
New methods for the preparation of nanoscale nickel phosphide catalysts for heteroatom removal reactions
Mark E.
Bussell

Department of Chemistry, Advanced Materials Science and Engineering Center, Western Washington University, MS-9150, 516 High Street, Bellingham, WA 98225, USA. E-mail: Mark.Bussell@wwu.edu; Tel: +1 360 650 3145
First published on 18th August 2017
Abstract
The removal of heteroatom impurities (S, N, O) from fossil fuel and biomass feedstocks is a critical step in the production of clean-burning transportation fuels. Nickel phosphide (Ni2P) has emerged as a leading candidate material on which to base a new class of catalysts to compete with the long established Ni–Mo and Co–Mo sulfides for hydrotreating of crude oil feedstocks. A recent assessment by researchers at ExxonMobil suggested that a Ni2P catalyst having a 3 nm particle size could compete with a state-of-the-art Co–Mo/Al2O3 catalyst for the hydrodesulfurization (HDS) of a straight-run gas oil feed. This article reviews recent advances in the low-temperature preparation of highly-active, nanoscale Ni2P catalysts.
 Mark E. Bussell | Mark Bussell joined the faculty at Western Washington University in 1990 where he is a Professor of Chemistry and Faculty Associate of the Advanced Materials Science and Engineering Center (AMSEC). Bussell received a B.A. in chemistry from Reed College and a Ph.D. in physical chemistry from the University of California, Berkeley. He was a NSF-NATO postdoctoral fellow at the Université Pierre & Marie Curie in 1988–89, and a postdoctoral research associate at the University of Washington in 1990. Bussell has held sabbatical or leave positions at the ETH-Zürich, Tufts University, the University of British Columbia and Yale-NUS College. |
Introduction
Hydrotreating catalysis
The development of highly-active catalysts for the hydrodesulfurization (HDS) and hydrodenitrogenation (HDN) processes is critical for the production of ultra-low sulfur transportation fuels, and these catalysts may also have utility for the hydrodeoxgenation (HDO) process used in the conversion of bio-oils to renewable biofuels.1,2 The combination of tightened regulations on sulfur content in transportation fuels and the need to process lower quality fossil fuel feedstocks necessitates the development of highly active hydrotreating catalysts for the removal of sulfur from refractory organosulfur compounds such as 4,6-dimethyldibenzothiophene (4,6-DMDBT), whose structure is shown below. The removal of sulfur from organosulfur compounds via HDS is inhibited by organonitrogen compounds in refinery feeds, necessitating that hydrotreating catalysts have high activity for HDS and HDN. The chemical structure of carbazole, often used as a model compound in HDN studies, is shown in Fig. 1b. | ||
| Fig. 1 Chemical structures of (a) 4,6-dimethyldibenzothiophene and (b) carbazole. | ||
Molybdenum sulfide and nickel phosphide hydrotreating catalysts
The current workhorse catalysts for commercial hydrotreating processes are based on molybdenum sulfide (MoS2), a highly anisotropic material in which molybdenum atoms are sandwiched between layers of sulfur atoms. The sulfur layers separating MoS2 sheets are only weakly interacting, which is responsible for the lubricating properties of bulk MoS2. For heterogeneous catalysis applications, such as the heteroatom removal reactions that comprise hydrotreating, MoS2 is dispersed in the form of nanoscale slabs on a metal oxide support such as gamma-alumina (γ-Al2O3), as depicted in Fig. 2a.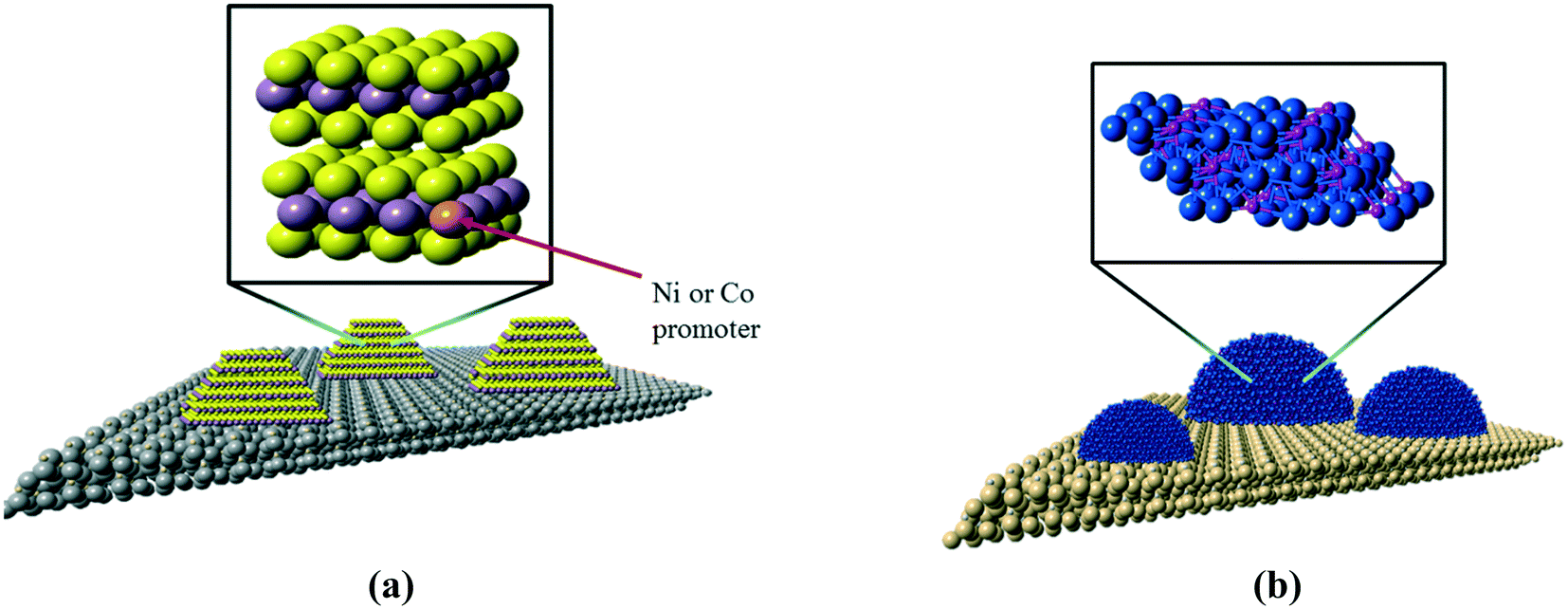 | ||
| Fig. 2 Schematic representations of (a) Ni- or Co–Mo/Al2O3 and (b) Ni2P/SiO2 catalysts. | ||
The active catalytic sites of MoS2 crystallites are located on the edge planes where Mo atoms are exposed; a consequence of the layered structure of MoS2 is a low exposure of active sites. The last twenty years have seen tremendous advances in the development of metal phosphide catalysts, to the extent that nickel phosphide (Ni2P) is now viewed as a candidate material for a new class of industrial hydrotreating catalysts.3,4 In contrast to MoS2, Ni2P is an isotropic solid that exposes metal atoms on all surface planes (see Fig. 2b), resulting in a higher density of active sites. A recent assessment of unsupported Ni2P catalysts suggested that nanoscale Ni2P materials having particles with average diameters of 3 nm would have HDS activities that are competitive with that of a state-of-the-art, cobalt-promoted Mo sulfide catalyst (Co–Mo/Al2O3).5
Recent advances in the preparation of nanoscale nickel phosphide catalysts
Background
The most widely employed method for preparing Ni2P catalysts, either in bulk or oxide-supported form, is temperature-programmed reduction (TPR) in flowing hydrogen of phosphate-based precursors prepared from nickel and phosphate salts (e.g. Ni(NO3)2 + NH4H2PO4).3 However, high temperatures (773–973 K) are needed to reduce phosphate-based precursors to Ni2P owing to the strong P–O bonds in PO43−.6 A consequence of carrying out TPR to these high temperatures is that Ni2P catalysts prepared from phosphate-based precursors have relatively large Ni2P particles (>10 nm) and wide particle size distributions. High reduction temperatures can also lead to reactions between surface P and aluminum in Al-containing supports (e.g. γ-Al2O3, amorphous SiO2–Al2O3 (ASA)) leading to formation of AlPO4, which results in poor catalytic performance.7,8 Finally, high synthesis temperatures also preclude the in situ preparation of metal phosphide catalysts as industrial hydrotreating reactors cannot withstand temperatures in excess of ∼673 K. This mini-review highlights some key advances in the low-temperature preparation of small Ni2P particles (∼3 nm) on metal oxide supports that are in the size range identified as being necessary for HDS and HDN activities competitive with state-of-the-art Co–Mo and Ni–Mo sulfide catalysts.Low temperature methods for preparing Ni2P catalysts
To obviate the need of high temperatures (773–973 K) in the reduction of oxidic precursors to Ni2P catalysts, alternate synthesis methods have been developed that enable reduction at substantially lower temperatures (473–673 K).3 One promising strategy has been to use P sources in the precursors that have lower oxidation states than phosphate, for which the P is in the +5 oxidation state. For example, phosphite (H2PO31−, P oxidation state: +3) and hypophosphite (H2PO21−, P oxidation state: +1), when used in Ni2P catalyst precursors, permit lower temperature reduction due to weaker P–O bonds.8–16 Phosphine (PH3, P oxidation state: −3) and its organophosphorus analogs (e.g. triphenyl phosphine, P(C6H5)3) can also be used for the phosphidation of Ni2+ in catalyst precursors, but the toxicity and high cost of these P sources makes their use in large-scale catalyst synthesis impractical.17–22 Alternatively, low temperature reduction of Ni2P catalyst precursors has been accomplished by employing a hydrogen plasma or microwave heating.23–25 While these recently reported preparation methods have achieved successful synthesis of bulk and supported Ni2P at temperatures considerably lower than those required by conventional TPR of phosphate-based precursors, the resulting Ni2P particle sizes have often been large (>10 nm). As a result, continued effort has focused on the development of synthesis methods that consistently yield small Ni2P particle sizes (<5 nm) and which are amenable for industrial hydrotreating applications.New developments in the synthesis of nanoscale Ni2P catalysts
Specific challenges that must be overcome for Ni2P-based catalysts to successfully compete with state-of-the-art Co–Mo and Ni–Mo sulfide catalysts for heteroatom removal reactions include small particles (<5 nm), versatility of oxide supports (e.g. SiO2, Al2O3, ASA), and a reduction protocol amenable for use in industrial reactors. Recently, we reported a new synthesis method involving the preparation of nickel hypophosphite (Ni(H2PO2)2) precursors and their reduction at 473–673 K to yield well dispersed, small Ni2P particles (3–4 nm) on oxide supports (Fig. 3).8 | ||
| Fig. 3 The synthesis of Ni(H2PO2)2 precursors and their conversion via TPR to oxide-supported Ni2P catalysts. | ||
Different from earlier reported syntheses that use nickel and hypophosphite salts (e.g. NiCl2 and NaH2PO2),12 this synthesis takes advantage of the acid–base chemistry between hypophosphorus acid (H3PO2) and nickel hydroxide (Ni(OH)2) to give nickel hypophosphite and water. Of particular note, the synthesis outlined in Fig. 3 reduces the number of synthesis steps necessary to prepare Ni2P catalysts as the precursors are not calcined and the final step (i.e. reduction of the precursor via TPR) can be carried out directly in the hydrotreating reactor at relatively low temperatures (≤673 K).8
The low temperature synthesis of Ni2P is enabled by the decomposition of the Ni(H2PO2)2 precursor on SiO2 and Al-containing supports (Al2O3, ASA) to give well dispersed, small Ni2P particles. The reactivity of Ni(H2PO2)2 on the oxide supports was probed with temperature-programmed reduction – mass spectrometry (TPR-MS) and, as shown in Fig. 4a, the reduction of a Ni(H2PO2)2/SiO2 precursor exhibited TPR-MS traces for PH3 (m/z = 34) and H2O (m/z = 18) in the effluent gases that indicate reaction of Ni(H2PO2)2 starting at ∼475 K.
 | ||
| Fig. 4 (a) TPR-MS traces for H2O (m/z = 18) and PH3 (m/z = 34) for a Ni(H2PO2)2/SiO2 precursor. (b) TEM image of a 15 wt% Ni2P/SiO2 catalyst prepared from the Ni(H2PO2)2/SiO2 precursor. The figures are taken with permission from ref. 8 (Copyright 2016 Elsevier). | ||
The temperature at which Ni(H2PO2)2 reacted shifted higher for Al2O3 and ASA (507 and 571 K, respectively), indicating stronger interactions between Ni(H2PO2)2 and these supports. Importantly, the reduction of Ni(H2PO2)2 yielded small, uniformly dispersed Ni2P particles on the SiO2, Al2O3 and ASA supports as confirmed by X-ray diffraction (XRD) and transmission electron microscopy (TEM); the TEM image in Fig. 4b reveals Ni2P particles of average diameter 3.2 ± 0.4 nm on the SiO2 support. TEM images of the Ni2P/Al2O3 and Ni2P/ASA catalysts prepared similarly from Ni(H2PO2)2 showed average Ni2P particle sizes of 3.9 ± 0.6 nm and 3.3 ± 0.5 nm, respectively, underscoring the utility of this new synthesis method for preparing small Ni2P particles on a range of oxide supports. Preparation of Ni2P on the same supports from phosphate-based precursors yielded significantly larger particles as indicated by average Ni2P crystallite sizes of 12–19 nm. Despite having substantially smaller particles, the Ni2P/SiO2 and Ni2P/Al2O3 catalysts prepared from hypophosphite-based precursors had similar or lower CO chemisorption capacities than did their counterparts prepared from phosphate-based precursors. However, as discussed later the catalysts derived from the hypophosphite precursors had similar or higher HDN and HDS activities, as well as significantly higher turnover frequencies (TOFs).
Lan et al. recently reported the results of a head-to-head comparison of the properties of Ni2P/SiO2 catalysts prepared from precursors composed of nickel nitrate co-impregnated with either H3PO2, H3PO3 or (NH4)2HPO4 (as the P source) onto the silica support.15 TPR-MS measurements of silica impregnated with H3PO2 revealed hypophosphite to decompose at 453 K as indicated by PH3 evolution, while the decomposition of phosphite was observed at a considerably higher temperature as indicated by PH3 evolution at 673–823 K. Not surprisingly, the production of PH3 from the hydrogenation of phosphate (HPO42−) occurred at an even higher temperature of 873 K. Since PH3 is a strong reducing (and phosphiding) reagent, the lower decomposition temperature of hypophosphite relative to phosphite and phosphate enables Ni2P formation at lower temperatures. TEM images of the Ni2P/SiO2 catalysts prepared using Ni nitrate and H3PO2, and reduced at 623, 723 and 823 K, are shown in Fig. 5 along with the corresponding particle size distributions. Following reduction at 623 K, small Ni2P particles (1.8 ± 0.3 nm) were apparent on the silica support; the particle size increased to 3.4 ± 0.7 nm for the same hypophosphite-based precursor reduced at 823 K. Only broad and very weak XRD peaks were apparent for the Ni2P/SiO2 catalysts reduced at 623–823 K, which is not surprising given the small particle sizes, but the peaks are at the expected positions for Ni2P with no evidence for additional phases being present.
 | ||
| Fig. 5 TEM images and particle size distributions for Ni2P/SiO2 catalysts (6.3 wt% Ni) prepared from a Ni(NO3)2 + H3PO2 precursor reduced at (a) 623 K, (b) 723 K and (c) 823 K. The figures are taken with permission from ref. 15 (Copyright 2017 Elsevier). | ||
Employing a different synthetic strategy, Liu et al. also used hypophosphite as the P source to synthesize Ni2P/Al2O3 catalysts, but relied on vapor transport of PH3 generated by the decomposition of the hypophosphite to carry out phosphidation of a Ni-impregnated Al2O3 precursor.13 The synthesis set-up was comprised of two porcelain containers positioned in a tubular reactor with the upstream container loaded with the hypophosphite compound and the downstream container holding the Ni-impregnated Al2O3 precursor. Nitrogen gas was flowed through the reactor during the synthesis. The authors explored a number of synthetic parameters, including the Ni salt impregnated onto the γ-Al2O3 support, the amount and type of hypophosphite compound loaded upstream and the phosphidation temperature. The optimized synthesis used Ni acetate-impregnated Al2O3, H3PO2 in a molar ratio of P/Ni = 2–3, and a reaction temperature of 573 K. The freshly prepared Ni2P/Al2O3 catalysts were passivated using a 10 mol% H2S/H2 flow prior to removal from the synthesis reactor. As in the study by Liu et al.,13 metal phosphide catalysts are most often prepared in a synthesis reactor and then transferred to a different reactor for catalytic studies. To protect the freshly prepared catalysts from deep oxidation, a dilute oxygen flow (e.g. 1 mol% O2/He) is typically used for passivation of metal phosphide catalysts, resulting in a thin oxide layer at the surface of the phosphide particles. It has been recently shown, however, that passivation with H2S may be more effective (i.e. higher HDS activity after subsequent reduction).26
Hypophosphite as a reagent for in situ generation of PH3
The studies discussed above highlight recent advances in the synthesis of oxide-supported Ni2P catalysts enabled by the use of hypophosphorus acid or hypophosphite salts (e.g. NaH2PO2, NH4H2PO2) as the P source. It is instructive, therefore, to discuss the current understanding of the reactivity of hypophosphite and its relevancy to the synthesis of metal phosphide catalysts. While studied by many researchers over the years, the thermal decomposition of NaH2PO2 has only recently been clarified by Liu et al.27 Upon heating, NaH2PO2 undergoes disproportionation to give reduced (PH3) and oxidized (P3O105−) P species according to the following overall reaction (eqn (1)).| 10NaH2PO2 → 4PH3 + 2Na5P3O10 + 4H2 | (1) |
The TPR-MS traces in Fig. 6a give insight into the decomposition of NaH2PO2 in flowing Ar.8 The evolution of PH3 at 535 K indicates the onset of the disproportionation of NaH2PO2, while a significantly smaller PH3 peak at 579 K and a high temperature shoulder (∼590 K) are assigned to the disproportionation of the intermediate compounds, Na2H2P2O5 and Na2HPO3.27 The results illustrate the usefulness of NaH2PO2 and other hypophosphites as a reagent for the in situ production of PH3, which can then be used directly for the phosphidation of oxide-supported metal precursors. Most importantly, the in situ generation of PH3via the disproportionation of hypophosphite obviates the need to transport, store and directly handle highly toxic PH3 for use as a reagent.
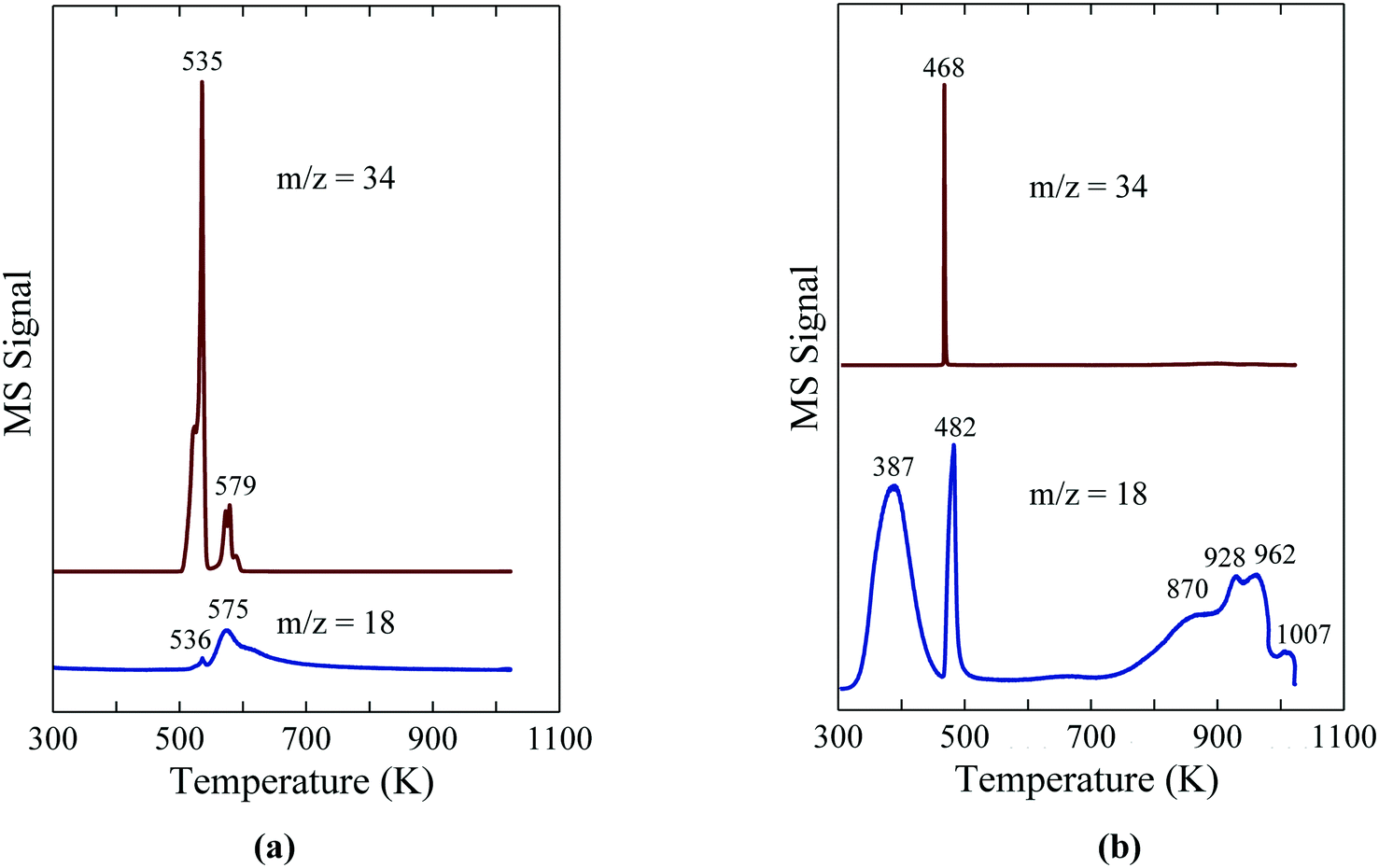 | ||
| Fig. 6 TPR-MS traces for H2O (m/z = 18) and PH3 (m/z = 34) for (a) NaH2PO2 and (b) Ni(H2PO2)2. The figures are taken with permission from ref. 8 (Copyright 2016 Elsevier). | ||
In using hypophosphite as a reagent for the phosphidation of catalyst precursors with PH3, one must be mindful of the additional products that result from the disproportionation reaction and their effect on catalyst properties. As indicated in eqn (1), the disproportionation of NaH2PO2 leaves sodium tripolyphosphate (Na2H2P2O5) as a solid residue, but a range of phosphate products are possible depending on the oxide support used and the hypophosphite salt impregnated. For example, reduction of a support impregnated with NiCl2 and NaH2PO2 is expected to yield NaCl and phosphate species as byproducts on the support surface in addition to the desired Ni2P. Water washing has been employed in some cases to remove solid residues left on Ni2P/SiO2 catalysts prepared using Ni salts and NaH2PO2.12,28 By separating hypophosphite and Ni-impregnated Al2O3 into separate containers, Liu et al. avoided the issue of solid residue deposition on the catalyst surface during reduction, but additional complexity is introduced into the catalyst synthesis procedure.13
By employing Ni(H2PO2)2 as the precursor impregnated onto the support, d'Aquino et al. minimized byproduct residue in the reduced catalyst.8 As probed with TPR-MS, unsupported Ni(H2PO2)2 reacts in flowing H2 to give Ni2P at 468 K as indicated by the evolution of PH3 and H2O in Fig. 6b. XRD analysis of the solid obtained following TPR to 500 K revealed the presence of Ni2P, but no evidence for crystalline phosphate phases (e.g. Ni3(PO4)2, Ni2P2O7 or Ni(PO3)2). Instead, the oxidized product from the disproportionation reaction was identified by IR spectroscopy to be H3PO4, indicating that the overall reaction for the decomposition of Ni(H2PO2)2 can be described with eqn (2).
| 12Ni(H2PO2)2 + 35H2 → 6Ni2P + 14PH3 + 32H2O + 4H3PO4 | (2) |
The evolution of water above 800 K in the TPR-MS trace is assigned to the hydrogenation of the H3PO4 residue. For the oxide-supported Ni2P catalysts, the H3PO4 byproduct from the reaction of Ni(H2PO2)2 likely remains on the catalyst surface to form Brønsted acid sites (P–OH) since the Ni2P catalysts are prepared at temperatures (≤673 K) below which the phosphate species undergo reduction. High surface densities of Brønsted acid sites on unsupported CoxNi2P were shown to correlate with high activity for the HDS of 4,6-dimethyldibenzothiophene.29
Recent advances in hydrotreating over nickel phosphide catalysts prepared using hypophosphite
The development of Ni2P-based materials as a new class of hydrotreating catalysts has been shown to require Ni2P particles having diameters of ∼3 nm if they are to be competitive with state-of-the-art Co–Mo and Ni–Mo sulfide catalysts.5 One synthetic approach for achieving small Ni2P particle sizes takes advantage of the low oxidation state of P and weak P–O bonds in hypophosphite to prepare Ni2P catalysts at relatively low temperatures, as described earlier. d'Aquino et al. prepared Ni2P on SiO2, Al2O3 and ASA supports having particle sizes of 3–4 nm, while Lan et al. prepared Ni2P/SiO2 catalysts having Ni2P particle sizes smaller than 3 nm.8,15 Not surprisingly, the catalysts comprised of small Ni2P particles on oxide supports exhibited high HDS and HDN activities. Fig. 7a compares carbzaole HDN conversions for Ni2P on SiO2, Al2O3 and ASA supports prepared using either H3PO2 or NH4H2PO4 as the P source. The Ni2P catalysts prepared using H3PO2 were reduced via TPR either in situ (“Hypo-in”) or ex situ (“Hypo-ex”) at 673 K, with the ex situ prepared catalysts then re-reduced at 673 K in the reactor to remove the passivation layer. The phosphate-based catalyst was reduced via TPR at 923 K and passivated, and then re-reduced at 673 K in the reactor. | ||
| Fig. 7 Carbazole HDN (a) conversions (WHSV = 49 h−1) and (b) turnover frequencies for Ni2P on SiO2, Al2O3 and ASA. Fig. 7a and b are based on data from ref. 8 (Copyright 2016 Elsevier). | ||
For the Ni2P/Al2O3 and Ni2P/ASA catalysts, the hypophosphite-based precursors reduced in situ had significantly higher carbazole HDN (and benzothiophene HDS) conversions than either the hypophosphite-based precursors reduced ex situ or the catalysts prepared from phosphate-based precursors. For Ni2P/SiO2, the hypophosphite-based catalyst reduced in situ and the phosphate-based catalyst both had high HDN (and HDS) conversions and, along with the in situ prepared Ni2P/Al2O3 catalyst, had significantly higher activities than did a commercially available Ni–Mo/Al2O3 catalyst (Shell 424; 4 wt% NiO, 19.5 wt% MoO3, 8 wt% P2O5). Additional evidence for the superior hydrotreating properties of the Ni2P catalysts prepared in situ comes from the carbazole HDN TOFs that are plotted in Fig. 7b. The Ni2P/SiO2 and Ni2P/Al2O3 catalysts prepared in situ from the hypophosphite-based precursors had higher TOFs than the catalysts prepared ex situ from either the hypophosphite- or phosphate-based precursors. The Ni2P/ASA catalysts were outliers in that the phosphate-based catalyst had the highest carbazole HDN TOF, but this catalyst had an exceptionally low CO chemisorption capacity (19 μmol g−1) compared to the others (52–113 μmol g−1). On a TOF basis, the Ni2P/SiO2 and Ni2P/Al2O3 catalysts prepared in situ surpassed the Ni–Mo/Al2O3 catalyst for HDN and HDS efficacy.
The HDS results of Lan et al. shown in Fig. 8 highlight the advantage of low temperature reduction of hypophosphite- and phosphite-based Ni2P/SiO2 catalysts in comparison to phosphate-based Ni2P/SiO2.15 While the phosphate-based Ni2P/SiO2 catalyst exhibited low HDS activity when the precursor was reduced at 723 K or lower, the hypophosphite- and phosphite-based Ni2P/SiO2 catalysts had high HDS activities when the precursors were reduced at a temperature as low as 623 K. Of the Ni2P/SiO2 catalysts investigated, the catalyst prepared from a hypophosphite-based precursor reduced at 723 K was the most active. Liu et al. compared the dibenzothiophene HDS properties of two Ni2P/Al2O3 catalysts prepared by TPR at 603 K: 1) Ni-impregnated Al2O3 and H3PO2 in separate containers (with phosphidation by vapor transport of PH3), and 2) Al2O3 co-impregnated with Ni(CH3COO)2 and H3PO2.13 The Ni2P/Al2O3 catalysts prepared by vapor transport of PH3 were 20–40% more active for dibenzothiophene HDS than the catalyst prepared from the co-impregnated precursor and, interestingly, water washing of the latter Ni2P/Al2O3 catalyst did not improve its HDS properties.
 | ||
| Fig. 8 Thiophene HDS activities at 643 K and 20 h on-stream for Ni2P/SiO2 catalysts as a function of the precursor reduction temperature. Fig. 8 is based on data from ref. 15 (Copyright 2017 Elsevier). | ||
Summary and future directions
Recent advances in the synthesis of Ni2P catalysts prepared from hypophosphite-based precursors and their promising HDN and HDS properties underscore the potential for a new class of hydrotreating catalysts based on nickel phosphide. In addition to enabling the preparation of small Ni2P particles (<5 nm), hypophosphite-based precursors facilitate preparation of highly active Ni2P catalysts on Al-containing supports (Al2O3 and ASA) as well as in situ reduction in hydrotreating reactors. To develop the potential of Ni2P catalysts further, it will be critical to better understand the role of Ni2P particle size on catalyst activity and selectivity for HDN and HDS reactions. The use of solution-prepared Ni2P nanoparticles of different sizes and narrow dispersity should provide an excellent system for probing these relationships.30 New insights into the nature of the active sites of Ni2P catalysts and the role of Brønsted acidity in determining catalytic properties will also aid in the design and optimization of the preparation of these catalysts from hypophosphite and other precursors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Science Foundation under grant number CHE-1361702. The author would also like to acknowledge the financial support of Yale-NUS College (Singapore).References
- V. C. Srivastava, RSC Adv., 2012, 2, 759–783 RSC.
- T. V. Choudhary and C. B. Phillips, Appl. Catal., A, 2011, 397, 1–12 CrossRef CAS.
- R. Prins and M. E. Bussell, Catal. Lett., 2012, 142, 1413–1436 CrossRef CAS.
- S. T. Oyama, T. Gott, H. Zhao and Y.-K. Lee, Catal. Today, 2009, 143, 94–107 CrossRef CAS.
- S. Soled, S. Miseo, J. Baumgartner, J. Guzman, T. Bolin and R. Meyer, Catal. Today, 2015, 246, 3–8 CrossRef CAS.
- J. A. Rodriguez, J.-Y. Kim, J. C. Hanson, S. J. Sawhill and M. E. Bussell, J. Phys. Chem. B, 2003, 107, 6276–6285 CrossRef CAS.
- S. J. Sawhill, K. A. Layman, D. R. Van Wyk, M. H. Engelhard, C. Wang and M. E. Bussell, J. Catal., 2005, 231, 300–313 CrossRef CAS.
- A. I. d'Aquino, S. J. Danforth, T. R. Clinkingbeard, B. Ilic, L. Pullan, M. A. Reynolds, B. D. Murray and M. E. Bussell, J. Catal., 2016, 335, 2014–2214 Search PubMed.
- J. A. Cecilia, A. Infantes-Molina, E. Rodríguez-Castellón and A. Jiménez-López, J. Catal., 2009, 263, 4–15 CrossRef CAS.
- J. A. Cecilia, A. Infantes-Molina, E. Rodríguez-Castellón and A. Jiménez-López, J. Phys. Chem. C, 2009, 113, 17032–17044 CAS.
- L. Song, S. Zhang and Q. Wei, Catal. Commun., 2011, 12, 1157–1160 CrossRef CAS.
- Q. Guan, W. Li, M. Zhang and K. Tao, J. Catal., 2009, 263, 1–3 CrossRef CAS.
- D. Liu, A. Wang, C. Liu and R. Prins, Catal. Today, 2017, 292, 133–142 CrossRef CAS.
- G. Shi and J. Shen, J. Mater. Chem., 2009, 19, 2295–2297 RSC.
- X. Lan, E. J. M. Hensen and T. Weber, Catal. Today, 2017, 292, 121–132 CrossRef CAS.
- J. Li, Y. Chai, B. Liu, Y. Wu, X. Li, Z. Tang, Y. Liu and C. Liu, Appl. Catal., A, 2014, 469, 434–441 CrossRef CAS.
- S. Yang, C. Liang and R. Prins, J. Catal., 2006, 237, 118–130 CrossRef CAS.
- S. Yang and R. Prins, Chem. Commun., 2005, 4178–4180 RSC.
- Z. Wang, L. Zhou, M. Zhang, W. Li and K. Tao, Chem. – Asian J., 2009, 4, 1794–1797 CAS.
- H. Song, M. Dai, H. Song, X. Wan, X. Xu, C. Zhang and H. Wang, Catal. Commun., 2014, 43, 151–154 CrossRef CAS.
- J. Wang, H. Chen, Y. Fu and J. Shen, Appl. Catal., B, 2014, 160–161, 344–355 CrossRef CAS.
- J. Chen, M. Han, S. Zhao, Z. Pan and Z. Zhang, Catal. Sci. Technol., 2016, 6, 3938–3949 CAS.
- A. Wang, M. Qin, J. Guan, L. Wang, H. Guo, X. Li, Y. Wang, R. Prins and Y. Hu, Angew. Chem., Int. Ed., 2008, 47, 6052–6054 CrossRef CAS PubMed.
- Y. Xue, Q. Guan and W. Li, RSC Adv., 2015, 5, 53623–53628 RSC.
- L. Ding, M. Zheng, A. Wang and T. Zhang, Catal. Lett., 2010, 135, 305–311 CrossRef CAS.
- X. Li, Z. Sun, A. Wang, Y. Wang, X. Duan and Y. Chen, Catal. Commun., 2014, 43, 21–24 CrossRef CAS.
- D. Liu, X. Li, W. Ling, T. Zhang, A. Wang, C. Liu and R. Prins, Dalton Trans., 2017, 46, 6366–6378 RSC.
- Q. Guan and W. Li, J. Catal., 2010, 271, 413–415 CrossRef CAS.
- I. I. Abu and K. J. Smith, J. Catal., 2006, 241, 356–366 CrossRef CAS.
- G. H. L. Savithra, R. H. Bowker, B. A. Carrillo, M. E. Bussell and S. L. Brock, Chem. Mater., 2013, 25, 825–833 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |