
Robust, non-fouling liters-per-day flow synthesis of ultra-small catalytically active metal nanoparticles in a single-channel reactor†
Wai Kuan
Wong
a,
Swee Kun
Yap
 a,
Yi Chen
Lim
a,
Saif A.
Khan
*a,
Frédéric
Pelletier
b and
Elena Cristina
Corbos
b
a,
Yi Chen
Lim
a,
Saif A.
Khan
*a,
Frédéric
Pelletier
b and
Elena Cristina
Corbos
b
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: saifkhan@nus.edu.sg
bJohnson Matthey Technology Centre, Blounts Court, Sonning Common, Reading RG4 9NH, UK
First published on 30th June 2017
Abstract
In this communication, we demonstrate the robust, non-fouling continuous synthesis of catalytically active palladium nanoparticles using a triphasic segmented flow in a hybrid milli–meso flow reactor, which not only allows us to completely eliminate fouling over extended operational duration, but also allows the achievement of ∼10 L per day volumetric productivity in a single-channel reactor. From the synthesis perspective, we select the harshest challenge for this demonstration – the aqueous flow synthesis of metal nanoparticles using the strong, gas-evolving reducing agent sodium borohydride. We also present comparative evaluations of the catalytic activities of flow-synthesized nanoparticles compared to their batch counterparts in a model hydrogenation reaction to highlight the consistency and quality of the nanoparticles produced by the scaled-up flow synthesis.
There is tremendous interest in the synthesis and application of ultra-small metal nanoparticles and nanoclusters (<5 nm in size) in diverse areas ranging from plasmonics to biosensors and chemical catalysis due to their fascinating, size-dependent physical and chemical properties.1–4 The translation of their tremendous potential into large-scale, high-impact applications crucially hinges on the availability of advanced manufacturing methods that enable robust, high-volume production with tightly controlled particle properties such as size and composition. Flow chemistry methods for the synthesis of metal-based nanomaterials have been extensively explored over the last decade, and while numerous demonstrations of various flow reactor strategies have been reported, there is no decisive demonstration of robust, high-volume synthesis on the liters per day scale yet.5–26 This is due to two interrelated reasons – the inherent challenge in scaling up small flow reactors (by parallelization), especially for flow chemistries that yield solids, and the crucial challenge of fouling, which leads to irreversible particle deposition on the channel walls, with dramatic losses in the reactor's productivity within minutes for the case of metal nanoparticles. In this work, we select the harshest challenge from this perspective – the aqueous flow synthesis of ultra-small metal nanoparticles using strong, gas-evolving reducing agents like sodium borohydride – to demonstrate a general, operationally simple flow methodology for robust, non-fouling synthesis of catalytically active palladium nanoparticles on an ∼10 L per day scale. To do this, we use a triphasic segmented flow in a hybrid milli–meso flow reactor, which not only allows us to completely eliminate fouling over extended operational duration, but also allows the achievement of ∼10 L per day productivity in a single-channel reactor.
Fig. 1(a) is a schematic of our flow reactor setup and Fig. 1(b)–(d) are photographs of the assembled system in operation (see the ESI† for a video of the reactor operation). The aqueous palladium precursor is co-injected with a small amount of fluorinated oil into a Y-junction, at which a core-annular flow is produced within a 0.5 mm ID PTFE tube. Next, an inert gas (nitrogen) is injected into this system at a T-junction to form a three-phase segmented flow (Fig. 1(e)). Finally, the reducing agent (aqueous sodium borohydride) is injected at a second downstream T-junction leading to a 3 m long PTFE tube of 1 mm ID, and this aqueous stream periodically merges with the train of precursor droplets arriving at the junction (Fig. 1(b) and (f)). The function of the inert, fluorinated oil is to lubricate the aqueous slugs, thereby preventing contact with the walls and subsequent fouling, as has been well demonstrated in previous works.8,17 The inert nitrogen gas injected into the system as bubbles serves as a sink for the molecular hydrogen evolved from the rapid decomposition of the added sodium borohydride,27 thereby preventing it from rapidly crossing the solubility threshold within the aqueous slug, leading to the uncontrolled generation of hydrogen bubbles and fouling within minutes, as seen in both single- and bi-phasic flows in such systems (see ESI† Fig. S1 and S2 and accompanying discussion for this comparison). Finally, after the rapid nucleation of palladium nanoparticles downstream of the second T-junction for ∼10 s, the droplets are led into a 9 m long PTFE tube with a larger cross-section (1.6 mm ID), where the nucleated nanoparticles are allowed to age and grow to their final size for ∼80 s. A continuous low-flow rate stream of fluorinated oil is also injected at the transition between the small and large tubes at the Y-junction (Fig. 1(c)) to ensure complete wetting of the channel walls, thereby preventing fouling in this second stage. This second ‘meso-scale’ section of the reactor serves two important purposes – it retains a high degree of convection and mixing within the aqueous slugs, while also allowing reactor operation at high flow rates and low pumping pressures. In effect, the first reactor section provides the requisite intensified mixing for rapid nanoparticle nucleation, as evidenced by the rapid induction of black color in the flowing droplets after the addition of the reducing agent, while the second section of the reactor allows the nucleated nanoparticles to age and consume the added precursor, at high flow rates that would have resulted in prohibitive pumping pressures when the entire reactor is composed of tubes of 1 mm cross-section. A highly stable, regular flow is obtained, with nearly monodisperse aqueous slug length distributions before and after the addition of the reducing agent and after the small-to-large tube transition (Fig. 2(a)–(c)). Overall, this reactor is able to stably operate at an aqueous flow rate of ∼300 mL per hour, with an extremely well-controlled flow pattern.
 | ||
| Fig. 1 (a) Schematic of the triphasic segmented flow reactor for ultra-small nanoparticle synthesis: a core-annular flow is first formed at a Y-junction, followed by the injection of nitrogen gas into the first T-junction to form a triphasic segmented flow and the subsequent introduction of a strong reducing agent into the second T-junction to initiate the nucleation and growth of nanoparticles. Photographs of (b) the triphasic flow after the injection of the reducing agent, (c) the transition between the reactor tubes of different cross-sectional dimensions (an additional fluorinated oil stream is injected into the reactor through the Y-junction to ensure complete wetting downstream) and (d) the triphasic flow downstream of the transition between reactor tubes. High-speed stereomicroscopy images of (e) the triphasic flow after the addition of nitrogen gas, (f) the triphasic flow in the microreactor after the injection of aqueous sodium borohydride, highlighting stable and undisrupted flow even with minor gas hold-up in the aqueous slugs, and (g) the triphasic flow in the 1.6 mm ID reactor tube. | ||
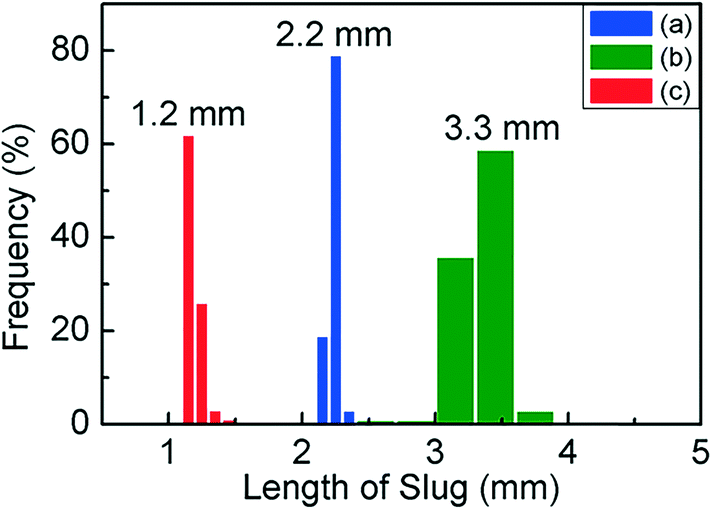 | ||
| Fig. 2 Histograms of aqueous slug length distributions: (a) prior to the addition of the reducing agent, (b) after the addition of the reducing agent, and (c) after the small-to-large reactor tube transition. | ||
Next, we characterize the palladium nanoparticles (PdNPs) produced from this reactor in terms of their size and catalytic activity and compare them to those produced by the equivalent small-scale (10 mL) batch synthesis carried out under optimal conditions. The PdNPs synthesized using the flow reactor were of a smaller average size (∼2.3 ± 0.3 nm) compared to the PdNPs produced in the batch synthesis (∼3.4 ± 0.9 nm), as seen in Fig. 3(a) and (b). The relative difference in mixing efficiencies of the two methods is a key factor underlying this difference in outcome. The volume of each aqueous droplet within the flow reactor is only ∼2.5 μL, which is 3 orders of magnitude smaller than the volume of the batch reactor (10 mL); this significantly smaller reaction volume in the flow reactor, when coupled with the advective mixing within the droplets,28,29 enables mixing within an estimated time of ∼0.15 s,30 which is two orders of magnitude smaller than the estimated mixing time of ∼10 s in the batch synthesis.31 Rapid, near-instantaneous mixing implies a sharp burst of Pd atom nucleation, leading to a smaller average size of PdNPs compared to the batch-synthesized PdNPs. Next, since PdNPs display exceptionally good catalytic properties in hydrogenation and dehydrogenation reactions, the catalytic activity of the synthesized PdNPs was assessed using a recently developed triphasic flow reactor for hydrogenation reactions,32,33 which enables the determination of the catalytic activity of PdNPs under minimal mass transfer limitations. In this reactor, as shown in Fig. 4, an organic–aqueous segmented flow was first formed at a T-junction before it was directed into a second T-junction where hydrogen gas was injected to obtain a triphasic flow consisting of alternating slugs of aqueous PdNPs and long hydrogen gas bubbles coated with a continuous, thin organic substrate film. The latter allows highly intensified diffusive transport of hydrogen and the organic substrate to the liquid–liquid interface surface, where the reaction occurs, as described by Yap et al.33 The catalytic activities of batch- and flow-synthesized PdNPs for 1-hexene hydrogenation and isomerization were ∼6.2 ± 1.1 molhexane molPdNPs−1 s−1 and ∼11.9 ± 0.7 molhexane molPdNPs−1 s−1, respectively. As expected, the flow-synthesized PdNPs are almost twice as active compared to the PdNPs produced in batch synthesis due to their smaller average NP size, hence providing a higher effective surface area to catalyse the reaction. Finally, to highlight the exceptionally controlled and robust operation of this flow reactor, we present results from an extended run of ∼6 hour duration in Fig. 5, in which we characterized the nanoparticle sizes and assessed the catalytic activity hourly. Approximately 1.8 L of aqueous 4 mM PdNP solution was produced during the 6 hour run, with a mean nanoparticle size of ∼2.2 ± 0.5 nm and a narrow particle size distribution for the sample assessed every hour (Fig. 5(a)); all seven samples displayed a consistent catalytic activity of ∼12.1 ± 1.1 molhexane molPdNPs−1 s−1 (Fig. 5(b)). After the 6 hour run, no deposition of nanoparticles on the reactor wall was observed. We note that the six hour duration does not represent a limit after which the process becomes unstable and/or fouling occurs; we have reused the reactor tubes more than five times for the extended-duration synthesis of Pd-nanoparticles with no noticeable effects on the outcome.
 | ||
| Fig. 3 (a) Particle size distribution and TEM image for batch-synthesized PdNPs with an average size of 3.4 ± 0.9 nm. (b) Particle size distribution and TEM image of flow-synthesized PdNPs with an average size of 2.3 ± 0.3 nm, highlighting smaller mean size and narrower particle size distribution compared to those produced using the small-scale batch reactor. | ||
 | ||
| Fig. 4 Schematic of the triphasic segmented flow reactor for hydrogenation reactions to investigate the catalytic activity of PdNPs under minimal mass transfer limitations.32 | ||
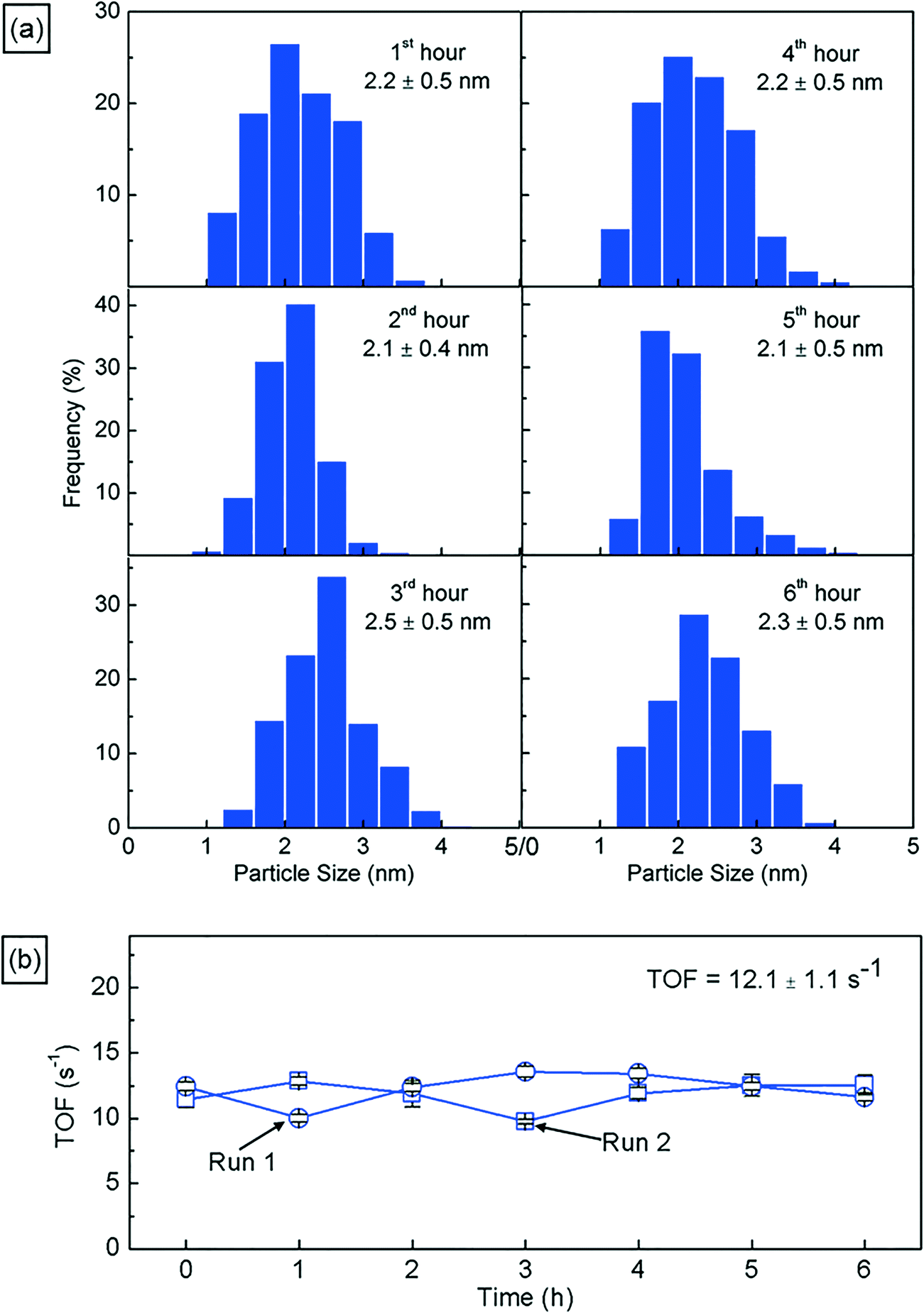 | ||
| Fig. 5 (a) Particle size distributions of the PdNP sample assessed hourly during a 6 hour production run, highlighting the consistency in the particle size for prolonged operation. (b) Catalytic activities of PdNPs in terms of the turnover frequency (TOF) for samples assessed hourly from two separate 6 hour nanoparticle production runs, highlighting the consistency in the quality of PdNPs over prolonged operation (see the ESI† for further information). | ||
In summary, we have demonstrated a flow chemistry strategy for the synthesis of metal nanoparticles that not only completely eliminates the all-pervasive problem of fouling, but also enables high-volume production within a single-channel system. This is enabled by the combination of a triphasic segmented flow on the one hand and a two-stage ‘milli–meso’ scale design on the other. We envision that this methodology can be applied directly to synthesize other ultra-small metallic nanoparticles and nanoclusters of other metals, such as Au, Ag, Pt, Rh and Ru and their bi/trimetallic mixtures, which involves the usage of strong reducing agents. Even further scale-up can easily be envisioned for the 100 L per day scale production with modest eight-fold parallelization of such reactor systems,14,34 which can likely meet or exceed the demand for their application in specialty and fine chemical industries, as well as the emerging biosensing and plasmonics fields.
Experimental
Materials
Octadecafluorodecahydronaphthalene, PFD (95%), 1H,1H,2H,2H-perfluoro-1-octanol, PO (97%), palladium(II) chloride (99%), 1 N hydrochloric acid and sodium borohydride (98%) from Sigma Aldrich Co. Ltd., Singapore and polyvinylpyrrolidone, PVP (M.W. = 40k Da), from Alfa Aesar were all used as-received without any further purification. Ultrapure water (18.2 MΩ at 25 °C) was obtained from a Milli-Q purifier.PdNP syntheses in flow and batch reactors
2 g of palladium(II) chloride was first dissolved in 22.5 mL of 1 N hydrochloride acid to form a metal complex in an aqueous form, dihydrogen tetrachloropalladate(II) (H2PdCl4). The aqueous Pd(II) precursor solution consists of 18 mL of H2PdCl4 solution, 500.1 mL of 1.8 M PVP solution and 981.9 mL of ultrapure water. Fluorinated oil was prepared by mixing 10% v/v PO in PFD. 48 mM aqueous sodium borohydride solution was prepared with ice cold water and its reservoir and fluidic damper were submerged in a cold ice water bath to suppress the decomposition of sodium borohydride. Peristaltic pumps were used to pump the Pd(II) precursor solution and sodium borohydride solution from the reagent reservoir through an inline hydraulic damper into the reactor system as shown in Fig. 1(a) at ∼3.3 mL min−1 and ∼1.7 mL min−1. The hydraulic damper of the Pd(II) precursor solution consists of 15 cm long, 4.76 mm ID Viton tube sandwiched between two 40 cm long, 0.5 mm ID PTFE tubes. The hydraulic damper of the aqueous sodium borohydride solution is composed of a 30 cm long, 254 μm ID fluorinated ethylene propylene (FEP) tube, a 10 cm long, 4.76 mm ID Viton tube and a 30 cm long, 254 μm ID FEP tube assembled in series. Since the flow rate of fluorinated oil is sufficiently low, it was delivered at 210 μl min−1 using two syringe pumps into two individual Y-junctions as shown in Fig. 1(a). Nitrogen gas was introduced into the reactor system through the first T-junction from a cylinder equipped with a two-stage pressure regulator through polyether ether ketone (PEEK) tubing (1 m long, 1 mm ID and 7.5 cm long, 100 μm ID) followed by a 30 cm long, 1 mm ID polytetrafluoroethylene (PTFE) tube. Subsequently, the resultant segmented flow entered a 3 m long, 1 mm ID PTFE tube with 8 bends (90°, 3 cm per side) to promote intra-slug mixing. The flowing three-phase mixture exited the reactor and immediately phase-separated into layered aqueous and oil phases in the collection vessel; the PdNP solution could be separated from the fluorinated oil by simple decantation. After the reactor reached an operating steady state, 10 ml samples were collected for further analysis. High-speed stereomicroscopic imaging (Basler piA640-210gm) at 200 frames per second was used to visualize the flow for flow stability and regularity assessment. The aqueous PdNP solution obtained by decantation was stored under refrigeration and diluted from 4 mM to 1 mM before its usage in the hydrogenation of 1-hexene, as noted below.For the batch synthesis of PdNPs, 7.294 mL of ultrapure water, 80 μL of H2PdCl4 solution and 2.222 mL of 16.7 mM PVP solution were pipetted into a 30 mL glass bottle with a magnetic stirrer. The solution was stirred at 1200 rpm to mix the precursor solution. After that, sodium borohydride solution was added rapidly into the glass bottle. The product was left to stir for 15 minutes before transferring it into a 50 mL centrifuge tube.
To assess the catalytic activity of PdNPs, the batch- and flow-synthesized palladium nanoparticles were diluted to their respective concentration prior to use in the hydrogenation flow experiment. 10% v/v 1-hexene in cyclohexane and diluted PdNP solution (1 mM) were infused into the first T-junction of the millifluidic reactor at 10 μL min−1 and 20 μL min−1 respectively by using separate syringe pumps. Subsequently, the resultant biphasic flow entered a 10 cm long (1 mm ID) PTFE tube. Hydrogen gas was introduced into the reactor through the second T-junction from a cylinder equipped with a two-stage pressure regulator through a series combination of a 1 m long (1 mm ID) PTFE tube, a 24.5 cm long (65 μm ID) PEEK tube and a 50 cm (1 mm ID) PTFE tube. The resultant triphasic flow entered a 2 m long (1 mm ID) PTFE tube. The reactor was allowed to run for 30 minutes to achieve a steady state and samples were collected over 20 minutes. The organic phase in the mixture from the reactor outlet was analyzed via gas chromatography (Shimadzu 2010Plus). To prepare samples for TEM imaging, the collected sample was diluted 100 times with ultrapure water by mixing 10 μL of sample with 990 μL of ultrapure water. A drop of this diluted sample was placed onto a 200 mesh copper grid, which was dried overnight and analysed using TEM (JEOL 2010, accelerating voltage 200 kV). Using ImageJ, the PdNP diameter and size distribution were determined from at least 500 particles.
References
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- J. F. Hainfeld, D. N. Slatkin and H. M. Smilowitz, Phys. Med. Biol., 2004, 49, N309–N315 CrossRef CAS PubMed.
- S. Hrapovic, Y. Liu, K. B. Male and J. H. Luong, Anal. Chem., 2004, 76, 1083–1088 CrossRef CAS PubMed.
- N. Yan, C. Xiao and Y. Kou, Coord. Chem. Rev., 2010, 254, 1179–1218 CrossRef CAS.
- S. Biswas, J. T. Miller, Y. Li, K. Nandakumar and C. S. Kumar, Small, 2012, 8, 687–698 CrossRef CAS PubMed.
- V. S. Cabeza, S. Kuhn, A. A. Kulkarni and K. F. Jensen, Langmuir, 2012, 28, 7007–7013 CrossRef PubMed.
- S. Duraiswamy and S. A. Khan, Small, 2009, 5, 2828–2834 CrossRef CAS PubMed.
- S. Duraiswamy and S. A. Khan, Nano Lett., 2010, 10, 3757–3763 CrossRef CAS PubMed.
- L. Gomez, V. Sebastian, S. Irusta, A. Ibarra, M. Arruebo and J. Santamaria, Lab Chip, 2014, 14, 325–332 RSC.
- H. Jun, T. Fabienne, M. Florent, P. E. Coulon, M. Nicolas and S. Olivier, Langmuir, 2012, 28, 15966–15974 CrossRef CAS PubMed.
- Y. H. Kim, L. Zhang, T. Yu, M. Jin, D. Qin and Y. Xia, Small, 2013, 9, 3462–3467 CrossRef CAS PubMed.
- A. Knauer, A. Csáki, F. Möller, C. Hühn, W. Fritzsche and J. M. Köhler, J. Phys. Chem. C, 2012, 116, 9251–9258 CAS.
- S. E. Lohse, J. R. Eller, S. T. Sivapalan, M. R. Plews and C. J. Murphy, ACS Nano, 2013, 7, 4135–4150 CrossRef CAS PubMed.
- A. M. Nightingale, J. H. Bannock, S. H. Krishnadasan, F. T. F. O'Mahony, S. A. Haque, J. Sloan, C. Drury, R. McIntyre and J. C. deMello, J. Mater. Chem. A, 2013, 1, 4067 CAS.
- M. T. Rahman, P. G. Krishnamurthy, P. Parthiban, A. Jain, C. P. Park, D. P. Kim and S. A. Khan, RSC Adv., 2013, 3, 2897 RSC.
- V. Sebastian, C. D. Smith and K. F. Jensen, Nanoscale, 2016, 8, 7534–7543 RSC.
- I. Shestopalov, J. D. Tice and R. F. Ismagilov, Lab Chip, 2004, 4, 316–321 RSC.
- M. Thiele, A. Knauer, A. Csáki, D. Mallsch, T. Henkel, J. M. Köhler and W. Fritzsche, Chem. Eng. Technol., 2015, 38, 1131–1137 CrossRef CAS.
- L. Zhang, G. Niu, N. Lu, J. Wang, L. Tong, L. Wang, M. J. Kim and Y. Xia, Nano Lett., 2014, 14, 6626–6631 CrossRef CAS PubMed.
- A. Singh, C. K. Malek and S. K. Kulkarni, Int. J. Nanosci., 2010, 09, 93–112 CrossRef CAS.
- Y. Ying, G. Chen, Y. Zhao, S. Li and Q. Yuan, Chem. Eng. J., 2008, 135, 209–215 CrossRef CAS.
- A. M. Karim, N. Al Hasan, S. Ivanov, S. Siefert, R. T. Kelly, N. G. Hallfors, A. Benavidez, L. Kovarik, A. Jenkins, R. E. Winans and A. K. Datye, J. Phys. Chem. C, 2015, 119, 13257–13267 Search PubMed.
- M. T. Rahman and E. V. Rebrov, Processes, 2014, 2, 466–493 CrossRef.
- J. Wagner, T. Kirner, G. Mayer, J. Albert and J. M. Köhler, Chem. Eng. J., 2004, 101, 251–260 CrossRef CAS.
- S. Sharada, P. L. Suryawanshi, R. P. Kumar, S. P. Gumfekar, T. B. Narsaiah and S. H. Sonawane, Colloids Surf., A, 2016, 498, 297–304 CrossRef CAS.
- M. Luty-Błocho, M. Wojnicki, J. Grzonka and K. J. Kurzydłowski, Arch. Metall. Mater., 2014, 59, 509–512 Search PubMed.
- S. A. Khan and S. Duraiswamy, Lab Chip, 2012, 12, 1807–1812 RSC.
- H. Song, D. L. Chen and R. F. Ismagilov, Angew. Chem., Int. Ed., 2006, 45, 7336–7356 CrossRef CAS PubMed.
- H. Song, J. D. Tice and R. F. Ismagilov, Angew. Chem., Int. Ed., 2003, 42, 768–772 CrossRef CAS PubMed.
- M. R. Bringer, C. J. Gerdts, H. Song, J. D. Tice and R. F. Ismagilov, Philos. Trans. R. Soc., A, 2004, 362, 1087–1104 CrossRef CAS PubMed.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- S. K. Yap, Y. Yuan, L. Zheng, W. K. Wong, J. Zhang, N. Yan and S. A. Khan, Green Chem., 2014, 16, 4654–4658 RSC.
- S. K. Yap, Y. Yuan, L. Zheng, W. K. Wong, N. Yan and S. A. Khan, J. Flow Chem., 2014, 4, 200–205 CrossRef.
- S. K. Yap, W. K. Wong, N. X. Y. Ng and S. A. Khan, Chem. Eng. Sci., 2016, 169, 117–127 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00072c |
| This journal is © The Royal Society of Chemistry 2017 |