
Ionic liquids, ultra-sounds and microwaves: an effective combination for a sustainable extraction with higher yields. The cumin essential oil case†
R.
Ascrizzi‡
 a,
J.
González-Rivera‡
b,
C. S.
Pomelli
a,
C.
Chiappe
a,
P.
Margari
a,
F.
Costagli
a,
I.
Longo
c,
M. R.
Tiné
b,
G.
Flamini‡
*a and
C.
Duce‡
*b
a,
J.
González-Rivera‡
b,
C. S.
Pomelli
a,
C.
Chiappe
a,
P.
Margari
a,
F.
Costagli
a,
I.
Longo
c,
M. R.
Tiné
b,
G.
Flamini‡
*a and
C.
Duce‡
*b
aDepartment of Pharmacy, University of Pisa, Via Bonanno 6, 56126 Pisa, Italy. E-mail: guido.flamini@unipi.it; Tel: +39 050 2219686
bDepartment of Chemistry and Industrial Chemistry, University of Pisa, Via G. Moruzzi 3, 56124 Pisa, Italy. E-mail: celia.duce@unipi.it; Tel: +39 050 2219311
cNational Institute of Optics (INO), National Research Council of Italy (CNR), Via G. Moruzzi 1, 56124 Pisa, Italy
First published on 6th July 2017
Abstract
This paper deals with the concept of process intensification applied to the extraction of essential oil (EO). Microwave hydrodistillation (MWHD) and simultaneous ultrasound MW-assisted hydrodistillation (US-MWHD) were intensified by coupling them with a green tool: ionic liquids (ILs). The yield and chemical composition of the cumin EO obtained by MWHD and US-MWHD were compared with those from conventional hydrodistillation (HD) using water and three mixtures of water with three different ILs synthesized ad hoc, as maceration and extraction media, and analysed by a multivariate statistical analysis approach. The cumin EO was chemically characterized by GC and GC-MS analysis. The interaction of the ILs and ILs–H2O mixtures with MW was experimentally investigated and discussed, while the ILs dipole moments and optimized geometry in vacuo and in water were calculated at the DFT level. The different approaches were also compared in terms of energy and time savings. All the data clearly showed that the most promising approach was US-MWHD using a 1,3-dimethylimidazolium dimethylphosphate mixture as maceration and extraction medium. A total yield increase was achieved of up to 75% and an energy saving of 46% compared to the classical HD. The proposed technology, using ILs as green solvents, which fits well with the MW and US technology, enabled a continuous-flow and batch extractor to be constructed which would be useful for industrial applications.
Introduction
Essential oils (EOs) are one of the most valuable products obtained from spices, with a well-established importance in the flavour, fragrance and pharmaceutical industries, as well as in traditional medicine.1,2 Their role as active agents in controlling plant bacterial diseases has also been increasingly studied, as they represent safe yet effective alternatives to hazardous synthetic chemicals.3 Emerging applications such as green chemical agents in metal nanoparticle synthesis have also made them very attractive compounds in nanotechnology and bionanotechnology.4Approximately two hundred different essential oils are traded internationally: volumes range from 20–30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes for orange oil to less than 100 kg for specialty flower extracts. Prices vary widely, but for the majority of oils traded in high volumes, they fall within the range of US$4–$60 per kg, and for specialist minor oils, the price can be many hundreds of US$ per kg.5,6
000 tonnes for orange oil to less than 100 kg for specialty flower extracts. Prices vary widely, but for the majority of oils traded in high volumes, they fall within the range of US$4–$60 per kg, and for specialist minor oils, the price can be many hundreds of US$ per kg.5,6
In this work the extraction of Cumin EO from Cuminum cyminum L. seeds was chosen as the benchmark for the proposed methodologies due to the high extraction yields and the cumin commercial value. Cumin is the second most popular spice in the world, and cumin seeds have a well-established use in traditional medicine due to their diuretic, carminative, antispasmodic and emmenagogic effects.7 The antibacterial activity of cumin EO has been widely reported.3,7
Along with the raw material itself, the extraction cost generally represents the major factor that determines the market price of essential oils. The EO yield is associated with the intrinsic nature of the raw materials. The highest EO yields can be obtained by hydrodistillation, the typical EO extraction approach, which usually requires a long processing time and high energy consumption, representing an expensive extraction process.
There is thus a clear need for cheaper and greener alternatives to conventional hydrodistillation, with higher yields and reduced energy consumption.
Besides hydrodistillation (HD), steam extraction (STE), is also traditionally used for EO production.6 Novel and greener extraction methods have recently been exploited,8 such as microwave assisted extraction (MWAE),9,10 supercritical fluid extraction (SFE)11 and ultrasound-assisted extraction.12
MW technology has also been proposed as a suitable methodology for obtaining EOs.8 There are several configurations using MW irradiation for classical hydrodistillation involving microwave-assisted hydrodistillation (MWHD),13 microwave steam distillation (MWSD),9 and microwave-assisted hydrodiffusion and gravity (MWHG).14 Most of these methods use expensive MW oven-type devices which are difficult to scale up.
In previous works, coaxial MW hydrodistillation extraction (coaxial MWHD) has been proposed as a viable method: a coaxial dipole antenna was used to apply the electromagnetic energy inside the aqueous extraction medium.10,15–17 This particular MW configuration overcomes the classical drawbacks of the closed-device MW oven type methodologies, and the extracted EOs show the same quality as those obtained by conventional techniques, but more rapidly and with energy savings.
Recently reported new extraction systems use the coaxial MW technology: a solventless MW-assisted extraction (SMWAE) approach and a simultaneous ultrasound and MW-assisted hydrodistillation (US-MWHD) method where MW and US can be irradiated simultaneously inside the extraction device.17 During the solventless extraction (dry processing), the use of MW accelerated both the induction and the extraction time, while water has become an exploitable product leading to cheaper and greener alternatives. The simultaneous uses of US and MW also provide an additional decrease in the extraction time. In fact, ultrasound improves the extraction efficiency by increasing the penetration of solvents into plant cells via cavitation, and preventing the degradation of extracts.18
The above findings then led to the development of improved intensification methods of EO extraction. Simultaneous US-MWHD and coaxial MWHD were therefore intensified by coupling them with an extra green tool: ionic liquids.
Ionic liquids (ILs) are widely used in various fields of science, because of their high dissolving power regarding biopolymers (such as lignocellulosic material, chitin, etc.), greenness, high thermal stability, and negligible vapour pressure.19 These features make them ideal additives in plant maceration and extraction of EOs. In addition, ILs strongly absorb MW due to their intrinsic dipole relaxation and ionic conduction.20
In two recent works, it has been reported that ILs can significantly enhance the EO extraction yield without affecting the composition. In the case of Rosmarinus officinalis L., the addition of a series of ILs, prior to conventional hydrodistillation, improved the EO yield by about 25%.21 The use of a mixture of 1:1 dimethylimidazolium dimethylphosphate during the maceration step, led to an increase in the essential oil yield of Cinnamomum verum by about 200%, with the composition unchanged.22
Consequently, the combination of MW and ILs during the coaxial MWHD and more especially the combination of ILs with the US-MWHD process seems a promising new environmentally-friendly approach to EO isolation. Due to the strong matching of ILs with MWs, a reduction in energy consumption, enhancement of the thermal profile and improved control of the extraction process are expected.
This paper outlines our new methods using an aqueous IL solution as the extraction phase and MW activation (IL-MWHD) or the simultaneous use of MW, US and ILs (IL-USMWHD) in an integrated cavity-less device. To the best of our knowledge, this is the first time that an intensified system (IL-USMWHD) has been reported with regard to EO extraction. The individual and combined contribution of these techniques and the assessment of possible synergistic effects are thus evaluated. The intensified approach is then compared with the conventional hydrodistillation (HD) and IL assisted hydrodistillation (IL-HD).
Experimental
Materials
Commercial raw Cuminum cyminum L. seeds (origin: India) by TRS Asia's Finest Foods (“Whole Jeera Cumin seeds” imported by TRS Wholesale CO. LTD. Southall, Middlesex, England) were bought from a local (Pisa, Italy) spice store. Seeds from different packages were thoroughly mixed to obtain a homogeneous starting material. The extractions were all performed using tap water as a solvent and using a solid–liquid dispersion of 1:20.
Synthesis of 3-(2-hydroxyethyl)-1-methylimidazolium chloride (IL1)
A mixture of 1-methylimidazole (365 ml, 4.698 mol) and 2-chloroethanol (378.1 ml, 5.64 mol) was heated, in an oil-bath, at 100 °C for 36 h. After cooling, the crude product, which crystallized upon cooling, was finely crushed, washed with Et2O (3 × 100 mL), and dried under reduced pressure for 10 h and further purified by hydrodistillation for 4 h. Considering the importance of IL purity,23 the absence of volatile impurities was assessed by HS-SPME, with a PDMS fiber. The yellowish liquid (706 g, 92.7%) was characterized by 1H NMR and 13C NMR, FTIR analysis and thermogravimetry (TG).TG: T = 80 °C, mass loss = 10% (water); T = 315 °C, mass loss = 88.5%; residue at 800 °C = 1.5%.
FTIR: wavenumber = 2954 and 2865 cm−1 (νas and νs of aliphatic C–H); wavenumber = 1158 cm−1 and 1065 cm−1 (in plane δ of CH3 and ν of C–C–O); wavenumber = 3070–3235 cm−1 (broad peak due to quaternary amine salt and ν of O–H of 2 hydroxyethyl substituent and of moisture water); wavenumber = 1650 cm−1 and 1432 cm−1 (ring stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN ring and δ of CH2); wavenumber = 1338 cm−1 (δ of OH); wavenumber = 760 cm−1 and 620 cm−1 (ν of C–N; wavenumber = 660 cm−1 out of plane δ of OH).
C and CN ring and δ of CH2); wavenumber = 1338 cm−1 (δ of OH); wavenumber = 760 cm−1 and 620 cm−1 (ν of C–N; wavenumber = 660 cm−1 out of plane δ of OH).
1H NMR (250 MHz, D2O) δ 9.66 (s, 2H), 8.39 (dt, J = 13.8, 1.8 Hz, 4H), 5.71–5.44 (m, 4H), 5.29–5.08 (m, 5H).
13C NMR (63 MHz, D2O) δ 136.02, 123.27, 122.13, 59.48, 51.24, 35.50.
Synthesis of 4-(2-hydroxyethyl)-4-methylmorpholinium dimethylphosphate (IL2)
A mixture of 4-(2-hydroxyethyl)morpholine (327.0 ml, 2.7 mol) and trimethyl phosphate (341.7 ml 2.92 mol) was heated, in an oil-bath, at 100 °C for 36 h. After cooling, the reaction mixture was washed with (50 ml ×3) diethyl ether and further purified by hydrodistillation for 4 h. Considering the importance of IL purity,23 the absence of volatile impurities was assessed by HS-SPME, with a PDMS fiber. The yellowish liquid (732.37 g, 95.2%) was characterized by 1H NMR and 13C NMR analysis, FTIR analysis and TG.TG: T = 80 °C, mass loss = 10% (water); T = 300 °C, mass loss = 70%; T = 530 °C, mass loss = 10%; residue at 800 °C = 10%.
FTIR: wavenumber = 2952 and 2840 cm−1 (νas and νs of aliphatic C–H); wavenumber = 1180 cm−1 and 1096 cm−1 (in plane δ of CH3 and ν of C–C–O); wavenumber = 3075–3379 cm−1 (broad peak due to quaternary amine salt and moisture water); wavenumber = 1650 cm−1 and 1462 cm−1 (ring stretching of CC and CN ring); wavenumber = 1233 cm−1 and 1034 cm−1 (ν PO and P–O, respectively); wavenumber = 774 cm−1 and 618 cm−1 (ν of C–N); wavenumber = 730 cm−1 (ν of P–O–P).
1H NMR (250 MHz, D2O) δ 4.01 (s, 6H), 3.70–3.37 (m, 12H), 3.25 (s, 3H).
13C NMR (63 MHz, D2O) δ 60.03, 59.93, 54.41, 52.43, 52.34.
Synthesis of 1,3-dimethylimidazolium dimethylphosphate (IL3)
A mixture of 1-methylimidazole (239.1 ml, 3 mol) and trimethylphosphate (378.12 ml, 3.3 mol) was heated, in an oil-bath, at 120 °C for 24 h. After cooling, the reaction mixture was washed with (150 ml ×3) diethylether and further purified by hydrodistillation for 4 h. Considering the importance of IL purity,23 the absence of volatile impurities was assessed by HS-SPME, with a PDMS fiber. The yellowish liquid (653.9 g, 98.1%) was characterized by 1H NMR and 13C NMR analysis, FTIR analysis and TG.TG: T = 80 °C, mass loss = 13% (water); T = 300 °C, mass loss = 66.5%; T = 530 °C, mass loss = 13%; residue at 800 °C = 7.5%.
FTIR: wavenumber = 2954 and 2840 cm−1 (νas and νs of aliphatic C–H); wavenumber = 1128 cm−1 and 1034 cm−1 (in plane δ of CH3 and ν of C–C–O); wavenumber = 3168 cm−1 (broad peak due to quaternary amine salt and ν of O–H of moisture water); wavenumber = 1650 cm−1 and 1472 cm−1 (ring stretching of CC and CN ring and δ of CH2); wavenumber = 1233 cm−1 and 1034 cm−1 (ν PO and P–O, respectively); wavenumber = 948 and 890 cm−1 ring vibrational modes; wavenumber = 786 cm−1 and 625 cm−1 (ν of C–N); wavenumber = 730 cm−1 (ν of P–O–P).
1H NMR (250 MHz, DMSO) 9.17 (s, 1H), 7.53 (s, 2H), 3.39 (s, 6H), 2.71–2.67 (d, 6H).
13C NMR (250 MHZ, DMSO) 137.60, 122.84, 50.66, 50.57, 34.41.
Computational details
The structures of all the ions and ion pairs involved in this study were optimized at the B3LYP/6-311++G(d,p) level using Gaussian 09 (ref. 24) using a Linux (Opensuse 12.3) workstation equipped with a Xeon E5-1660 Intel CPU and 32Gb RAM. All the Gaussian default parameters were used, except for the integration grid which was Ultrafine instead of Fine.Cumin seed EO extraction
The cumin seeds essential oil was obtained by different methods. Conventional hydrodistillation (HD), Microwave assisted hydrodistillation (MWHD) and simultaneous ultrasound and microwave assisted hydrodistillation (US-MWHD) with a combination of these three approaches with three different ionic liquids (ILs) were all carried out after a pre-hydrodistillation step in which 30 g of cumin seeds were macerated for 24 h in a glass bottle and kept in a mechanical agitator at 100 rpm with:600 ml of distilled water (blank)
600 ml of distilled water NaCl saturated
75 ml of distilled water and 50 ml of EtOH MIM Cl (IL1)
75 ml of distilled water and 50 ml of EtOH MMORF DMP (IL2)
75 ml of distilled water and 50 ml of DMI DMP (IL3)
In the case of hydrodistillation with ionic liquids, 475 ml of distilled water were added to obtain the final 600 ml volume just before hydrodistillation. Each method of extraction was repeated in triplicate.
Conventional hydrodistillation
Conventional hydrodistillations (HD) were performed in a Clevenger-type apparatus, equipped with an electric mantle heater for the thermal activation. Briefly, after the maceration step, the mixture was transferred to a 2000 mL glass beaker connected to a Clevenger condenser. The HD started after 15 min and was kept under static conditions for 2 h. In order to explore the ionic strength of the extraction media, HD was also performed using a NaCl saturated solution as the solvent, under the same experimental extraction conditions (same conditions as the pre-hydrodistillation and HD steps).The HDs with ionic liquids (ILs-HD) were performed using the same Clevenger-type apparatus as the HD. Briefly, after the maceration step, the mixtures containing the ILs (EtOH MIM Cl (IL1), EtOH MMORF DMP (IL2) and DMI DMP (IL3)) were then transferred to a 2000 mL glass beaker connected to a Clevenger condenser. A total of 475 ml of distilled water were then added to obtain the final 600 ml volume before the HD. The ILs-HD started after 15 min and was kept under static conditions for 2 h.
Coaxial microwave assisted hydrodistillations (MWHD)
Microwave assisted hydrodistillation was carried out in a modified Clevenger-type extractor in which the MW-assisted thermal activation was performed by direct MW irradiation into the extraction media by an open-ended coaxial applicator. A full description of the experimental set-up can be found in our recent work reported elsewhere.16 A total of 30 g of cumin were macerated in water and ILs/water solutions, as reported above. Each macerated aqueous substrate dispersion was then loaded into a 1000 mL MW assisted flask vessel. The vessel was covered with a metallic grid to prevent the loss of MW irradiation out of the reaction medium and to ensure safe operating conditions. The extraction vessel was also fully covered with thermal isolation material. A magnetic stirrer was placed on the base of the MW-assisted extractor device. MW energy was applied by a coaxial dipole antenna immersed in the extraction medium, protected by a glass tube. The MW source was a magnetron oscillator which supplied up to 1200 W of continuous MW irradiation power at a frequency of 2450 MHz. The substrate dispersion was heated using 500 W of MW power for 11 min (induction time) and stirred at 300 rpm. Once the hydrodistillation had started, the power was reduced to 300 W and kept at steady state conditions for 2 h. A power meter was used to continuously monitor the stray radiation and ensure it was always within the safe region.Ultrasound coaxial microwave-assisted hydrodistillations (US-MWHD)
The simultaneous US-MWHD device used in this work is described in a previous paper.17 A Vibra cell sonics (VCX 750) sonicator with a frequency of 20 kHz and a tip diameter of 13 mm, with a maximum power level of 300 W, was used to apply the US irradiation. The MW source was the same as that for the MWHD. The experiments were performed using the same solid–liquid dispersion of 1:20. 30 g of cumin were macerated in water and ILs/water solutions as mentioned above. Each macerated aqueous substrate dispersion was then loaded into the coaxial US-MWHD modified Clevenger extractor. A total of 40% of the maximum nominal power of both US and MW was then applied (500 W of MW power and 40% amplitude US) during the induction time (10 min). Once the hydrodistillation had started, the MW energy was reduced to 300 W. While the MW irradiation was being continuously applied (2 h), the US irradiation was performed by cycles of 30 s on/off throughout the extraction process (effective US irradiation applied of 1 h).
MW heating of ILs and ILs-water mixtures
The interaction between the ILs, ILs–H2O mixture (1:11 v/v) and MW was evaluated by the determination of the temperature profile of a weighted quantity (100 mg) of pure ILs and of their water mixtures under different constant applied MW power (from 30 to 50 W) using a commercial MW generator SAIREM, Mod. GMP 03 K/SM which supplies up to 300 W of continuous MW irradiation power at a frequency of 2.45 GHz for 1 min. For these experiments, the liquid sample was loaded into a quartz tube and held in a microstrip line. The temperature was measured using an optical fiber thermometer placed in the middle of the sample.
Gas chromatography – mass spectrometry analyses
The hydrodistilled essential oils were diluted to 5% in n-hexane HPLC grade and then injected into a GC-MS apparatus. Gas chromatography-electron impact mass spectrometry (GC-EI-MS) analyses were performed with a Varian CP-3800 gas chromatograph equipped with a DB-5 capillary column (30 m × 0.25 mm; coating thickness 0.25 μm) and a Varian Saturn2000 ion trap mass detector. Analytical conditions were as follows: injector and transfer line temperatures 220 and 240 °C, respectively; oven temperature programmed from 60 to 240 °C at 3 °C min−1; carrier gas helium at 1 ml min−1; injection of 0.2 μl (5% n-hexane HPLC grade solution); split ratio 1:30. Identification of the constituents was based on a comparison of the retention times with those of the authentic samples, comparing their linear retention indices relative to the series of n-hydrocarbons. Computer matching was also used against commercial (NIST 14 and ADAMS) and laboratory-developed library mass spectra built up from pure substances and components of known oils and MS literature data.25–29
Statistical analyses
The statistical analyses were carried out using the JMP software package (SAS Institute, Cary, NC, USA). Extraction yields (% w/w) replicates were transformed using arcsine square root (arcsin √x) for normalization and then subjected to analysis of variance (ANOVA) to obtain mean values and confidence intervals (α = 0.05). Averages were separated by Tukey's b post hoc test. P < 0.05 was used for the significance of differences between means. The hierarchical cluster analysis (HCA) was performed with the Ward's method.SPME
Supelco SPME (Solid Phase Micro-extraction) devices coated with polydimethylsiloxane (PDMS, 100 μm) were used to sampling the headspace of the ionic liquid inserted into a 4 ml glass septum vial and allowed to equilibrate for 30 min. SPME sampling was performed using the same fibre, preconditioned according to the manufacturer instructions, for all the analyses. After the equilibration time, the fibre was exposed to the headspace for 20 min. Once sampling was finished, the fibre was withdrawn into the needle and transferred to the injection port of the GC-MS system. All the SPME sampling and desorption conditions were identical for all the samples. Furthermore, blanks were performed before each first SPME extraction and randomly repeated during each series. GC-EI MS analyses were performed using the same conditions described for essential oils, except that the splitless injection mode was used and the injector temperature was 250 °C.Thermogravimetry (TG)
The TG experiments were performed using a Q5000IR TA Instruments Thermobalance equipped with Pt crucibles. Scan rate was set at 10 °C min−1, from 30 °C to 800 °C under nitrogen flow (25 ml min−1). The amount of sample in each measurement varied between 4 and 5 mg.FTIR spectroscopy
Infrared spectra were recorded using a FT-IR Agilent Technologies Spectrophotometer model Cary 640, equipped with a universal attenuated total reflectance accessory (ATRU). Few micrograms of dry samples were used with the following spectrometer parameters; resolution: 4 cm−1, spectral range: 600–4000 cm−1, number of scans: 128. Agilent spectrum software was used to process FTIR spectra.NMR spectroscopy
Unless otherwise specified, the reagents were used without any purification. 1H NMR spectra were recorded in appropriate solvents using a Bruker Avance II operating at 250.13 MHz. 13C NMR spectra were recorded using the spectrometers operating at 62.9 MHz. The following abbreviations are used: s = singlet, d = doublet, dd = double doublet, t = triplet, m = multiplet, br = broad. The temperature was controlled to ±0.1 °C. 1H and 13C NMR chemical shifts (ppm) referred to TMS as the external standard.Results and discussion
The first part of this work describes the results of the cumin seed EO extraction by different innovative MW and MW-US assisted methods using water or water–IL mixtures as extraction media. A full characterization of the isolated EOs is then shown and analysed with a multivariate statistical analysis approach. The second part deals with the study of the MW interaction of ILs and IL–H2O mixtures as an efficient MW absorbing extraction media, and the DFT calculations of ILs dipole moments depending on ILs geometry optimized in vacuo and in water in order to better investigate the role of these salts in EO extraction. Finally, energy savings and time reductions of the proposed methodologies are discussed and the potential application in the green industry of these intensified novel processes (including safe operation points, control process parameters and scalability) is presented.EO extraction: kinetics and yield analyses
Fig. 1 shows the kinetic investigations of the percentage yield of cumin seed EO obtained by HD, MWHD, US-MWHD in water. All the yields obtained with the different hydrodistillation approaches (HD, MWHD, US-MWHD in water and ILs/water mixtures) are reported in Fig. 2. Table S1 of the ESI† reports the extraction yields for all the methods studied.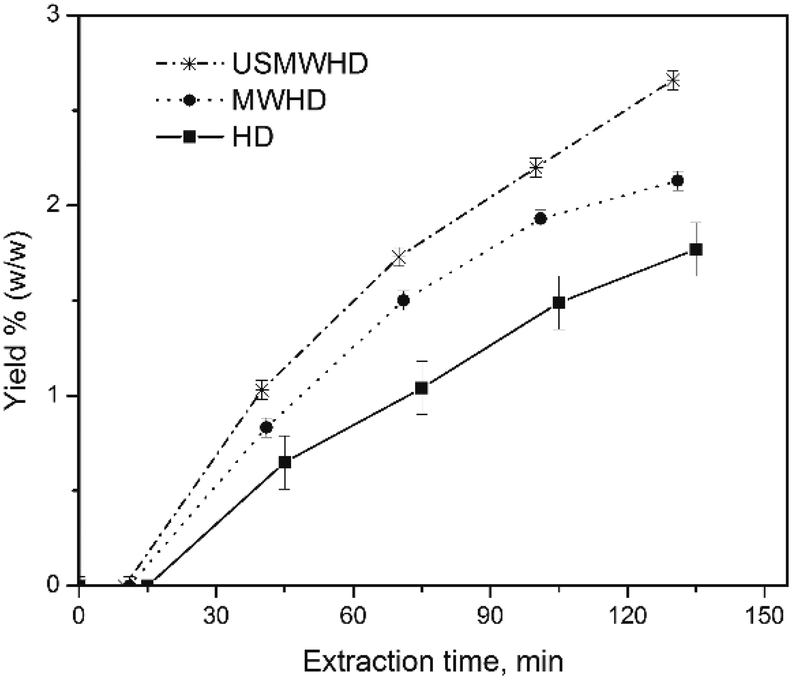 | ||
| Fig. 1 Kinetic investigation of the percentage yields of cumin seeds EO obtained by (■) HD, (●) MWHD, and (*) US-MWHD in water. | ||
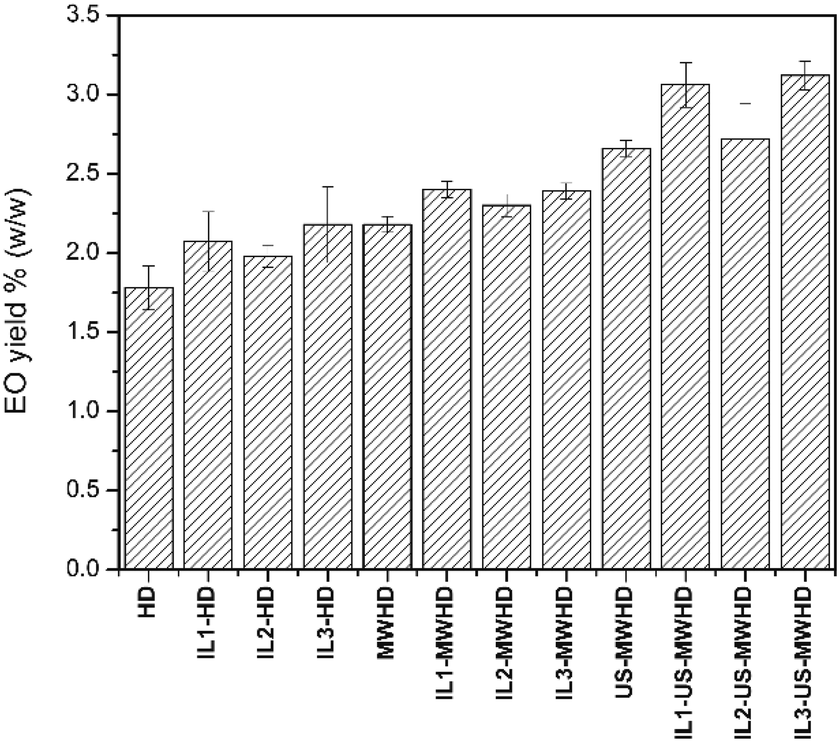 | ||
| Fig. 2 Essential oil yields (% w/w) of extractions performed with the various combinations of methods. | ||
The kinetic investigation in water was performed in order to assess the extraction time for all the experiments.
The kinetic curves present the classical hydrodistillation behaviour with time period of induction (tind), where the extraction mixture achieves the boiling point and the volatile compounds begin to be released into the extraction media from exogenous secreting glands without actually separating (zero yield), and a second time period (textrac) where the EO is separated and the yield increases.17,30 The total extraction time (ttotal) is the sum of tind and textrac.
The tind was dependent on the approach used. The HD took the longest, where tind = 15 min, and US-MWHD (10 min) ≈ MWHD (11 min) were faster.
In two hours, all the kinetic curves in water reached a plateau, so the textrac was set at two hours for all the experiments. The MW and US-MW assisted hydrodistillations (MWHD; US-MWHD) gave higher EO yields in water than conventional hydrodistillation (HD). The extraction efficiency was HD < MWHD < US-MWHD, comparing the data for the same liquid medium. (Table S1,†Fig. 1) Comparing the experimental data at a fixed yield value, the use of MW and US-MW speeds up the extraction (US-MWHD > MWHD > HD).
The cumin EO yields vary considerably depending on the geographical origin of the seeds, chemotype, pre-extraction treatment and extraction method. Table S2 of the ESI† summarizes the literature on cumin seeds EOs by different approaches. The reported yields range between 0.63% (ref. 31) with a solvent free microwave extraction and 5.4% (ref. 32) for a Bulgarian EO extracted with a conventional HD method. The reports on distillations of whole seeds32–34 report higher extraction yields compared to grounded samples.35–39 Chinese36 and Bulgarian32 cumin seeds have been shown to yield higher amounts of EO.
In this work, higher yields of cumin seeds EO (from 1.78 to 2.66 wt%) were obtained with a lower extraction time with all the extraction approaches (HD, MWHD and US-MWHD).
Water–IL mixtures as extraction media were then tested. Three ad hoc synthesized ILs have been used: IL1 3-(2-hydroxyethyl)-1-methylimidazolium chloride; IL2 4-(2-hydroxyethyl)-4-methylmorpholinium dimethylphosphate; and IL3 1,3-dimethylimidazolium dimethylphosphate (see Fig. 3). These ILs have been selected considering two important aspects that significantly could affect eventual large scale applications; IL price and IL environmental impact. All three ILs can be prepared starting from largely available compounds through single step processes, avoiding expensive anion metathesis reactions. Furthermore, the introduction of long alkyl chains on cation, that generally increase IL toxicity towards the aquatic compart, has been avoided. In particular, IL1 and IL2 have also a hydroxyl group on the alkyl chain that can further reduce toxicity leading to an increased biodegradation.40
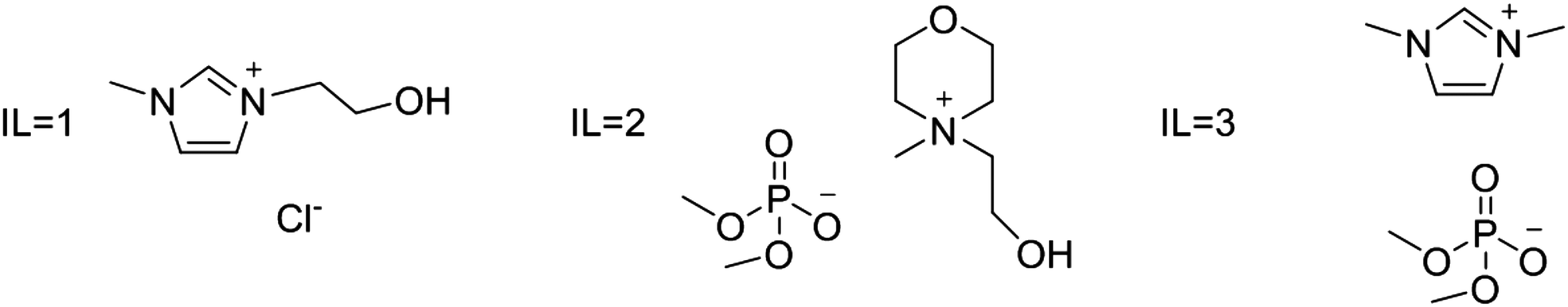 | ||
| Fig. 3 Ionic liquids used in the present study IL1 3-(2-hydroxyethyl)-1-methylimidazolium chloride ([HOEMIM]+Cl−); IL2 4-(2-hydroxyethyl)-4-methylmorpholinium dimethylphosphate ([HOEMMor]+[(CH3)2PO4]−); IL3 1,3-dimethylimidazolium dimethylphosphate ([MMIM]+[(CH3)2PO4]−). | ||
Furthermore, IL1 resulted the best additive in the case of Rosmarinus officinalis L.21 The ability of this IL to favor cell wall modification through hydrogen-bonding interactions between both IL ions (chloride and IL cation) and cell wall constituents (mainly cellulose) was considered the principal factor determining plant tissue disruption and oil release. Consequently, IL1 was chosen to be used also in this investigation. The screening was then extended to dimethyphosphate based ILs, considering that also these ILs can be included in the relatively short list of the ionic media able to dissolve biological macromolecules, such as cellulose, wool and feathers. This anion, analogously to chloride, presents indeed a strong hydrogen bond acceptor activity, and is able to favour biopolymers dissolution through the formation of hydrogen bonds with the hydroxyl groups of biopolymers.41,42 Nonetheless, dimethylphosphate based ILs have a high thermal and chemical stability and belong to the halide-free ionic liquid class, a feature which makes them more environmentally friendly.
The addition of ILs to water increased the extraction yields, maintaining the ranking among the extraction methods HD < MWHD < US-MWHD with equal liquid medium. The lowest yield increase was obtained with IL2, which accounted for 11.24%. A 16.29% yield increase was obtained with IL1, whilst the best performing was IL3, which led to a 22.47% increase in yield (Table S1,†Fig. 2).
This result shows that ILs are effective both during the maceration step (yield increase for HD) and in the extraction step (yield increase for MWHD and US-MWHD), where they respond differently to MW and US-MW applications. The highest yields were obtained when ILs, MW and US were used simultaneously, US-MWHD accounting for 49.4% of the extraction efficacy. The best combination was IL3-US-MWHD, with a total yield increase of 75.28% against the blank. A slightly lower increase was obtained with the IL1-US-MWHD method, which showed an increase of 71.91% against the blank.
The plant extraction process is intensified using US negative pressure cavitation effects. One proposed mechanism17,18 highlights that when a bubble collapses near the surface of a plant leaf, very high energy solvent jets, released by this collapse, are directed towards the surface, creating high local temperatures and pressures at a large number of reaction sites which are normally related to the enhanced reaction rates in cavitation systems. Given this mechanism, cell membranes are disrupted by the hammer-like action of the produced solvent jets. Comparing the effect of each single IL on the EO yields with equal extractive methods (HD, MWHD, US-MWHD), the extractive power of the mixture was IL3/water ≥ IL1/water > IL2/water > water. It is however noteworthy that in the proposed process ILs not only benefit the EO extraction through chemical effects exerted on plant tissues but they can also improve the efficiency through an enhanced MW absorption of the extraction media. As reported in the next section, this rank matched with the heating curves obtained under 50 W of MW irradiation of IL1/water, IL2/water and water, but not of the IL3/water mixture. Note that the heating behaviour of a mixture under MW irradiation is only one of the factors that determines the extractive power of a mixture. The EO yield in the IL3/water mixture was also the highest for HD, thus revealing that IL3 performs better at dissolving biopolymers.
An HD in NaCl saturated water was also performed in order to evaluate the effect of the water ionic strength factor in the extraction process. Under these conditions, the extraction yield decreased by 24%, thus confirming that the increase in EO yields when extraction is performed using the IL/water mixture is not due to a “salting-out” effect.
Multivariate statistical analysis
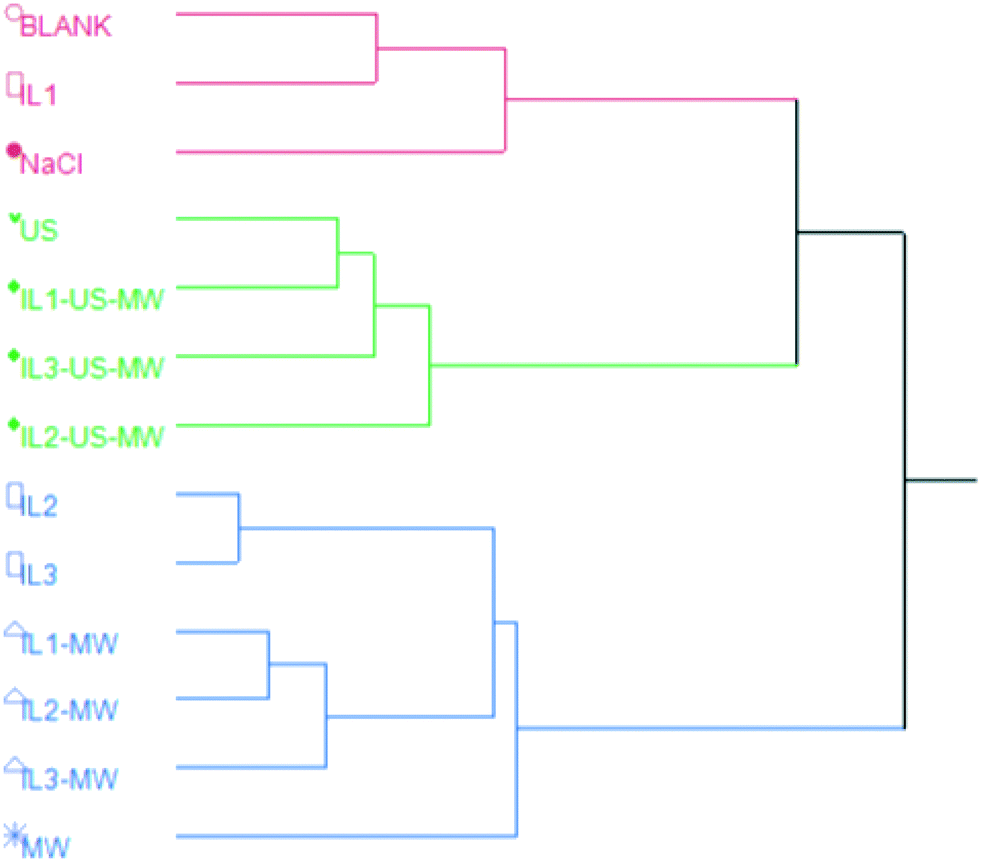 | ||
| Fig. 4 HCA dendrogram for the total composition of the EOs based on the method of extraction. | ||
The HCA revealed three major clusters: the red cluster comprises extractions performed without the aid of MW and/or US; the green one groups together all the EOs extracted with methods using US; the blue one contains EOs obtained from MW-aided extractions, along with IL2- and IL3-HD. This shows the pattern in the chemical composition of the EOs induced by the extraction method. The relative abundances of oxygenated VOCs are red < green < blue. Notably, MW- and US-aided distillations are grouped separately from each other.
The same distribution patterns were highlighted by the principal component analysis (PCA). Fig. 5 shows the PCA score plot: the considered covariance data matrix was a 50 × 13 matrix (50 VOCs identified in total × 13 samples = 650 data). The PCA was performed by selecting the two highest PCs obtained by the linear regressions: the PC1 and PC2 chosen cover 48.00 and 32.87% of the variance, respectively, for a total explained variance of 80.87%. The loading plot is reported in Fig. S1 of ESI.†
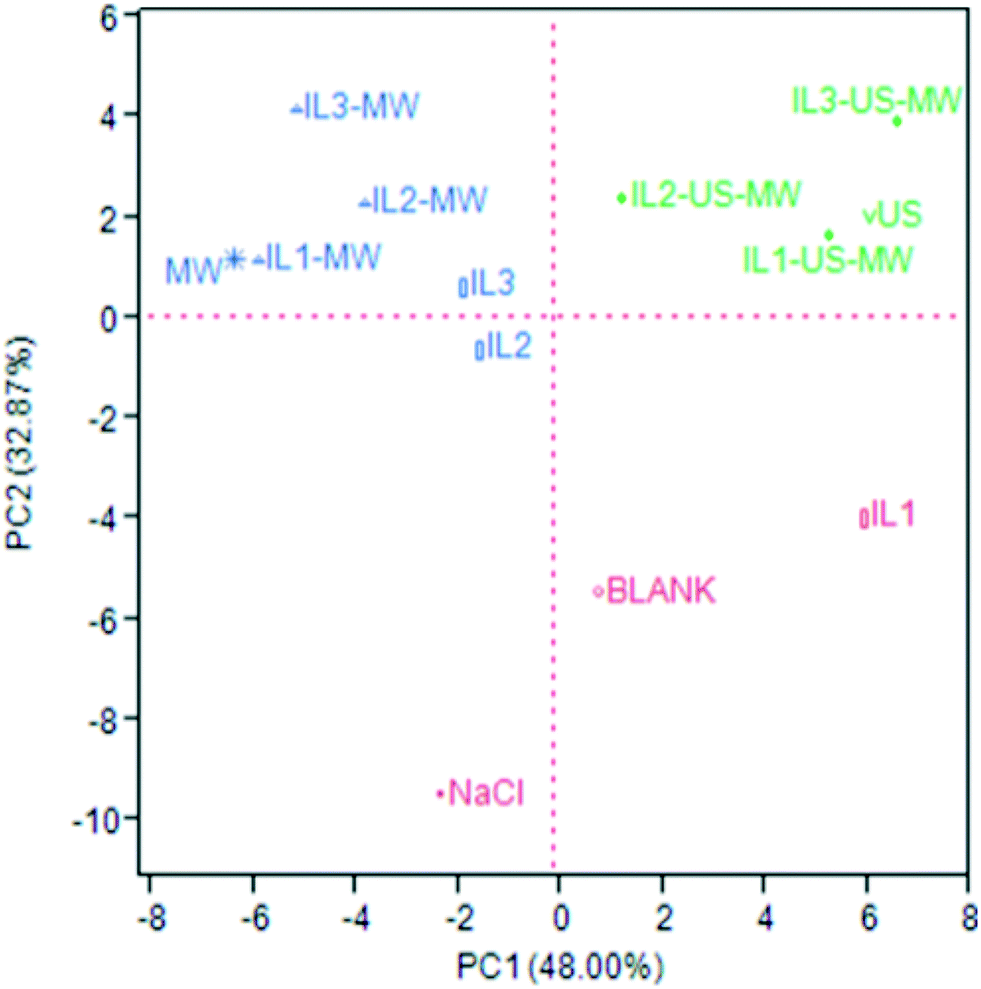 | ||
| Fig. 5 PCA score plot for the total composition of the EOs based on the method of extraction. | ||
The red cluster is all plotted in the negative PC2 area, where there is only one blue sample (IL2). As Fig. S1† shows, the major contributions to this plotting are the relative abundances of α-terpinen-7-al, p-cymene and sabinene, which in these samples are more significant than the others. The EOs of this group are only constituted by monoterpenes.
The green cluster, comprising all the US-aided extraction samples, is all plotted in the positive PC1 and PC2 area, in the upper right quadrant: β-pinene, γ-terpinene and limonene are more abundant in these samples, where they are grouped on their own.
The blue cluster, which comprises the MW-aided extraction (non-US coupled) and two ILs-aided extraction samples, is plotted in the negative PC1 and, except for IL2, positive PC2 areas. These samples are characterized by higher relative abundances of trans-pinocarveol, cuminaldehyde, γ-terpinen-7-al and, in general, oxygenated compounds. As Fig. S1† shows, these compounds are responsible for the evidenced plotting. Table S4 (ESI†) shows the full chemical characterization obtained by GC-MS.
Yields of extraction
Fig. 6 shows the HCA dendrogram of the samples according to their extraction yields.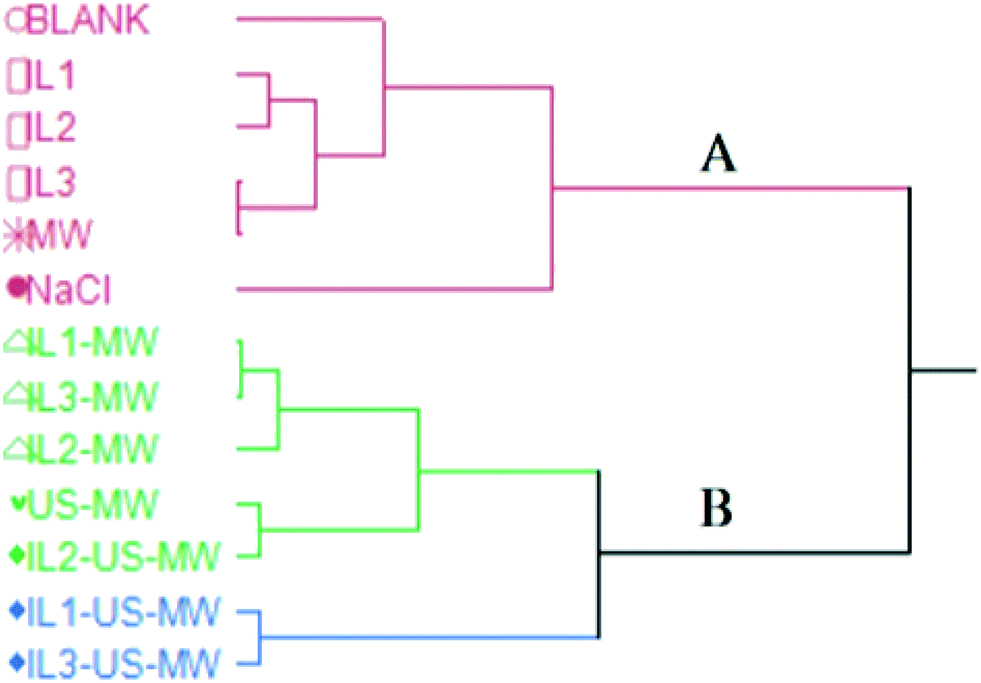 | ||
| Fig. 6 HCA dendrogram for the EO yields based on the method of extraction. | ||
The two macro-clusters A and B sharply divide the MW and/or US-aided extractions (except for MW) and the unaided extractions. The B macro-cluster is further divided into two clusters: green and blue. The green cluster comprises all the ILs and MW aided extractions in a smaller sub-group, whilst the US coupled ones are clustered together. The blue cluster contains the two best-performing extractions in terms of yield: IL1-US-MWHD and IL3-US-MWHD.
Both the US and MW contributions to the extraction yields are significant enough to represent a different statistical macro-cluster. The dendrogram clearly shows the efficacy of the US and MW-aided extractions, in comparison with those extractions assisted solely by the ILs.
ILs/MW interactions
There are several approaches to investigate the MW interaction with a material. From an electromagnetic point of view, the power per unit volume, PV absorbed by a material submitted to microwave heating, is given by eqn (1):43| PV = ε0ε′′ωErms2 | (1) |
δ = ε′′/ε′ instead of ε′′ as the characterizing parameter is very misleading, as in some chemical physics textbooks, because the imaginary and the real part of the complex permittivity are both functions of temperature, but with a different behavior. It is also only possible to calculate the time necessary to heat up a material with microwaves if the dependence of ε′′ on temperature is known. Although in hydrothermal processes, water is commonly considered as a strong absorber of microwaves, the ε′′ of water actually diminishes as the temperature increases, its value in proximity to the boiling point being about eight times lower than that at room temperature. The imaginary part of the relative dielectric permittivity of strongly polar solvents, such as ionic liquids on the other hand, is generally an increasing function of temperature.44 This is another reason why ionic liquids are particularly suited to hydrothermal microwave plant extraction.
In this work, in order to evaluate the MW interaction with the extraction media, a simple experimental approach was used, based on recording the temperature changes of the medium under a constant MW power applied during a fixed time interval, as proposed in the literature.20,45 Under such conditions, the MW interaction is indirectly inferred: a poor MW absorber material does not show temperature changes (transparent to MW), while a continuous temperature increasing profile is shown by a strong absorber material.45 Further insights into IL–water interactions were also explored by the IL dipole moments and their optimized geometry in vacuo and in water through DFT calculations.
The temperature profile changes of ILs and their aqueous mixtures with the MW power was investigated by applying increasing MW powers (30, 40 and 50 W) to the samples.
All the temperature profiles of ILs, water and their aqueous mixtures at different MW powers are reported in Fig. S2 and S3 of the ESI,† respectively. Each curve shows two main steps: 1) a temperature increase, during the first 60 s due to the MW absorption and, 2) the naturally cooling profile, once the MW applied power is stopped (from 60 to 120 s). Table 1 shows the maximum temperatures reached by pure ILs, water and their aqueous mixtures after 60 s of MW irradiation for MW power = 30, 40 and 50 W.
:H2O, 1:11 v/v) after 60 s of MW irradiation for MW power = 30, 40 and 50 W
| Applied MW power (W) | T max,60s (°C) of IL1 pure (IL1/water mixture) | T max,60s (°C) of IL2 pure (IL2/water mixture) | T max,60s (°C) of IL3 pure (IL3/water mixture) | T max,60s (°C) of water |
|---|---|---|---|---|
| 30 | 100 | 42 | 95 | 34 |
| (57) | (46) | (45) | ||
| 40 | 143 | 64 | 141 | 38 |
| (76) | (56) | (57) | ||
| 50 | 179 | 92 | 172 | 42 |
| (98) | (67) | (68) |
As an example of IL–MW and IL–H2O–MW interaction, Fig. 7 compares the temperature profiles under a constant MW power of 50 W of the pure IL3, water and of IL3/H2O mixture at the same weight ratio as that used in the cumin seed EO extraction. Water is the traditional reference material as the strong MW absorber (at room temperature) and green chemical solvent.
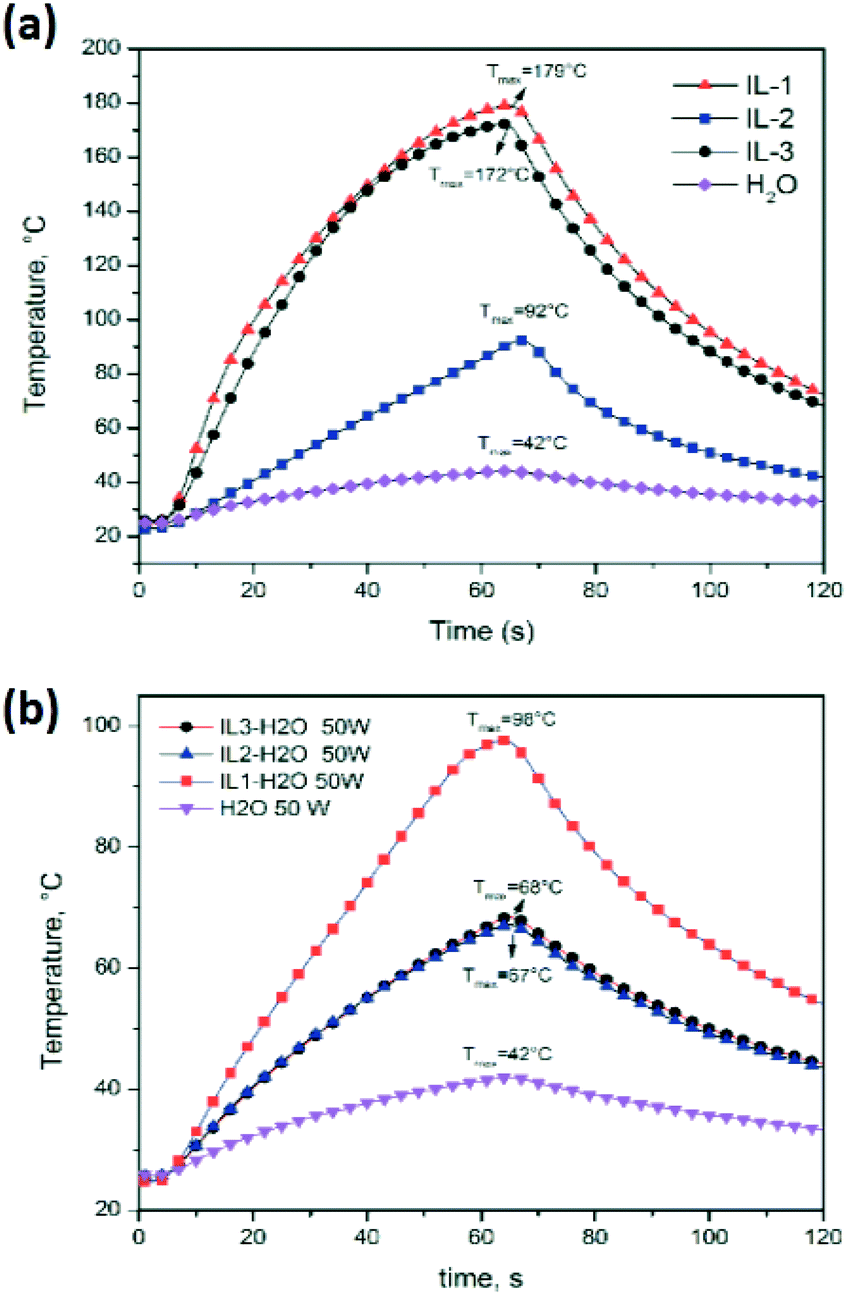 | ||
| Fig. 7 Temperature profiles of water and (a) ILs under 50 W of MW irradiation, and (b) ILs/H2O mixtures used in the cumin seed EO extraction. | ||
The experiments clearly showed a higher MW absorption of pure ILs compared to water (Tmax at 60 s = 42 °C) with the order IL1 > IL3 > IL2 > water (Table 1 and Fig. 7a). ILs/water mixtures (Tmax at 60 s = 98 °C for IL1/water, and 68 °C for IL2/water and IL3/water) also showed a higher MW absorption compared to water (Table 1 and Fig. 7b, however the order was IL1/water > IL2/water = IL3/water > water.
Increasing the applied MW power from 30 to 50 MW, the pure ILs and IL water mixtures reached higher temperatures at 60 s, however the order between ILs and their water mixtures was maintained.
Besides the MW power, the MW absorption of an IL depends on different parameters such as anion and cation structure, cation molar mass and volume used.20IL1, IL2 and IL3 have different anions and different cation structures, molar masses and volume, thus preventing a rationalization of IL1, IL2, IL3 experimental MW absorption ranking. With a high water content, the MW absorption of IL/water mixtures seems to be governed by the IL anion/water interactions. In fact, IL2 and IL3 have the same phosphate anion, while IL1 has a chlorine anion.
ILs dipole moment and water solvation by DFT calculations
Since the dipole moment is the key parameter for the energy transfer from microwaves to molecules, we performed various DFT calculations in order to understand whether there was any correlation between the extraction efficiency and the molecular structure. It is noteworthy that the investigated mixtures are actually complex heterogeneous systems: a water solution of the selected ionic liquid is indeed mixed with a solid material containing the compounds that we wanted to extract.Consequently, the initial question is: in an aqueous solution, what form do ionic liquids take? Are they single ions, ion pairs or something more organized?
Here we considered single ions and ion pairs. The structures of all the ions and ion pairs involved in this study were optimized at the B3LYP/6-311++G(d,p) level using Gaussian 09.24 This level of calculation has been successfully tested to study ionic liquid ions.46 First, they were optimized in vacuo, i.e. without any environment effect, then in water using the IEFPCM continuum solvent scheme.47
Table 2 shows some useful values extracted from these calculations.
| Ion or pair | Dipole/D | ΔE/(kJ mol−1) | k/(mdyne Å−1) | |||
|---|---|---|---|---|---|---|
| In vacuo | Water | In vacuo | Water | In vacuo | Water | |
| Cl− | 0.00 | 0.00 | — | — | — | — |
| [(CH3)2PO4]− | 4.95 | 7.22 | — | — | — | — |
| [MMIM]+ | 0.77 | 1.05 | — | — | — | — |
| [HOEMIM]+ | 4.29 | 6.24 | — | — | — | — |
| [HOEMMor]+ | 3.12 | 3.96 | — | — | — | — |
| [HOEMIM]+Cl− (IL1) | 12.32 | 18.59 | −418.16 | 83.93 | 0.0573 | 0.0556 |
| [HOEMMor]+[(CH3)2PO4]− (IL2) | 11.01 | 21.39 | −413.06 | 68.71 | 0.0092 | 0.0041 |
| [MMIM]+[(CH3)2PO4]− (IL3) | 10.66 | 15.65 | −393.43 | 73.94 | 0.0076 | 0.0070 |
We used in vacuo values as a reference to estimate the effect of the water solvation on ionic liquids. This situation is actually quite unusual: IL are generally employed as solvents rather than solutes. An analysis of the dipole values suggests that water solvation enhances the dipole values of ions (except for the spherical chloride ion) and ion pairs. In particular, the dipoles of ion pairs consistently exceed the sum of the dipoles of the constituting ions. This is not surprising: the dipole of a polar ion that arises from an internal charge distribution cannot compete with the dipole of two neat charged species arranged at a specific distance.
The ion pair stabilization values show that in vacuo ion pairs are more stable than separate ions (opposite charges in vacuo attract each other without any shield effect). However, solvated ion pairs are less energetically stable than separate ions. In other words, the separate solvation of oppositely charged ions is more favorable than the solvation of a dipolar but neutral ionic pair. Thus, in water optimized ion pairs are local minima: they have a higher energy than the constituting ions at an infinite distance. A solvated free ion, however, is a realistic representation of a solution at an infinite dilution. A situation significantly different from our system, which is instead a concentrated IL water solution containing solid components. In fact, our ions have a limited rather than an infinite space to move apart from each other.
Therefore, in our scenario, the relative energy is not an useful parameter to compare the three ionic liquids. Useful parameters need to be related to short range interactions. Table 2 reports the spring constant of ion–ion stretching i.e. the normal mode that mainly corresponds to a variation in distance between ions. However, since the two ions are not completely rigid during this motion the harmonic approximation might be not completely satisfactory in this range of frequencies (between 30 and 150 cm−1). Therefore, these values can be considered as a semi-quantitative index of the short distance strength of the ion pairs. Optimized ion pair structures in both environments (in vacuum and water) are reported in Fig. 8.
 | ||
| Fig. 8 Structures of ionic pairs. From top to bottom: [MMIM]+[(CH3)2PO4]−, [HOEMIM]+Cl, [HOEMMor]+[(CH3)2PO4]−. On the left, in vacuo structures, on the right, in water structures. The distances between the interacting pair of atoms (dotted lines) are written next to them. | ||
The ion pair graphical representation, also considering the spring constant values, highlights that the [HOEMMor]+[(CH3)2PO4]− ion pair is the only one characterized by a significant different disposition in the two environments.
In water, the chelated structure present in vacuo is disrupted with a consequent considerable reduction in the spring constant. Conversely, in the other two cases, the ions are more spaced in water (this can be also deduced from the dipole values) although the geometry is very similar.
[HOEMMor]+[(CH3)2PO4]− therefore has a lower ability to give ion pairs in water and the equilibrium shifts more towards separate ions. In a pure IL framework, there is no water (or only a limited amount) that can separate ions and, probably, the ion pairs have a lifetime. All the three ionic liquids presented here have a chelated ionic pair structure when pure salts. In heating experiments of pure ionic liquids, IL1 and IL3, presented a similar behavior and allowed to reach temperatures significantly higher than water, whereas IL2 produced only a moderate increase.
A reasonable hypothesis is that the presence of the imidazolium ring leads to ILs with longer ion pair lifetimes. When ion pairs do not exist or rapidly dissociate/reassociate, they are not available for energy transfer. On the other hand, in water –OH group are solvated and thus distracted from ion–ion interactions: IL2 where the OH group plays a fundamental role in ion pair easily dissociates. On the contrary, the acidic hydrogen at the C2 position of the imidazolium ring of IL1 can maintain its interaction with the small chloride anion also in water. Since the dipole is the key parameter for microwave/solution energy transfer, a more dissociated ionic liquid loses the temperature enhancement effect which arises from the dipole associated to the IL organization as ion pairs. Thus, the energy transfer is less efficient as consequently so is the extractive capacity.
Extraction intensification: advantages of the combined ILs-MWHD and ILs-US-MWHD approaches
Process intensification is currently a hot research topic in chemical engineering due to its potential to obtain more sustainable processes.48 There are several ways to intensify a chemical process: at operation, task or phenomena level, all involving several tools to obtain overall enhancements. In this work, we achieved the intensification of operations and tasks by coupling two different operation units (US-assisted extraction and MW-assisted extraction) resulting in a simultaneous US-MWHD extraction. Considering the phenomena scale, by introducing ILs to the intensified extraction, we developed a greener and better processing alternative (ILs-USMWHD) which reduced costs by performing US and MW irradiation by means of a single piece of equipment. While productivity enhancements were achieved by chemical processing using ILs, the energy demands were reduced due to the MW and US fast processing and good thermal coupling of MW + ILs. In the following sections, we present the results of the energy involved in these processes, some comments on the safety operations, together with industrial scalability projections.Energy demands
Since in most chemical processes energy requirements represent the major costs involved, the energy consumed (Econsumed) in each step of the EO extraction was evaluated. Table 3 summarizes the Econsumed during the EO extractions for each extraction approach and the intensified processes with IL3 as an example and the best results obtained (due to heating and yield improvements). The results of all the extraction approaches used in this work are presented in Table S3 of ESI.†| Extraction approach | Induction period | Extraction periodb | E consumed,tot./g EO (kW h g−1) | Carbon footprintd (kgCO2 g−1 OE) | ||||
|---|---|---|---|---|---|---|---|---|
| t ind (s) | Effective powera (W) | E consumed,ind (kW h) | Effective powera (W) | E consumed,extract (kW h) | ||||
| a Effective power consumed (≈70% of nominal power). b Extraction time: 120 min. c Experimental value. d Emission factor 0.527 kg kW−1 h−1, 48 including an allowance for the 7.5% of losses on the national grid. | ||||||||
| HD | 900 | 500 | 0.125 | 380 | 0.76 | 1.66 | 0.87 | |
| IL3-HD | 900 | 500 | 0.125 | 380 | 0.76 | 1.35 | 0.71 | |
| MWHD | 660 | 350 | 0.11 | 200 | 0.67 | 1.18 | 0.62 | |
| IL3-MWHD | 660 | 350 | 0.11 | 200 | 0.67 | 1.08 | 0.57 | |
| US-MWHD | 600 | MW | 350 | 0.10 | 200 | 0.67 | 1.04 | 0.55 |
| USc | 0.01 | USc | 0.06 | |||||
| IL3-USMWHD | 600 | MW | 350 | 0.10 | 200 | 0.67 | 0.89 | 0.47 |
| USc | 0.01 | USc | 0.06 | |||||
E consumed and total energy consumption per unit mass of products Econsumed,tot./g EO are calculated according to the methodology reported elsewhere:17 using eqn (2) for Econsumed; where P = effectively applied power (from a mains plug, W) and t = time period (s) and eqn (3) for Econsumed,tot./g EO:
| Econsumed = P × t | (2) |
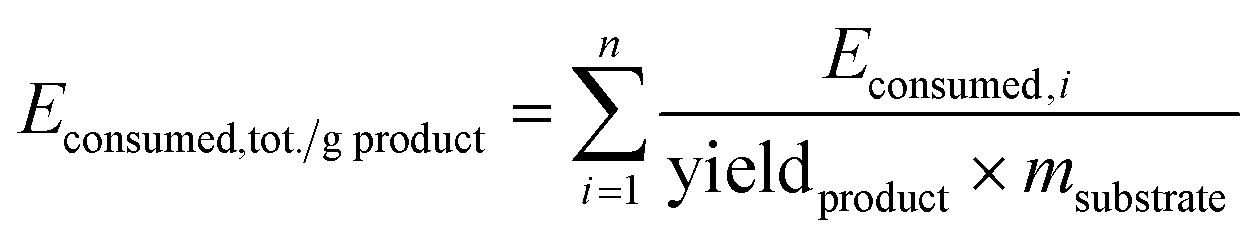 | (3) |
The experimental Econsumed,tot./g EO data showed that MWHD and US-MWHD presented more than 30% energy savings compared to the HD. Better results were observed by the extraction with IL3. We obtained energy savings of 18%, 35% and 46% for IL3-HD, IL3-MWHD and IL3-USMWHD compared with the HD, respectively.
Compared to the conventional HD, the use of ILs alone led to a reduction of 18.39% in the carbon footprint of the process, while MW alone reduced the emitted kg of CO2 by 28.74%. The combination of these extraction methods led to a reduction of as much as 34.48% in terms of carbon footprint. US-MWHD led to a similar reduction of IL3-MWHD, while the use of the three extraction methods combined (IL3-US-MWHD) showed the best carbon footprint reduction profile. The reduction in the emitted kg of CO2 accounted for over 45% compared to HD.
Safety and operation considerations and industrial scalability
The proposed technology enables a continuous-flow to be constructed or a useful batch extractor for industrial applications. A number of MW dipoles (each emitting up to 1 kW at 2450 MHz) can be placed inside the reactor of whatever shape, volume and wall material, at a few centimeters from each other, thus enabling the operator to control both the MW power locally applied and the temperature distribution.In addition, an array of US emitters can be placed in the same reactor, thereby obtaining a simultaneous MW and US activation. The presence of the US-emitting titanium horn has a very small influence on the MW emission inside the reactor, due to the fact that the MW electric field radiated by the dipole coaxial antenna is nearly perpendicular to the axis of the perturbing metal rod. This configuration ensures safe working conditions, without sparks or discharges, with a very good control of the MW absorbed by the sample. It also ensures that a consistent volume of sample is simultaneously submitted to the high power MW and US.
Another advantage of using ILs in plant extractions is the possibility of working at temperatures higher than 100 °C. A variety of ILs with a high thermal stability and with onset temperatures ranging up to 400 °C have been summarized in the literature.49 In principle these ILs could be used to perform extractions at temperatures well above the water boiling point inside a non-pressurized reactor.
Conclusions
In the present study, the suitability of MWHD and US-MWHD approaches using ILs/water mixtures as extraction media to obtain a completely comparable product (in terms of EO composition) with higher yields and in a more sustainable and less expensive (in terms of costs and time) way has been demonstrated. The quality of the obtained essential oils is also stable and comparable to the conventional extraction method. MW applied without US led to the extraction of EOs richer in oxygenated compounds in comparison with other methods. The MW and US applications also enriched the EO bouquet of small relative abundances of other chemical classes of VOCs (i.e. sesquiterpenes) which are not detected in the unimplemented extractions.The experimental temperature profiles of ILs and ILs/water mixtures, showing their higher MW absorption compared to water, highlight the use of ILs as green solvents, which are well matched with the MW and US technology in intensifying sustainable extraction processes.
The proposed technology enables a continuous-flow to be constructed or a useful batch reactor for industrial applications. The use of ILs with a high thermal stability and onset temperatures could enable extraction processes to be performed at temperatures well above the water boiling point inside a non-pressurized reactor.
Acknowledgements
This work was supported by FIRB 2012 (No. RBFR12ETL5), funded by the Italian Ministry of University and Research.Notes and references
- K.-H. Kubeczka, in Handbook of Essential oils, Science, technology and applications, ed. K. Hüsnü Can Başer and G. Buchbauer, Taylor and Francis Group, LLC, Boca Raton FL., 1st edn, 2010, pp. 3–28 Search PubMed.
- C. Franz and J. Novak, in Handbook of Essential oils, Science, technology and applications, ed. K. Hüsnü Can Başer and G. Buchbauer, Taylor and Francis Group, LLC, Boca Raton FL., 1st edn, 2010, pp. 39–82 Search PubMed.
- N. S. Iacobellis, P. Lo Cantore, F. Capasso and F. Senatore, J. Agric. Food Chem., 2005, 53, 57–61 CrossRef CAS PubMed.
- J. González-Rivera, C. Duce, V. Ierardi, I. Longo, A. Spepi, M. R. Tiné and C. Ferrari, ChemistrySelect, 2017, 2, 2131–2138 CrossRef.
- U. N. Industrial Development Organization and Food and Agriculture Organization, Herbs, spices and essential oils. Post-harvest operations in developing countries, 2005 Search PubMed.
- E. Schmidt, in Handbook of Essential oils, Science, technology and applications, ed. K. Hüsnü Can Başer and G. Buchbauer, Taylor and Francis Group, LLC, Boca Raton FL., 1st edn, 2010, pp. 83–120 Search PubMed.
- H. Hajlaoui, H. Mighri, E. Noumi, M. Snoussi, N. Trabelsi, R. Ksouri and A. Bakhrouf, Food Chem. Toxicol., 2010, 48, 2186–2192 CrossRef CAS PubMed.
- Y. Li, A.-S. Fabiano-Tixier and F. Chemat, in Green Chemistry for Sustainability, Springer International Publishing, Cham, 2014, pp. 9–21 Search PubMed.
- F. Chemat, M. E. Lucchesi, J. Smadja, L. Favretto, G. Colnaghi and F. Visinoni, Anal. Chim. Acta, 2006, 555, 157–160 CrossRef CAS.
- G. Flamini, M. Tebano, P. L. Cioni, L. Ceccarini, A. S. Ricci and I. Longo, J. Chromatogr. A, 2007, 1143, 36–40 CrossRef CAS PubMed.
- G. Wenqiang, L. Shufen, Y. Ruixiang, T. Shaokun and Q. Can, Food Chem., 2007, 101, 1558–1564 CrossRef.
- J. M. Roldán-Gutiérrez, J. Ruiz-Jiménez and M. D. Luque de Castro, Talanta, 2008, 75, 1369–1375 CrossRef PubMed.
- M.-T. Golmakani and K. Rezaei, Food Chem., 2008, 109, 925–930 CrossRef CAS PubMed.
- M. A. Vian, X. Fernandez, F. Visinoni and F. Chemat, J. Chromatogr. A, 2008, 1190, 14–17 CrossRef CAS PubMed.
- J. González-Rivera, J. Tovar-Rodríguez, E. Bramanti, C. Duce, I. Longo, E. Fratini, I. R. Galindo-Esquivel and C. Ferrari, J. Mater. Chem. A, 2014, 2, 7020 Search PubMed.
- J. Gonzalez Rivera, C. Duce, D. Falconieri, C. Ferrari, L. Ghezzi, A. Piras and M. R. Tiné, Innovative Food Sci. Emerging Technol., 2016, 33, 308–318 CrossRef CAS.
- J. Gonzalez-Rivera, A. Spepi, C. Ferrari, C. Duce, I. Longo, D. Falconieri, A. Piras and M. R. Tiné, Green Chem., 2016, 18, 6482–6492 RSC.
- I. A. Saleh, M. Vinatoru, T. J. Mason, N. S. Abdel-Azim, E. A. Aboutabl and F. M. Hammouda, Ultrason. Sonochem., 2016, 31, 330–336 CrossRef CAS PubMed.
- M. Gericke, P. Fardim and T. Heinze, Molecules, 2012, 17, 7458–7502 CrossRef PubMed.
- J. Hoffmann, M. Nüchter, B. Ondruschka and P. Wasserscheid, Green Chem., 2003, 5, 296–299 RSC.
- G. Flamini, B. Melai, L. Pistelli and C. Chiappe, RSC Adv., 2015, 5, 69894–69898 RSC.
- L. Pistelli, S. Giovanelli, P. Margari and C. Chiappe, RSC Adv., 2016, 6, 52421–52426 RSC.
- U. Domańska and R. Bogel-Łukasik, J. Phys. Chem. B, 2005, 109, 12124–12132 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- E. Stenhagen, S. Abrahamsson and F. W. McLafferty, Registry of Mass spectral data, Wiley & Sons, New York, NY, 1974 Search PubMed.
- Y. Masada, Analysis of essential oils by gas chromatography and mass spectrometry, John Wiley & Sons, Inc., New York, NY, 1976 Search PubMed.
- W. Rödel, Nahrung, 1982, 26, 830 CrossRef.
- N. W. Davies, J. Chromatogr., 1990, 503, 1–24 CrossRef CAS.
- R. P. Adams, Identification of essential oil components by gas chromatography/quadrupole mass spectroscopy, Allured Publishing Corporation, Carol Stream, Illinois, USA, 1995 Search PubMed.
- S. Milojevic, D. Radosavljevic, V. Pavicevic, S. Pejanovic and V. Veljkovic, Hem. Ind., 2013, 67, 843–859 CrossRef CAS.
- M. E. Lucchesi, F. Chemat and J. Smadja, Flavour Fragrance J., 2004, 19, 134–138 CrossRef CAS.
- L. Jirovetz, G. Buchbauer, A. S. Stoyanova, E. V. Georgiev and S. T. Damianova, Int. J. Food Sci. Technol., 2005, 40, 305–310 CrossRef CAS.
- V. D. Zheljazkov, A. Gawde, C. L. Cantrell, T. Astatkie and V. Schlegel, PLoS One, 2015, 10, 0144120 Search PubMed.
- H. B. Sowbhagya, B. V. Sathyendra Rao and N. Krishnamurthy, J. Food Eng., 2008, 84, 595–600 CrossRef.
- S. H. Beis, N. Azcan, T. Ozek, M. Kara and K. H. C. Baser, Chem. Nat. Compd., 2000, 36, 265–268 CrossRef CAS.
- R. Li and Z. T. Jiang, Flavour Fragrance J., 2004, 19, 311–313 CrossRef CAS.
- I. Bettaieb, S. Bourgou, J. Sriti, K. Msaada, F. Limam and B. Marzouk, J. Sci. Food Agric., 2011, 91, 2100–2107 CrossRef CAS PubMed.
- I. Bettaieb Rebey, I. Jabri-Karoui, I. Hamrouni-Sellami, S. Bourgou, F. Limam and B. Marzouk, Ind. Crops Prod., 2012, 36, 238–245 CrossRef CAS.
- N. Hashemian, A. G. Pirbalouti, M. Hashemi, A. Golparvar and B. Hamedi, Aust. J. Crop Sci., 2013, 7, 1752–1760 Search PubMed.
- A. Jordan and N. Gathergood, Chem. Soc. Rev., 2015, 44, 8200–8237 RSC.
- R. Ferreira, H. Garcia, A. F. Sousa, M. Petkovic, P. Lamosa, C. S. R. Freire, A. J. D. Silvestre, L. P. N. Rebelo and C. S. Pereira, New J. Chem., 2012, 36, 2014 RSC.
- H. Garcia, R. Ferreira, M. Petkovic, J. L. Ferguson, M. C. Leitão, H. Q. N. Gunaratne, K. R. Seddon, L. P. N. Rebelo and C. Silva Pereira, Green Chem., 2010, 12, 367 RSC.
- A. C. Metaxas and R. J. Meredith, Industrial microwave heating (IEE Power Series), P. peregrinus on behalf of the Institution of Electrical Engineers, London, 1993 Search PubMed.
- J. Robinson, S. Kingman, D. Irvine, P. Licence, A. Smith, G. Dimitrakis, D. Obermayer and C. O. Kappe, Phys. Chem. Chem. Phys., 2010, 12, 4750–4758 RSC.
- J. González-Rivera, I. R. Galindo-Esquivel, M. Onor, E. Bramanti, I. Longo and C. Ferrari, Green Chem., 2014, 16, 1417 RSC.
- C. Chiappe and C. S. Pomelli, Phys. Chem. Chem. Phys., 2013, 15, 412–423 RSC.
- B. Mennucci, E. Cancès and J. Tomasi, J. Phys. Chem. B, 1997, 101, 10506–10517 CrossRef CAS.
- D. K. Babi, M. Sales Cruz and R. Gani, in Process Intensification in Chemical Engineering, ed. J. G. Segovia-Hernández and A. Bonilla-Petriciolet, Springer International Publishing, 1st edn, 2016, pp. 7–30 Search PubMed.
- J. L. Anderson, R. Ding, A. Ellern and D. W. Armstrong, J. Am. Chem. Soc., 2005, 127, 593–604 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00075h |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2017 |