elsevier). | |
Droplet morphologies in microflows
From the perspective of traditional chemical engineering, excellent mixing, mass and heat transfer performances, and precise reaction time have led microflow reactors to be close to ideal PFRs. Therefore, many chemical processes can be implemented with high working efficiency. In addition to carrying out reactions for synthesizing chemicals, microflow devices are also effective platforms for preparing functional materials.40 Owing to the various flow patterns of multiphase microflows, it is easy to use dispersed droplets as templates to prepare materials with complicated structures.89 For example, microflow devices can generate multi-emulsion droplets, as shown in Fig. 6(a).90 These multilayer droplets can then be solidified into hollow spheres or microcapsules through UV-photo or heat-induced polymerization.91,92 Further improved multilayer droplets can give very thin liquid films as a middle phase (Fig. 6(b)), which is useful for the preparation of core–shell particles.93 If interfacial tensions between phases do not obey the rule γoi > γom + γmi, where o, i, and m represent outer, inner, and middle phase, respectively, the droplets form combined fluid particles (Fig. 6(c)),94 which are called Janus droplets.95 These droplets are good templates for preparing asymmetric particles.96 To generate irregular fluid particles, other flow patterns are also useful, such as three-phase flow contained squeezed bubbles and droplets, as shown in Fig. 6(d).97 These morphologies allow microflow reactors to be used as production lines for particle materials.
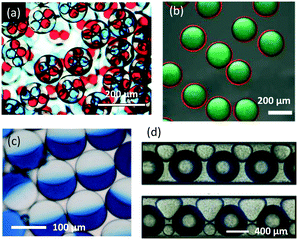 |
| | Fig. 6 Various shapes and structures of microdroplets. (a) Optical microscopy images of double emulsion droplets (adapted from ref. 90 with permission of the Royal Society of Chemistry). (b) Multi-layer droplets with ultrathin oil shells as the middle phase (adapted from ref. 93 with permission of the Royal Society of Chemistry). (c) Optical image of Janus droplets (adapted from ref. 96 with permission of Springer). (d) Squeezed droplets and bubbles flowing in microchannels (adapted from ref. 97 with permission of Springer). | |
Roles of liquid–liquid microflow in chemical reaction and material preparation
The fast development of microflow research is a result of the practicality of the technology. During recent years, more and more chemists have realized that microflow systems are good platforms for discovering new chemicals, establishing new synthetic methods, and preparing new materials.3,25,98 As a number of chemical processes are difficult multiphase reactions, improvements in conversion, yield, and safety by using new reaction devices continues to attract attention from academia and industry. In addition to intrinsic multiphase reactions, deliberately constructed heterogeneous phase reactions sometimes bring more advantages in mixing and online purification. In this section, we present typical applications of liquid–liquid microflow reactions, classified by function of liquid–liquid phases during reactions.
Continuous chemical synthesis and reaction intensification
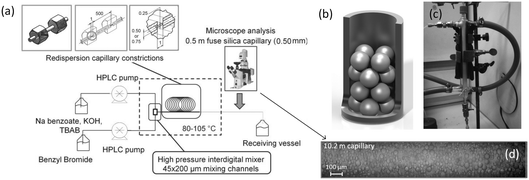
An important reason for using a continuous flow reactor is to improve the working efficiency of the process. Many studies have shown that conversion and yield in microflow reactors are better than those in traditional batch reactors.21,99 For example, H2O2 is widely used in chemical synthesis as a clean oxidation agent. However, when utilizing H2O2, it is usually controlled to low concentrations, mild temperature and low pressure owing to its high activity and strong exothermal effect.3 In fact, unsatisfactory reaction results are usually caused by bad contact of reactants or weak heat removal competence of a reactor. For many H2O2 oxidation reactions, organic reactants in oils are weakly soluble in aqueous solution of H2O2, and therefore, the reactions mainly take place in regions close to liquid–liquid interface. Increasing contact area of phases is a key for allowing the oxidation reaction to occur quickly, reducing decomposition of H2O2, and preventing excessive oxidation.100,101 Using Zn-substituted polyoxotungstate and tributylammonium hydrogen sulfate (TBAHS) as the catalyst (Scheme 1), 98.8% 1-phenylethanol conversion and 100% ketone selectivity was achieved in a chip reactor within 5 min in a recent study.102 The reaction productivity was then increased by using Corning AFRs (advanced flow reactors) without sacrificing mass and heat transfer performance. With the help of an advanced phase separator, emulsification was also avoided after the reaction was finished. In another example, adipic acid was prepared in a packed bed micro-reactor from cyclohexene using Na2WO4·2H2O in H2O2 solution as the oxidation catalyst and [CH3(n-C8H17)3N]HSO4 in cyclohexene as the phase transfer catalyst (Scheme 2).103 Smaller glass beads in the reactor resulted in better yields of adipic acid, which reached 50% in 20 min residence time at 100 °C. This clearly shows that smaller droplets increase the reaction yield by mass transfer enhancement.
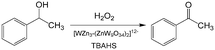 |
| | Scheme 1 H2O2 oxidation of 1-phenylethanol to acetophenone catalyzed by polyoxotungstate and TBAHS.102 | |
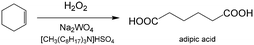 |
| | Scheme 2 H2O2 oxidation of cyclohexene to adipic acid catalyzed by Na2WO4 and [CH3(n-C8H17)3N]HSO4.103 | |
Similar to the H2O2 oxidation reactions, some other phase transfer reactions have also been improved with microflow reactions. For example, etherification reaction of benzyl bromide and phenoxide with tetrabutylammonium bromide (TBABr) as the catalyst (Scheme 3) achieved 95% phenoxide conversion in a tubular microreactor within 1 min residence time, whereas 180 min was required in a batch reactor.104 Using the constrictive tube to break droplets to smaller ones, an increase in reactant conversion from 38% to 95% with a residence time of 20 s was obtained for the esterification of sodium benzoate by using benzyl bromide (Scheme 4).75 In another example, selective alkylation of phenylacetonitrile in a 250 μm-diameter capillary employed large specific surface area droplets, and the circulation significantly promoted mass transfer between phases, as shown in Fig. 7.27 Both the conversion and selectivity of the monoalkylated products were larger than those in a stirred reactor, which employed droplets with an average diameter of 500 μm.
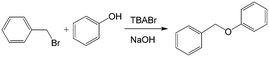 |
| | Scheme 3 Reaction of benzyl bromide and phenol to benzyl phenyl ether catalyzed by TBABr.104 | |
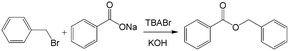 |
| | Scheme 4 Esterification reaction of sodium benzoate and benzyl bromide catalyzed by tetra-n-butylammonium bromide.75 | |
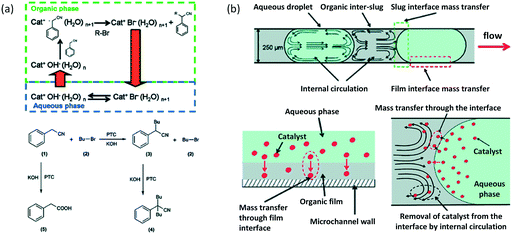 |
| | Fig. 7 Phase transfer catalyzed reactions in microflows. Selective alkylation by slug flow reaction in microchannel. (a) Mass transfer and kinetic steps in phase-transfer alkylation of phenylacetonitrile. Cat: triethylbenzylammonium chloride (TEBA). (b) Mass transfer through the interface in a slug flow. Internal circulation moves the catalyst away from the interface, thus increasing area of reaction (adapted from ref. 27 with permission of the American Chemical Society). | |
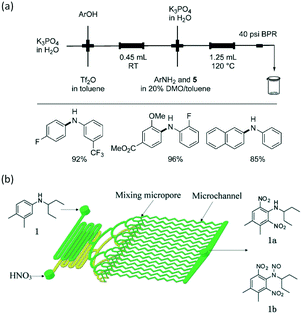
C–C and C–N cross-coupling reactions are very important in pharmaceutical industry. For the Suzuki–Miyaura cross-coupling reaction of aryl triflate and aryl boronic acid, a basic solution was required (Scheme 5) owing to the weak stability of intermediate aryl triflates. Since it is easy to induce phase separation between a highly concentrated salty solution and the organics after reaction, bubbly flow with tiny droplets is a good choice to enhance reactant mixing and reduce reaction time. A packed bed microreactor was therefore used in the reaction, and >96% yield was finally achieved for many biaryl compounds.105 Similar to C–C cross-coupling reaction, C–N cross-coupling reactions have also been successfully carried out in packed bed microreactors with high yields.106 As shown in Fig. 8(a), a mixture containing aryl triflates from the first step were directly fed into the second reactor after mixing with aryl amine substrates, XPhos precatalyst, and additional K3PO4, realizing high yields of >85% for biarylamines. N,N-Dimethyloctanamide (DMO) was also proved to be a good co-solvent that enables synthesis of (hetero)arylamines. In contrast to a previous report using in situ generated aryl triflates,105 intermediate purification was not required in this process.
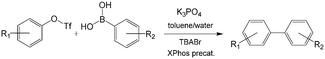 |
| | Scheme 5 Suzuki–Miyaura cross-coupling between aryl triflate and aryl boronic acid in a toluene/water system with K3PO4 as the base. XPhos ligands and XPhos precatalysts catalyze the reaction. TBABr is the phase transfer catalyst.105 Tf = trifluoromethanesulfonyl. | |
 |
| | Fig. 8 C–N cross-coupling and nitration reaction in microreactors. (a) Two-step conversion of phenols to biarylamines in consecutively connected packed bed microreactors (adapted from ref. 106 with permission of John Wiley and Sons). (b) Dinitration of N-(1-ethylpropyl)-3,4-xylidine in a multichannel microreactor (adapted from ref. 108 with permission of the Royal Society of Chemistry). | |
In addition to high reaction conversion and yield, which are preferable from the perspective of economy, microflow reactors are good platforms for carrying out hazardous process safely. For example, nitration reaction is a classical method for introducing nitrogen atoms into organics, but it should be carefully operated to prevent uncontrollable heat release and explosion.107 Microchannel reactors also provide large heat transfer areas and safe reaction environments. For example, a selective approach to prepare dinitro herbicides through a one-step nitration was safely carried out in a multi-channel microreactor, as shown in Fig. 8(b).108 The main products were obtained in >97% selectivity (1a and 1b in Fig. 8(b)). Unlike conventional batch processes, this reaction was conducted in a solvent-free condition owing to the excellent heat exchange performance of the microreactor. Similarly, 2-isopropoxy-5-nitrobenzaldehyde (Scheme 6, an important ligand building block for the catalysts of Hoveyda–Grubbs metathesis reaction) was synthesized by nitrating 2-isopropoxybenzaldehyde with a high productivity of 13 g h−1.109 Selectivity in the microflow reaction process reached an excellent value of 87% with fuming HNO3 as the reactant. In another example, nitration for the synthesis of 5-bromo-4-methyl-3-nitro-pyridin-2-ylamine was transferred from kg scale to 100 kg scale in a plant with a jacketed tubular flow reactor.110 This work proved that high heat of reaction can be managed in a microflow system.
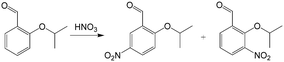 |
| | Scheme 6 Nitration of 2-isopropoxybenzaldehyde to 2-isopropoxy-5-nitrobenzaldehyde with 2-isopropoxy-3-nitrobenzaldehyde as side product.109 | |
Similar to nitration reactions using fuming nitric acid, reactions containing fuming sulfuric acid are also strongly exothermic and hazardous. Recently, reports have shown that Beckmann rearrangement of cyclohexanone oxime (Scheme 7) could realize >99% yield of ε-caprolactam by changing conventional homogeneous reaction to a liquid–liquid reaction by introducing n-octane as the solvent and a heat storage medium in the microflow reactor.111,112 Another approach to prepare ε-caprolactam using 65% SO3 in fuming sulfuric acid as the reactant was also safely implemented in a tubular reactor with >97% intermediate selectivity, owing to the rapid heat exchange between droplets and the solvent.88,113
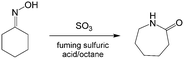 |
| | Scheme 7 Beckmann rearrangement of cyclohexanone oxime to prepare ε-caprolactam with 20% SO3 in fuming sulfuric acid as the catalyst.112 | |
Chemical processes with inline purification
Pharmaceutical and fine chemical syntheses are key areas for the application of microflow reaction. To obtain complicated organic products with high yield and high working efficiency, multistep reactions are necessarily implemented, which can be realized by consecutive microreactors.114 As each reaction should be precisely controlled, intermediate purification is sometimes inevitable to remove excess reactants or unused byproducts.7,14 Of all separation technologies, liquid–liquid extraction is one of the most convenient approaches for use in microflow processes. This approach has the advantage of removing one phase out of the system by using an inline phase separator115 and fast mass transfer between phases ensures rapid purification.116 Different from running reactions, these kinds of liquid–liquid microflow processes are auxiliaries of reactions.
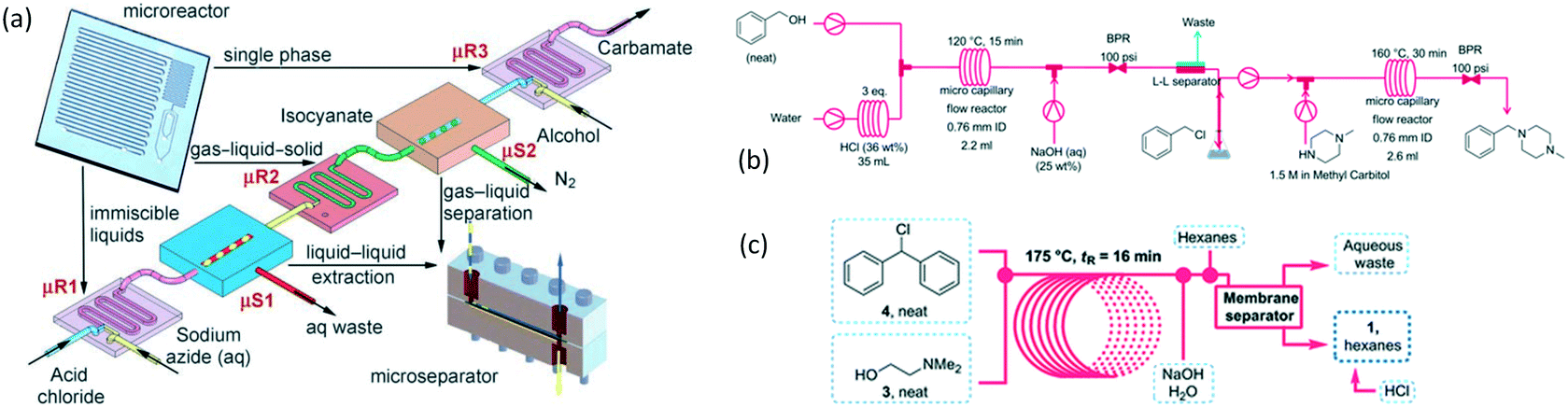
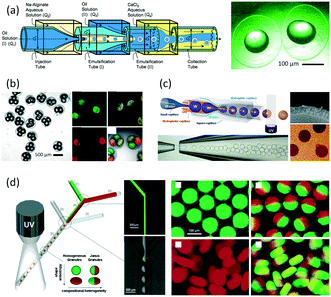
As an early example of combining reaction and extraction in one microflow process, a continuous multistep synthesis consisting of three reactions with intermediate separations for purification of intermediate products was implemented by using consecutive chip microreactors and inline phase separators, as shown in Fig. 9(a).117 After diazotization, the aqueous phase was separated from the reaction system by a mini phase separator to remove inorganic salts. This work demonstrated the concept of mini factory on table and explored safe syntheses of a family of chemical products with in situ synthesis and consumption of hazardous intermediates, shown by Scheme 8. In a similar research work, a two-step reaction for benzylmethylpiperazine was implemented with intermediate purification. As large amounts of HCl from the first reaction would make the subsequent reaction difficult, NaOH addition and inline phase separation were necessary to remove excess HCl between two reaction steps.118 Similar to this HCl removal process, HBr generated by halogenation reactions can also be removed by using inline liquid–liquid extraction.119 In another example, inline extraction was used in a Heck reaction (Scheme 9) with a microreactor system.120 After the first step, the base in the CH2Cl2 (DCM) solution was removed with an acidic wash in a mini extractor, and then the organic phase with aryl triflate intermediate was fed into the second step to achieve solvent exchange. In addition to washing or extraction by water, organic solvents have also been used in many microflow purification processes. As the example shown in Fig. 9(c),121 diphenhydramine was first separated and purified from an aqueous solution by hexane, and diphenhydramine hydrochloride was then precipitated by a HCl–isopropanol solution added into the organic phase.
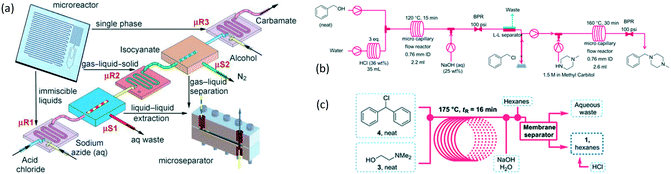 |
| | Fig. 9 Consecutive microreaction and liquid–liquid extraction. (a) Consecutive microreactors and phase separators for carbamate synthesis (adapted from ref. 117 with permission of John Wiley and Sons). (b) Schematic for benzylmethylpiperazine synthesis using a microflow system with inline phase separation (adapted from ref. 118 with permission of John Wiley and Sons). (c) Extraction for separation and purification of tertiary amines by using hexane in a microflow system (adapted from ref. 121 with permission of the Royal Society of Chemistry). | |
 |
| | Scheme 8 Carbamate synthesis from acid chloride.117 | |
 |
| | Scheme 9 Two-stage Heck reaction of phenol with vinyl ether. DIEA = diisopropylethylamine, Tf = trifluoromethanesulfonyl, DPPP = 1,3-bis(diphenylphosphino)propane.120 | |
Microliter droplet reactors
Multiphase reactions usually employ immiscible reactants. If the reactants are miscible, the reactions are usually carried out in homogeneous solutions. However, manually changing a homogeneous reaction system to a heterogeneous reaction system by introducing an inert phase may achieve additional advantages. As we discussed in the second section, if an inert medium segments a homogeneous fluid into dispersed units,58 the segmented microliter droplets can be used as mini batch reactors, which have good mixing performance, narrow RTD, and fast heat exchange ablity.122 The droplet reactors in microchannels were initially reported in about 10 years.123 Using a mixture of three fluorinated oils as the carrier fluid, droplets were continuously generated in a microchannel, as shown in Fig. 10(a). Rapid single-turnover kinetics of ribonuclease A (RNase A) was measured in this microchannel device with better than millisecond time resolution owing to the ideal plug flow condition. Although the reactants were stored in droplets, consecutive reactions could still be implemented just by applying droplet coalescence, as the example shown in Fig. 10(b).124 A two-stage amplification reaction (Scheme 10) to detect nanomolar concentrations of Co2+, an environmental pollutant, was implemented in a chip reactor.125 Obvious color change was recorded in the experiment, showing the advantages of this droplet-based reactor in contrast to a single-phase reaction in the same device. More interestingly, as shown in Fig. 10(c), if the fluid feeding equipment could be programmed, the reaction in droplet could also be programmed. Therefore, different experimental conditions could be tested in one study, as the microflow device was equipped with an inline detector.126 Different droplets in the reaction could be labeled by indexing droplets that contain different colors, which are located beside the reacting droplets, as shown in Fig. 10(c). Owing to the versatility of droplet reactors, this method is very useful for analysis of reactions that consume low volumes of chemicals.127,128
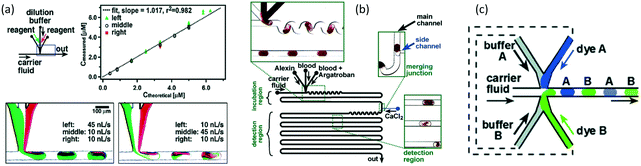 |
| | Fig. 10 Application of droplet reactors. (a) Inline solution dilution and mixing assisted by internal circulation of droplets (adapted from ref. 123 with permission of the American Chemical Society). (b) Schematic of a droplet-based microfluidic device with droplet coalescence structures for feeding the reactant (adapted from ref. 124 with permission of the American Chemical Society). (c) Schematic of an indexed droplet reactor with various concentrations (adapted from ref. 126 with permission of John Wiley and Sons). | |
 |
| | Scheme 10 Reaction for Co2+ concentration amplification.125 | |
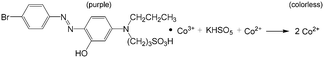
Inspired by the concept of droplet reactor from the area of analytical chemistry, recent reports have also shown that droplet reactors are beneficial for the preparation of colloidal nanoparticles.129 Besides excellent performance with respect to heat exchange, mixing, and residence time control, droplet reactors can well isolate particles from the microchannel, thus avoiding particle adsorption and deposition in devices with large specific areas.130 Using multistage microflow devices, as in the example shown in Fig. 11(a),131 Pd (Fig. 11(b)), Au, metallic and Pd–M (M = Au, Pd, or Ag) bimetallic nanocrystals and Ag nanoprisms have been synthesized with high quality.131,132 The characteristic of all these processes is that the chemicals or the seeds of metal nanocrystals as well as the clusters were mixed in the first T-connector and then the mixed solutions were segmented by oil in the following reaction tube. This continuous synthesis method is believed to be an effective approach for scaling up the production of nanocrystals with stable nucleation and growth conditions. In addition to the separated mixing and droplet generation method in Fig. 11(a), another approach is to generate droplets containing some of the reactants first and then add other reactants after the droplets are fed into the second connecter. This method is suitable for systems that need to initially protect some reactant by the inert phase. Using this method, Au/Ag/Au double-shell nanoparticles with narrow size distribution have been obtained by the reduction of AgNO3 and HAuCl4 at Au seed surfaces.133 The mixing and droplet generation processes can also be integrated, as shown in Fig. 11(c).134 Syntheses of Au, Ag, and Pt nanocrystals have been tried in this kind of microflow device, realizing particles with uniform sizes and morphologies as a local constrictive tube was used to enhance mixing.134,135 During the preparation of nanoparticles, the particle concentration is usually very low (on mg L−1 level); therefore, the engineering characteristics of the microflow process, such as RTD and mass/heat transfer rate, almost do not change after the particles are generated. Droplet morphology is also similar to that of working systems for chemical synthesis; however, droplet surface may vary for particle adsorption, which should be carefully considered. On one hand, interfacial adsorption can be used to generate asymmetric nanocrystals, and on the other hand, it is better to mitigate this effect to obtain nanocrystals of uniform size.136 Some surfactants, like Triton X-100, have proven to be effective for alleviating interfacial adsorption of Ag and Au–Ag nanoparticles without affecting the capping agent used for shape-control, as shown in Fig. 11(d).136
 |
| | Fig. 11 Nanoparticle synthesis in microflow and effect of mixing and interfacial adsorption. (a) Schematic of a microflow device to generate droplets for reactions to prepare metal nanocrystals. (b) Pt nanocrystals prepared using the device in (a) (adapted from ref. 131 with permission of the American Chemical Society). (c) Microflow devices with dyes in droplets and the effect of mixing on particles (adapted from ref. 134 with permission of John Wiley and Sons). (d) Effect of Triton X-100 on alleviating interfacial adsorption in Ag and Au–Ag particle synthesis (adapted from ref. 136 with permission of the American Chemical Society). | |
Apart from metal nanocrystals, other nanoparticles, such as semiconductors and metal oxides, can also be synthesized by using these slug flow reactors. For example, controlled and reproducible synthesis of monodispersed lead chalcogenide (PbS, PbSe) nanocrystal quantum dots (QDs) was achieved in a microflow reactor system with real-time monitoring of the reaction using NIR fluorescence spectroscopy.137 The liquid–liquid slug flow realized stable synthesis of PbS with a high photoluminescence quantum yield of 28%. In another example, a slug flow microreactor was proposed to prepare inorganic nanomaterials, including porous ZSM-5, γ-AlOOH, and β-FeOOH nanorods, which are difficult to produce.138 The reaction time was reduced from days to tens of minutes by combining the advantages of both ionic liquids and microflow process.
Similar to inorganic particle syntheses, polymerization reactions involving highly vicious fluids or polymer particles are also challenging for microflow reactors. Using droplets to proceed reactions, the sticky wall problem can be solved and the inert carrier fluid is able to remove reaction heat. As a model system, polymerization of styrene and methyl methacrylate were tested, showing narrow molecular weight distributions.19 Poly(butyl acrylate) synthesized in a tubular reactor was also proved to provide higher molecular weights and much lower polydispersity indexes (PDI) than a traditional batch reactor.85 Simulation results demonstrated that the polymerization conducted in droplets proceeded in nearly isothermal conditions. Droplet reactors can also be used to produce complicated polymer pariticles.139 In the synthesis of polymethyl methacrylate (PMMA), the presence of polyvinylpyrrolidone (PVP) in the carrier fluid affected the assembly of initial particles.140 Flower structures were found at lower PVP concentrations, as the surfaces of the particles were less covered. When hybrid surfactants (PVP–SDS or PVP–CTAB) were used, the shapes of PMMA particles were determined by competition of surfactant adsorption. If the surfactants were exchanged with polyelectrolyte molecules with high surface activities, more interesting rod, ellipsoidal, dumbbell, and necklace particles could be prepared.141
Templates for microparticles
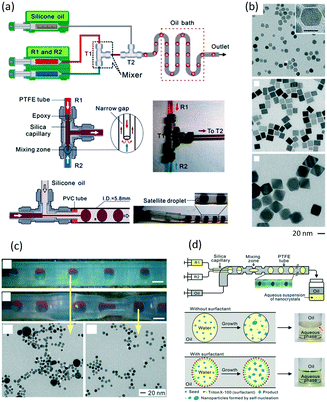
In addition to nanoparticles, microparticles are also products of liquid–liquid microflow reactions, especially polymer and ceramic microparticles.142 In a non-wetting microchannel or microtube for dispersed droplets, droplet morphologies will be fixed after solidification by using UV or temperature induced polymerization, as shown in Fig. 12(a).143 These particles could be disks, rods, or ellipsoids, and adjustment between these shapes can be realized in one device. Although particle output in a microfluidic device is not as common as seeding polymerization in batch reactors, uniform size and well controlled shapes are outstanding advantages of microflow method. Such particles can be used to standardize particles in granularity analysis.144Fig. 12(b) shows uniform fluorescent particles that are continuously synthesized in a glass capillary.145 These particles are a good supporting material for reaction and separation applications. For example, large amounts of specific binding sites can be created in the particles by adding imprinting molecules in the preparation process. After UV solidification, the obtained PMMA particles can be used for adsorbing radioligands.146
 |
| | Fig. 12 Principle of microparticle preparation in microflow device and uniform polymer and ceramic particles (a) schematic of particle shape molding by microchannel structure and solidification process (adapted from ref. 143 with permission of John Wiley and Sons). (b) Microscopic image and laser confocal fluorescence image of poly-1,6-hexanediol diacrylate particles with rhodamine B (adapted from ref. 144 with permission of Elsevier). (c) SEM images of silica particles and pores on the surface and within the body of particles (adapted from ref. 147 with permission of the American Chemical Society). | |
Apart from polymer materials, some ceramic particles containing mesopores can also be prepared using microflow devices. Using silica sol containing methylcellulose (MC) and polyethylene glycol (PEG) as the dispersed phases and paraffin as the carrier fluid, uniform silica spheres (Fig. 12(c)) with large pore volume have been prepared after removing template agents, and the pore size in particles can be adjusted by varying the amount of MC and PEG added.147 The prepared silica particles, which have large pore volumes and an average mesoporous diameter of >12 nm, can be used to adsorb bovine serum albumin (BSA) at a high speed. When using these BSA-loaded microparticles for chiral separation of tryptophan, an enantiomer excess (e.e.) value as high as 100% is achieved in the first 10 min in a packed column, and the particles can be regenerated by a buffer at pH = 5.0.148
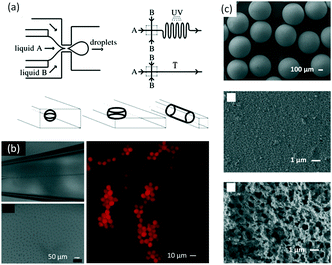
Based on multilayer droplets, complicated microparticles with various hollow structures can be prepared. As shown in Fig. 13(a), a microfluidic method was developed to prepare alginate microcapsules with oil cores, which had the potential to become a new type of vehicle for encapsulating, storing, and transferring drugs and active chemicals.149 The alginate droplets were generated from a capillary microfluidic device with three dispersion stages, and alginate was then solidified by complexation with Ca2+. The microflow device was easily assembled from commercial glass or crystal capillaries. Both the alginate microcapsule diameter and shell thickness of alginate were controlled by modulating dimensions of the microfluidic device and the flow rates of solutions. Using a similar method, different small droplets could be embedded into bigger droplets to prepare multiple-core particles,150 like multicolor QD-encoded core–shell particles shown in Fig. 13(b).151 Owing to the protection of a hydrogel shell, leakage of QDs was avoided and fluorescent stability was enhanced. By embedding different QDs into different cores, interaction between different color QDs was avoided and the fluorescence spectrum of each kind of QD was recorded separately. If the outside droplet could be solidified using a stimulation-response material, the particle could be further modulated by changing the local environment.152 For example, poly(N-isopropylacrylamide) (PNIPAM) micro gels could be prepared by using microfluidic technology with a dissolving template.153 The formed porous micro gels responded faster to changes in temperature than their void-less counterparts.
 |
| | Fig. 13 Complicated microparticles prepared by microflow reaction. (a) Schematic of a capillary microfluidic device, and a picture of oil-core-containing alginate microcapsules (adapted from ref. 149 with permission of Elsevier). (b) QD-encoded core–shell particles (adapted from ref. 151 with permission of the American Chemical Society). (c) Schematic of a microflow device for production of double-emulsion droplets with an ultrathin shell and in situ polymerization of the shell, as well as an optical microscopic image showing the generation of monodisperse double-emulsion droplets (adapted from ref. 156, an open access article from the American Chemical Society). (d) Schematic of a sheath microflow for producing monodisperse colloid-filled hydrogel granules with Janus structures and images of the prepared particles (adapted from ref. 157 with permission of the American Chemical Society). | |
When reactions in microflows occur close to the interface, interfacial polymerization or supramolecular self-assembly can be further introduced to form microcapsules with very thin shells using microflow method. A typical example is the host–guest system of polymer–Au nanoparticle composites held by cucurbit[8]uril ternary complexes.154 The advantage of this method is that highly stable microstructures on the droplet surface can be loaded in one step during capsule formation and that microcapsules are amenable to on-demand encapsulant release. In another example, polyurea microcapsules are prepared in a microflow device through an interfacial reaction between an oil-soluble isocyanate and a water-soluble amine.155 Different morphologies of micro-capsule are formed and the thickness of the shell can be adjusted using amine solubility in the oil. In addition, the ultrathin middle layers of double-emulsion droplets are also effective templates for preparing microcapsule structures. The microfluidic strategy in Fig. 13(c) allows for the successful preparation of semipermeable microcapsules with an ultra-thin shell using UV polymerization and subsequent phase separation between the resin and oil.156
Another application of microflow technologies in making microparticles is to generate asymmetrical structures. As the example shown in Fig. 13(d), the microparticles have an asymmetrical distribution of components, as immiscible or very slowly miscible fluids are used in the microchannel.157 Red and green fluorescent dyes are used to show different parts of the Janus particles in Fig. 13(d). These dyes did not mix because of low diffusion rate and weak circulation in tiny droplets. Using this method, cells and magnetic beads can be separately embedded into a hydrogel microparticle, and the negative effect of contact with magnetic substance on cells can be reduced. Alginate cell capsules prepared from this method can be manipulated by external magnetic fields and do not require specific surface modification.158 After Janus microparticles are generated, one part of the particles can be further removed by dissolution or decomposition, and special particles like meniscus are prepared.159,160 Unlike chemical synthesis and nanoparticle preparation reactions, the rheological behavior of droplet changes greatly during solidification, and it may affect the final shape of particle, internal circulation and mass/heat transfer performance. Nevertheless, the solids do not further take part in the reactions, and the continuous phases are usually inert. Therefore, flow patterns of final liquid–solid flow will not be so different from the original liquid–liquid flow.
Scale-up of microflow reactors and commercial applications
As an important technology for chemical synthesis, the scale-up of microflow reactors is attracting increasing attention from industry. Researchers have promoted numbering-up of the core structures in microreactors, such as heat exchange channels, mixing units, and catalyst beds,161 because the use of repeated microstructures can maintain all characteristics of microflow reactors on lab scale, which usually have one microchannel or one capillary tube. This scaling up is necessary for processes very sensitive to flow path size, such as preparation of uniform microparticles.162 To ensure that droplets from every microchannel have the same diameter, all flow rates should be consistent in every unit of droplet generation. Therefore, the key point is the design of fluid distributors. These distribution structures usually consist of several big chambers with nearly stationary fluid inside163,164 or bifurcated microchannels with symmetrical branches.165,166 Regardless of the kind of fluid distributors, all inlets of the droplet generation units should have the same pressure. To realize high productivity of microparticles, hundreds of microchannels are required to integrate in one chip. For simplification of the connecting pumps and particle collectors, round chips are more preferred by researchers.167Fig. 14(a) shows typical chips with an annular distribution chamber, an annular distribution network with a tree-like structure, and a fractal tree-like distribution network.168 Droplets with average diameter of 100 to 240 μm and size distribution <6% have been prepared in chips with production rates as high as 1 L h−1.
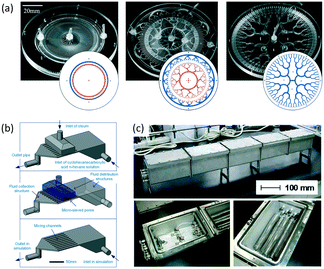 |
| | Fig. 14 Scale-up of microflow reactors. (a) Distribution networks for preparing uniform droplets with a volume of liters per hour. In the insets, blue represents feeding lines of the aqueous phase and orange denotes path of the oil phase (adapted from ref. 168 with permission of the Royal Society of Chemistry). (b) Distribution chambers and micro sieve dispersion structures for reactions with a treating capacity of 10 L min−1 cyclohexanecarboxylic acid solution (adapted from ref. 171 with permission of John Wiley and Sons). (c) Pictures of tubular reaction devices with heat exchange baths for polymerization of methyl methacrylate (adapted from ref. 175 with permission of the American Chemical Society). | |
For the application of microreactors in chemical synthesis, good mixing and mass/heat transfer performance are more preferable than narrow droplet size distribution. Therefore, enlarging the flow path is acceptable if the mixing and mass/heat transport performance are maintained.169 For example, mass transfer performance of Corning AFR is similar to that of a 500 μm-height microchannel, which has a 50-fold decrease in treating capacity.64 A micro sieve dispersion structure is a scale-up of the T-junction microchannel for mixing liquids by creating a large amount of tiny droplets in the continuous phase.170 In this device, tens of micro sieve pores can generate droplets together, and thus, the capacity of a millimeter-width mixing channel is greatly increased for preparing bubbly flows. Further numbering-up of the mixing channels with fluid distribution chambers can provide more than 100 times higher productivity, as shown in Fig. 14(b).171 Then, a reaction tube flowing through these mixing channels will allow completion of the reaction. During scale-up of the mixing channels, the mixing performance cannot be reduced. Therefore, if droplets are easy to coalesce in a large flow path, additional obstacles or constrictive structures can be added to break the coalesced droplets.172,173 Another limitation on the enlargement of the microflow unit is heat exchange performance.174 If a reaction can be implemented under adiabatic conditions, the combination of a scaled micromixer and a heat isolation tube can be more easily applied in industrial processes.171 However, many reactions have to be operated at certain temperatures. Therefore, the numbering-up of small reaction tubes should be considered to maintain heat transfer areas provided by the tube wall, as shown in Fig. 14(c).175
As an important technology used to improve chemical processes in applications, there are also many commercialized microflow devices. For example, the industrial grade Corning reactor reaches to a capacity of 3500 t per year for one plate and the same plate can be further integrated in a shelf to increase productivity.176 Ehrfeld (Bayer) has pushed-off an industrial grade shell and tubular reactor, which has a capacity of 10 kL h−1.177 This modularized reactor can be combined to fit for different reactions. The wide application of commercialized microflow reactor will promote the in-depth development of microflow technology. Many transnational chemical and pharmaceutical companies, such as LG, Sinopec, BASF and Novartis, have engaged in research and development of microflow reaction technology. The F3 (Flexible, Fast, and Future) Factory project is a typical technical validation in Europe.178 But there are still few reports about the detailed technology progress for commercial chemical processes. Because of the growing market and environmental pressure, the microflow reaction technology develops fast in developing Asian countries.9 As far as we know, many fine chemical companies have built pilot-plant platforms to test performances of microflow reactors in synthesizing fine chemical products, such as fire retardants, vulcanizing agents, pesticides, resins, and sulfonate surfactants in China. This highly efficient chemical engineering technology will lead chemical industry to a new era in the near future.
Conclusions and outlook
We gave a brief introduction to the fundamental principles and applications of microflow technology in this review. As a highly efficient chemical technology, liquid–liquid microflow device provides narrow RTD, which is important for precise reaction control. Liquid–liquid slug microflow can be used in droplet reactors. Mixing and mass transfer of chemicals are much faster in microflow devices than in traditional chemical equipment. Fast heat exchange between phases or contact with the tube wall provides a more controllable reaction temperature. In confined microspace, dispersed phases exhibit a number of morphologies that can be used as templates for preparing particle materials. Based on these advantages, liquid–liquid microflow devices provide ideal environments for chemical reactions, as well as improved yields and efficiency, for many processes, such as clean oxidation, nitrification, etherification, esterification, C–C and C–N coupling, and Beckmann rearrangement reactions. Additionally, liquid–liquid extraction can be applied to purify intermediate products and to separate final products, which are beneficial for implementing multistep reactions in continuous microreactors. Manually changing a homogeneous reaction into a heterogeneous reaction by adding an inert component as the carrier fluid sometimes allows improvement in control of reaction. Moreover, liquid–liquid microflow systems are good platforms for preparing metal, semiconductor, ceramic, and polymer particles with sizes ranging from nano to micrometers.
Despite extensive research on liquid–liquid microflow reaction processes, further studies are still anticipated for developing an in-depth understanding of this area. Some directions have shown good prospects. For example, in the fundamental study of microflows, some dynamic characteristics, such as dynamic interfacial tension induced by chemical reactions, are crucial for exploring unknown aspects of surface chemistry,179 which is believed to be an important meso-scientific issue between chemical processes on the molecular scale and multiphase fluids in macroscopic reactors.180 For transport studies, more widely applicable mass and heat transfer equations and highly accurate computational fluid dynamics (CFD) models are required for various applications of microflow process.28 In addition, new working systems, such as reactions with nanoemulsions,181 will increase the vitality of microflow reaction technology. To achieve a more “flexible” and “intelligent” platform for chemical synthesis, automated microflow systems have been used to screen and optimize reaction path.182 Moreover, the preparation of functional nano- or microparticles for medical diagnosis and treatment and scaled production are significant.183 As a brief introduction to liquid–liquid microflow reaction, this review is mainly presented from lab research. However, as an engineering technology, the microflow device and system has strong potential ability to be used in industrial processes. The spread and application of industrial microflow reaction devices are undoubtedly the priority subject in this area.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the support from the National Natural Science Foundation of China (91334201, U1302271, U1463208), the Foundation for the Author of National Excellent Doctoral Dissertation of the People's Republic of China (FANEDD 201349), and the Tsinghua University Initiative Scientific Research Programs (20131089196, 20151080361).
References
- K. Jahnisch, V. Hessel, H. Lowe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406–446 CrossRef PubMed.
- K. F. Jensen, Chem. Eng. Sci., 2001, 56, 293–303 CrossRef CAS.
- H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noel, Chem. Soc. Rev., 2016, 45, 83–117 RSC.
- O. Worz, K. P. Jackel, T. Richter and A. Wolf, Chem. Eng. Technol., 2001, 24, 138–142 CrossRef CAS.
- T. M. Squires and S. R. Quake, Rev. Mod. Phys., 2005, 77, 977–1026 CrossRef CAS.
- T. Illg, P. Lob and V. Hessel, Bioorg. Med. Chem., 2010, 18, 3707–3719 CrossRef CAS PubMed.
- J. I. Yoshida, A. Nagaki and T. Yamada, Chem. – Eur. J., 2008, 14, 7450–7459 CrossRef CAS PubMed.
- H. Hisamoto, T. Horiuchi, M. Tokeshi, A. Hibara and T. Kitamori, Anal. Chem., 2001, 73, 1382–1386 CrossRef CAS.
- K. F. Jensen, AIChE J., 2017, 63, 858–869 CrossRef CAS.
- V. Hessel, D. Kralisch, N. Kockmann, T. Noel and Q. Wang, ChemSusChem, 2013, 6, 746–789 CrossRef CAS PubMed.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS.
- S. D. Schaber, S. C. Born, K. F. Jensen and P. I. Barton, Org. Process Res. Dev., 2014, 18, 1461–1467 CrossRef CAS.
- J. P. McMullen and K. F. Jensen, Org. Process Res. Dev., 2011, 15, 398–407 CrossRef CAS.
- T. Tsubogo, H. Oyamada and S. Kobayashi, Nature, 2015, 520, 329–332 CrossRef CAS PubMed.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67 CrossRef CAS PubMed.
- M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef CAS PubMed.
- M. J. Shang, T. Noel, Q. Wang and V. Hessel, Chem. Eng. Technol., 2013, 36, 1001–1009 CrossRef CAS.
- L. Xie, Y. Shen, D. Franke, V. Sebastian, M. G. Bawendi and K. F. Jensen, J. Am. Chem. Soc., 2016, 138, 13469–13472 CrossRef CAS PubMed.
- Y. Okubo, T. Maki, F. Nakanishi, T. Hayashi and K. Mae, Chem. Eng. Sci., 2010, 65, 386–391 CrossRef CAS.
- V. Hessel, H. Lowe and F. Schonfeld, Chem. Eng. Sci., 2005, 60, 2479–2501 CrossRef CAS.
- J. Yoshida, A. Nagaki, T. Iwasaki and S. Suga, Chem. Eng. Technol., 2005, 28, 259–266 CrossRef CAS.
- T. Shimanouchi, Y. Kataoka, T. Tanifuji, Y. Kimura, S. Fujioka and K. Terasaka, AIChE J., 2016, 62, 2135–2143 CrossRef CAS.
- A. Ufer, M. Mendorf, A. Ghaini and D. W. Agar, Chem. Eng. Technol., 2011, 34, 353–360 CrossRef CAS.
- F. Bally, C. A. Serra, V. Hessel and G. Hadziioannou, Chem. Eng. Sci., 2011, 66, 1449–1462 CrossRef CAS.
- T. Wirth, Angew. Chem., Int. Ed., 2017, 56, 682–684 CrossRef CAS PubMed.
- P. Watts and S. J. Haswell, Chem. Eng. Technol., 2005, 28, 290–301 CrossRef CAS.
- J. Jovanovic, E. V. Rebrov, T. A. Nijhuis, V. Hessel and J. C. Schouten, Ind. Eng. Chem. Res., 2010, 49, 2681–2687 CrossRef CAS.
- K. Wang and G. S. Luo, Chem. Eng. Sci., 2017, 169, 18–33 CrossRef CAS.
- G. S. Luo, L. Du, Y. J. Wang, Y. C. Lu and J. H. Xu, Particuology, 2011, 9, 545–558 CrossRef CAS.
- A. Ufer, D. Sudhoff, A. Mescher and D. W. Agar, Chem. Eng. J., 2011, 167, 468–474 CrossRef CAS.
- A. Liedtke, F. Bornette, R. Philippe and C. de Bellefon, Chem. Eng. J., 2013, 227, 174–181 CrossRef CAS.
- K. Olivon and F. Sarrazin, Chem. Eng. J., 2013, 227, 97–102 CrossRef CAS.
- S. Y. Teh, R. Lin, L. H. Hung and A. P. Lee, Lab Chip, 2008, 8, 198–220 RSC.
- C. X. Zhao and A. Middelberg, Chem. Eng. Sci., 2011, 66, 1394–1411 CrossRef CAS.
- S. Hardt and T. Hahn, Lab Chip, 2012, 12, 434–442 RSC.
- Y. Chen, W. Gao, C. Zhang and Y. Zhao, Lab Chip, 2016, 16, 1332–1339 RSC.
- Q. Zhou, Y. Sun, S. Yi, K. Wang and G. S. Luo, Soft Matter, 2016, 12, 1674–1682 RSC.
- S. L. Anna, Annu. Rev. Fluid Mech., 2016, 48, 285–309 CrossRef.
- P. Zhu and L. Wang, Lab Chip, 2016, 17, 34–75 RSC.
- J. K. Nunes, S. Tsai, J. Wan and H. A. Stone, J. Phys. D: Appl. Phys., 2013, 46 Search PubMed.
- C. N. Baroud, F. Gallaire and R. Dangla, Lab Chip, 2010, 10, 2032–2045 RSC.
-
O. Levenspiel, Chemical Reaction Engineering, John Wiley & Sons, New York, 3rd edn, 1999 Search PubMed.
- D. Parida, C. A. Serra, D. K. Garg, Y. Hoarau, R. Muller and M. Bouquey, Macromol. React. Eng., 2014, 8, 597–603 CrossRef CAS.
- Z. Dong, S. Zhao, Y. Zhang, C. Yao, Q. Yuan and G. Chen, AIChE J., 2017, 63, 1404–1418 CrossRef CAS.
- A. Gunther and K. F. Jensen, Lab Chip, 2006, 6, 1487–1503 RSC.
- C. Yao, Z. Dong, Y. Zhang, Y. Mi, Y. Zhao and G. Chen, AIChE J., 2015, 61, 3964–3972 CrossRef CAS.
- L. Yang, M. J. Nieves-Remacha and K. F. Jensen, Chem. Eng. Sci., 2017, 169, 106–116 CrossRef CAS.
- V. Sebastian Cabeza, S. Kuhn, A. A. Kulkarni and K. F. Jensen, Langmuir, 2012, 28, 7007–7013 CrossRef CAS PubMed.
- S. Kuhn, R. L. Hartman, M. Sultana, K. D. Nagy, S. Marre and K. F. Jensen, Langmuir, 2011, 27, 6519–6527 CrossRef CAS PubMed.
- S. Oskooei and D. Sinton, Lab Chip, 2010, 10, 1732–1734 RSC.
- J. Bico and D. Quere, EPL, 2000, 51, 546–550 CrossRef CAS.
- F. Sarrazin, T. Bonometti, L. Prat, C. Gourdon and J. Magnaudet, Microfluid. Nanofluid., 2008, 5, 131–137 CrossRef.
- X. Y. Lin, K. Wang, J. S. Zhang and G. S. Luo, Chem. Eng. Sci., 2015, 127, 60–71 CrossRef CAS.
- J. D. Tice, H. Song, A. D. Lyon and R. F. Ismagilov, Langmuir, 2003, 19, 9127–9133 CrossRef CAS.
- C. Simonnet and A. Groisman, Phys. Rev. Lett., 2005, 94, 134501 CrossRef PubMed.
- L. Zhang, M. Geng, P. Teng, D. Zhao, X. Lu and J. X. Li, Ultrason. Sonochem., 2012, 19, 250–256 CrossRef CAS PubMed.
- W. Tanthapanichakoon, N. Aoki, K. Matsuyama and K. Mae, Chem. Eng. Sci., 2006, 61, 4220–4232 CrossRef CAS.
- K. Matsuyama, W. Tanthapanichakoon, N. Aoki and K. Mae, Chem. Eng. Sci., 2007, 62, 5133–5136 CrossRef CAS.
- K. Y. Tung, C. C. Li and J. T. Yang, Microfluid. Nanofluid., 2009, 7, 545–557 CrossRef CAS.
- F. Sarrazin, L. Prat, N. Di Miceli, G. Cristobal, D. R. Link and D. A. Weitz, Chem. Eng. Sci., 2007, 62, 1042–1048 CrossRef CAS.
- N. Deng, J. Sun, W. Wang, X. Ju, R. Xie and L. Chu, ACS Appl. Mater. Interfaces, 2014, 6, 3817–3821 CAS.
- N. Deng, S. Sun, W. Wang, X. Ju, R. Xie and L. Chu, Lab Chip, 2013, 13, 3653–3657 RSC.
- D. Jaritsch, A. Holbach and N. Kockmann, J. Fluids Eng., 2014, 136, 91211 CrossRef.
- A. Woitalka, S. Kuhn and K. F. Jensen, Chem. Eng. Sci., 2014, 116, 1–8 CrossRef CAS.
- Y. H. Su, Y. C. Zhao, G. W. Chen and Q. A. Yuan, Chem. Eng. Sci., 2010, 65, 3947–3956 CrossRef CAS.
- N. Harries, J. R. Burns, D. A. Barrow and C. Ramshaw, Int. J. Heat Mass Transfer, 2003, 46, 3313–3322 CrossRef CAS.
- A. L. Dessimoz, L. Cavin, A. Renken and L. Kiwi-Minsker, Chem. Eng. Sci., 2008, 63, 4035–4044 CrossRef CAS.
- J. G. Susanti, M. Winkelman, B. Schuur, H. J. Heeres and J. Yue, Ind. Eng. Chem. Res., 2016, 55, 4691–4702 CrossRef CAS.
- Y. Chen, Z. Chen and H. Wang, Lab Chip, 2012, 12, 4569–4575 RSC.
- P. Mary, V. Studer and P. Tabeling, Anal. Chem., 2008, 80, 2680–2687 CrossRef CAS PubMed.
- Y. H. Choi, Y. S. Song and D. H. Kim, J. Chromatogr. A, 2010, 1217, 3723–3728 CrossRef CAS PubMed.
- F. Zhao, Y. C. Lu, K. Wang and G. S. Luo, Solvent Extr. Ion Exch., 2016, 34, 60–73 CrossRef CAS.
- L. Bai, S. Zhao, Y. Fu and Y. Cheng, Chem. Eng.
J., 2016, 298, 281–290 CrossRef CAS.
- O. Jafari, M. Rahimi and F. H. Kakavandi, Chem. Eng. Process.: Process Intesif., 2016, 101, 33–40 CrossRef CAS.
- J. Jovanovic, W. Hengeveld, E. V. Rebrov, T. A. Nijhuis, V. Hessel and J. C. Schouten, Chem. Eng. Technol, 2011, 34, 1691–1699 CrossRef CAS.
- R. Langsch, J. Zalucky, S. Haase and R. Lange, Chem. Eng. Process., 2014, 75, 8–18 CrossRef CAS.
- X. Wang, K. Wang, A. Riaud, X. Wang and G. S. Luo, Chem. Eng. J., 2014, 254, 443–451 CrossRef CAS.
- N. Assmann and P. R. von Rohr, Chem. Eng. Process., 2011, 50, 822–827 CrossRef CAS.
- Y. H. Su, G. W. Chen, Y. C. Zhao and Q. Yuan, AIChE J., 2009, 55, 1948–1958 CrossRef CAS.
- K. Wang, Y. C. Lu, K. Qin, G. S. Luo and T. Wang, Chem. Eng. Technol., 2013, 36, 1047–1060 CrossRef CAS.
- N. Aoki, R. Ando and K. Mae, Ind. Eng. Chem. Res., 2011, 50, 4672–4677 CrossRef CAS.
- E. Lobry, T. Lasuye, C. Gourdon and C. Xuereb, Chem. Eng. J., 2015, 259, 505–518 CrossRef CAS.
- J. J. John, S. Kuhn, L. Braeken and T. Van Gerven, Chem. Eng. Process., 2016, 102, 37–46 CrossRef CAS.
-
F. Incropera, A. Lavine and D. DeWitt, Fundamentals of heat and mass transfer, John Wiley & Sons, Hoboken, 2011 Search PubMed.
- Z. D. Liu, Y. C. Lu, B. D. Yang and G. S. Luo, Ind. Eng. Chem. Res., 2011, 50, 11853–11862 CrossRef CAS.
-
G. Q. Liu, L. X. Ma and J. Liu, Chemistry and chemical property data manual, Chemical Industry Press, Beijing, 2002 Search PubMed.
- K. Wang, Y. C. Lu, H. W. Shao and G. S. Luo, Ind. Eng. Chem. Res., 2008, 47, 9754–9758 CrossRef CAS.
- K. Wang, Y. C. Lu, Y. Xia, H. W. Shao and G. S. Luo, Chem. Eng. J., 2011, 169, 290–298 CrossRef CAS.
- W. Wang, R. Xie, X. J. Ju, T. Luo, L. Liu, D. A. Weitz and L. Y. Chu, Lab Chip, 2011, 11, 1587–1592 RSC.
- L. L. A. Adams, T. E. Kodger, S. Kim, H. C. Shum, T. Franke and D. A. Weitz, Soft Matter, 2012, 8, 10719–10724 RSC.
- P. Clegg, J. W. Tavacoli and P. Wilde, Soft Matter, 2015, 12, 998–1008 RSC.
- Y. Zhao, H. C. Shum, H. Chen, L. L. A. Adams, Z. Gu and D. A. Weitz, J. Am. Chem. Soc., 2011, 133, 8790–8793 CrossRef CAS PubMed.
- N. Deng, W. Wang, X. Ju, R. Xie, D. A. Weitz and L. Chu, Lab Chip, 2013, 13, 4047–4052 RSC.
- C. H. Chen, R. K. Shah, A. R. Abate and D. A. Weitz, Langmuir, 2009, 25, 4320–4323 CrossRef CAS PubMed.
- X. Ge, J. Huang, J. Xu, J. Chen and G. S. Luo, Soft Matter, 2016, 12, 3425–3430 RSC.
- T. Nisisako and T. Hatsuzawa, Microfluid. Nanofluid., 2010, 9, 427–437 CrossRef.
- K. Wang, Y. C. Lu, J. Tan, B. D. Yang and G. S. Luo, Microfluid. Nanofluid., 2010, 8, 813–821 CrossRef CAS.
- H. U. Kim, D. G. Choi, Y. H. Roh, M. S. Shim and K. W. Bong, Small, 2016, 12, 3463–3470 CrossRef CAS PubMed.
- J. Kobayashi, Y. Mori and S. Kobayashi, Chem. - Asian J., 2006, 1, 22–35 CrossRef CAS PubMed.
- I. Q. Penate, G. Lesage, P. Cognet and M. Poux, Chem. Eng. J., 2012, 200, 357–364 CrossRef.
- E. Santacesaria, A. Renken, V. Russo, R. Turco, R. Tesser and M. Di Serio, Ind. Eng. Chem. Res., 2012, 51, 8760–8767 CrossRef CAS.
- M. Peer, N. Weeranoppanant, A. Adamo, Y. Zhang and K. F. Jensen, Org. Process Res. Dev., 2016, 20, 1677–1685 CrossRef CAS.
- M. Shang, T. Noel, Q. Wang and V. Hessel, Chem. Eng. Technol., 2013, 36, 1001–1009 CrossRef CAS.
- J. Ji, Y. Q. Zhao, L. P. Guo, B. H. Liu, C. Ji and P. Y. Yang, Lab Chip, 2012, 12, 1373–1377 RSC.
- T. Noel, S. Kuhn, A. J. Musacchio, K. F. Jensen and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 5943–5946 CrossRef CAS PubMed.
- J. C. Yang, D. Niu, B. P. Karsten, F. Lima and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 2531–2535 CrossRef CAS PubMed.
- C. E. Brocklehurst, H. Lehmann and L. La Vecchia, Org. Process Res. Dev., 2011, 15, 1447–1453 CrossRef CAS.
- Y. Z. Chen, Y. C. Zhao, M. Han, C. B. Ye, M. H. Dang and G. W. Chen, Green Chem., 2013, 15, 91–94 RSC.
- P. Knapkiewicz, K. Skowerski, D. E. Jaskolska, M. Barbasiewicz and T. K. Olszewski, Org. Process Res. Dev., 2012, 16, 1430–1435 CrossRef CAS.
- J. R. Gage, X. Guo, J. Tao and C. Zheng, Org. Process Res. Dev., 2012, 16, 930–933 CrossRef CAS.
- J. S. Zhang, K. Wang, Y. C. Lu and G. S. Luo, AIChE J., 2012, 58, 3156–3160 CrossRef CAS.
- J. S. Zhang, K. Wang, Y. C. Lu and G. S. Luo, AIChE J., 2012, 58, 925–931 CrossRef CAS.
- K. Wang, J. Zhang, C. Zheng, C. Dong, Y. C. Lu and G. S. Luo, AIChE J., 2015, 61, 1959–1967 CrossRef CAS.
- J. Britton and C. L. Raston, Chem. Soc. Rev., 2017, 46, 1250–1271 RSC.
- A. Adamo, P. L. Heider, N. Weeranoppanant and K. F. Jensen, Ind. Eng. Chem. Res., 2013, 52, 10802–10808 CrossRef CAS.
- S. K. Kurt, I. Vural Gürsel, V. Hessel, K. D. P. Nigam and N. Kockmann, Chem. Eng. J., 2016, 284, 764–777 CrossRef CAS.
- H. R. Sahoo, J. G. Kralj and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 5704–5708 CrossRef CAS PubMed.
- S. Borukhova, T. Noël and V. Hessel, ChemSusChem, 2016, 9, 67–74 CrossRef CAS PubMed.
- P. Xie, K. Wang, P. Wang, Y. Xia and G. S. Luo, AIChE J., 2017, 63, 1002–1009 CrossRef CAS.
- R. L. Hartman, J. R. Naber, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2010, 49, 899–903 CrossRef CAS PubMed.
- D. R. Snead and T. F. Jamison, Chem. Sci., 2013, 4, 2822–2827 RSC.
- H. Song, J. D. Tice and R. F. Ismagilov, Angew. Chem., Int. Ed., 2003, 42, 768–772 CrossRef CAS PubMed.
- H. Song and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 14613–14619 CrossRef CAS PubMed.
- H. Song, H. W. Li, M. S. Munson, T. G. Van Ha and R. F. Ismagilov, Anal. Chem., 2006, 78, 4839–4849 CrossRef CAS PubMed.
- C. J. Gerdts, D. E. Sharoyan and R. F. Ismagilov, J. Am. Chem. Soc., 2004, 126, 6327–6331 CrossRef CAS PubMed.
- B. Zheng, J. D. Tice and R. F. Ismagilov, Anal. Chem., 2004, 76, 4977–4982 CrossRef CAS PubMed.
- L. S. Roach, H. Song and R. F. Ismagilov, Anal. Chem., 2005, 77, 785–796 CrossRef CAS PubMed.
- B. Zheng, J. D. Tice, L. S. Roach and R. F. Ismagilov, Angew. Chem., Int. Ed., 2004, 43, 2508–2511 CrossRef CAS PubMed.
- G. Niu, A. Ruditskiy, M. Vara and Y. Xia, Chem. Soc. Rev., 2015, 44, 5806–5820 RSC.
- S. Duraiswamy and S. A. Khan, Small, 2009, 5, 2828–2834 CrossRef CAS PubMed.
- L. Zhang, G. Niu, N. Lu, J. Wang, L. Tong, L. Wang, M. J. Kim and Y. Xia, Nano Lett., 2014, 14, 6626–6631 CrossRef CAS PubMed.
- L. Zhang, Y. Wang, L. Tong and Y. Xia, Langmuir, 2013, 29, 15719–15725 CrossRef CAS PubMed.
- A. Knauer, A. Thete, S. Li, H. Romanus, A. Csáki, W. Fritzsche and J. M. Köhler, Chem. Eng. J., 2011, 166, 1164–1169 CrossRef CAS.
- L. Zhang and Y. Xia, Adv. Mater., 2014, 26, 2600–2606 CrossRef CAS PubMed.
- Y. H. Kim, L. Zhang, T. Yu, M. Jin, D. Qin and Y. Xia, Small, 2013, 9, 3462–3467 CrossRef CAS PubMed.
- L. Zhang, Y. Wang, L. Tong and Y. Xia, Nano Lett., 2014, 14, 4189–4194 CrossRef CAS PubMed.
- I. Lignos, L. Protesescu, S. Stavrakis, L. Piveteau, M. J. Speirs, M. A. Loi, M. V. Kovalenko and A. J. DeMello, Chem. Mater., 2014, 26, 2975–2982 CrossRef CAS.
- P. H. Hoang, H. Park and D. Kim, J. Am. Chem. Soc., 2011, 133, 14765–14770 CrossRef CAS PubMed.
- N. Visaveliya and J. M. Köhler, Small, 2015, 11, 6435–6443 CrossRef CAS PubMed.
- N. Visaveliya and J. M. Koehler, Langmuir, 2014, 30, 12180–12189 CrossRef CAS PubMed.
- N. Visaveliya and J. M. Köhler, ACS Appl. Mater. Interfaces, 2014, 6, 11254–11264 CAS.
- H. C. Shum, A.
R. Abate, D. Lee, A. R. Studart, B. G. Wang, C. H. Chen, J. Thiele, R. K. Shah, A. Krummel and D. A. Weitz, Macromol. Rapid Commun., 2010, 31, 108–118 CAS.
- S. Q. Xu, Z. H. Nie, M. Seo, P. Lewis, E. Kumacheva, H. A. Stone, P. Garstecki, D. B. Weibel, I. Gitlin and G. M. Whitesides, Angew. Chem., Int. Ed., 2005, 44, 724–728 CrossRef CAS PubMed.
- P. Dong, J. Xu, H. Zhao and G. S. Luo, Chem. Eng. J., 2013, 214, 106–111 CrossRef CAS.
- Y. K. Li, K. Wang, J. H. Xu and G. S. Luo, Chem. Eng. J., 2016, 293, 182–188 CrossRef CAS.
- M. Zourob, S. Mohr, A. G. Mayes, A. Macaskill, N. Perez-Moral, P. R. Fielden and N. J. Goddard, Lab Chip, 2006, 6, 296–301 RSC.
- Z. Zhai, Y. J. Wang, Y. C. Lu and G. S. Luo, Ind. Eng. Chem. Res., 2010, 49, 4162–4168 CrossRef CAS.
- Z. Zhai, Y. Chen, Y. J. Wang and G. S. Luo, Chirality, 2009, 21, 760–768 CrossRef CAS PubMed.
- P. Ren, X. Ju, R. Xie and L. Chu, J. Colloid Interface Sci., 2010, 343, 392–395 CrossRef CAS PubMed.
- X. Q. Gong, W. J. Wen and P. Sheng, Langmuir, 2009, 25, 7072–7077 CrossRef CAS PubMed.
- Y. Chen, P. Dong, J. H. Xu and G. S. Luo, Langmuir, 2014, 30, 8538–8542 CrossRef CAS PubMed.
- J. Wei, X. J. Ju, R. Xie, C. L. Mou, X. Lin and L. Y. Chu, J. Colloid Interface Sci., 2011, 357, 101–108 CrossRef CAS PubMed.
- L. Y. Chu, J. W. Kim, R. K. Shah and D. A. Weitz, Adv. Funct. Mater., 2007, 17, 3499–3504 CrossRef CAS.
- J. Zhang, R. J. Coulston, S. T. Jones, J. Geng, O. A. Scherman and C. Abell, Science, 2012, 335, 690–694 CrossRef CAS PubMed.
- I. Polenz, S. S. Datta and D. A. Weitz, Langmuir, 2014, 30, 13405–13410 CrossRef CAS PubMed.
- B. Kim, T. Y. Jeon, Y. Oh and S. Kim, Langmuir, 2015, 31, 6027–6034 CrossRef CAS PubMed.
- R. F. Shepherd, J. C. Conrad, S. K. Rhodes, D. R. Link, M. Marquez, D. A. Weitz and J. A. Lewis, Langmuir, 2006, 22, 8618–8622 CrossRef CAS PubMed.
- L. B. Zhao, L. Pan, K. Zhang, S. S. Guo, W. Liu, Y. Wang, Y. Chen, X. Z. Zhao and H. L. W. Chan, Lab Chip, 2009, 9, 2981–2986 RSC.
- W. J. Lan, S. W. Li, J. H. Xu and G. S. Luo, Lab Chip, 2011, 11, 652–657 RSC.
- Z. H. Nie, S. Q. Xu, M. Seo, P. C. Lewis and E. Kumacheva, J. Am. Chem. Soc., 2005, 127, 8058–8063 CrossRef CAS PubMed.
- R. Schenk, V. Hessel, C. Hofmann, J. Kiss, H. Lowe and A. Ziogas, Chem. Eng. J., 2004, 101, 421–429 CrossRef CAS.
- W. Li, E. Young, M. Seo, Z. Nie, P. Garstecki, C. A. Simmons and E. Kumacheva, Soft Matter, 2008, 4, 258–262 RSC.
- T. Femmer, A. Jans, R. Eswein, N. Anwar, M. Moeller, M. Wessling and A. J. C. Kuehne, ACS Appl. Mater. Interfaces, 2015, 7, 12635–12638 CAS.
- M. Al-Rawashdeh, F. Yue, N. G. Patil, T. A. Nijhuis, V. Hessel, J. C. Schouten and E. V. Rebrov, AIChE J., 2014, 60, 1941–1952 CrossRef CAS.
- Y. Su, G. Chen and E. Y. Kenig, Lab Chip, 2015, 15, 179–187 RSC.
- C. T. Riche, E. J. Roberts, M. Gupta, R. L. Brutchey and N. Malmstadt, Nat. Commun., 2016, 7, 10780 CrossRef CAS PubMed.
- T. Nisisako and T. Torii, Lab Chip, 2008, 8, 287–293 RSC.
- D. Conchouso, D. Castro, S. A. Khan and I. G. Foulds, Lab Chip, 2014, 14, 3011–3020 RSC.
- D. Kirschneck and G. Tekautz, Chem. Eng. Technol., 2007, 30, 305–308 CrossRef CAS.
- K. Wang, Y. C. Lu, J. H. Xu and G. S. Luo, Microfluid. Nanofluid., 2011, 10, 1087–1095 CrossRef CAS.
- K. Wang, Y. C. Lu and G. S. Luo, Chem. Eng. Technol., 2014, 37, 2116–2122 CrossRef CAS.
- R. Gerardy, M. Winter, A. Vizza and J. M. Monbaliu, React. Chem. Eng., 2017, 2, 149–158 CAS.
- A. Nazir, R. M. Boom and K. Schroen, Chem. Eng. Sci., 2013, 92, 190–197 CrossRef CAS.
- N. Kockmann and D. M. Roberge, Chem. Eng. Process., 2011, 50, 1017–1026 CrossRef CAS.
- T. Iwasaki, N. Kawano and J. Yoshida, Org. Process Res. Dev., 2006, 10, 1126–1131 CrossRef CAS.
-
X. Wu, Chemical Industry 4.0: Microchannel Reaction Technology in Micro Chemical Reaction Technology and Intelligent Manufacturing Industry Conference, Qingdao, June, 2017 Search PubMed.
-
Q. Liu, Application of Microreaction Technology in Fine Chemical Production in Micro Chemical Reaction Technology and Intelligent Manufacturing Industry Conference, Qingdao, June, 2017 Search PubMed.
-
F3 factory final report to European commission research and innovation, http://www.f3factory.com/scripts/pages/en/newsevents/F3_Factory_final_report_to_EC.pdf (accessed August 11, 2017) Search PubMed.
- B. Riechers, F. Maes, E. Akoury, B. Semin, P. Gruner and J. Baret, React. Chem. Eng., 2016, 113, 11465–11470 CAS.
-
K. Wang, J. H. Xu, G. T. Liu and G. S. Luo, Advances in Chemical Engineering, ed. G. B. Marin and J. H. Li, Academic Press, Burlington, 2015, ch. 3, pp. 164–191 Search PubMed.
- T. Gu, E. W. Q. Yeap, A. Somasundar, R. Chen, T. A. Hatton and S. A. Khan, Lab Chip, 2016, 16, 2694–2700 RSC.
- M. Abolhasani, N. C. Bruno and K. F. Jensen, Chem. Commun., 2015, 51, 8916–8919 RSC.
- S. A. Peyman, J. R. McLaughlan, R. H. Abou-Saleh, G. Marston, B. R. G. Johnson, S. Freear, P. L. Coletta, A. F. Markham and S. D. Evans, Lab Chip, 2016, 16, 679–687 RSC.
|
| This journal is © The Royal Society of Chemistry 2017 |

 ,
Liantang
Li
,
Pei
Xie
and
Guangsheng
Luo
*
,
Liantang
Li
,
Pei
Xie
and
Guangsheng
Luo
*