Continuous flow synthesis of poly(acrylic acid) via free radical polymerisation
Received
12th May 2017
, Accepted 14th July 2017
First published on 14th July 2017
Abstract
The free radical polymerisation of aqueous solutions of acrylic acid (1) has been studied using a continuous flow reactor to quickly screen reaction parameters such as temperature, residence time, monomer- and initiator concentration. The experimental data sets produced established a theoretical basis for conducting scale up processes to efficiently produce larger quantities of poly(acrylic acid) delivered with good control over the molecular weight and dispersity.
1 Introduction
Since the early 1990s there has been a steady growth within both academia and industry with regards to the use of flow reactors for the synthesis of chemical compounds due to the increase in reaction control afforded.1–7 In general, flow chemical synthesis offers several advantages over batch chemistry. For example, the reactors provide excellent heat transfer meaning reaction temperatures can be rapidly changed including accessing different sequentially linked temperature zones or facilitating super heating of solvents within an easily pressurised reactor. Mixing within flow channels can also be additionally enhanced generating more consistent reaction domains leading to better control and improvements in yield and purity. Automation of flow reactions enables better process control expedited via direct in-line analysis which can also be used in reaction feedback loops (RFL) and design of experiment (DoE) routines.8–16 Additional aspects of enhanced safety and the flexibility to conduct multi-step syntheses through integrated processing sequences leading to more complex chemical architectures are also advantageous.17,18 It should however be acknowledged that flow chemistry also has some drawbacks. The key issues are often associated with high investment costs in both equipment and training; difficulties in compensating for varying kinetics, dilution effects and the compatibility of the potentially different solvents in continuous multi-step sequences. Problems can also be encountered relating to precipitation of intermediates/product or increases in viscosity during reactions. In addition having to process sufficient material to achieve steady-state operation can use up valuable resources. Although these aspects can cause processing difficulties increasingly they are being shown to be less restrictive if planned for in advance as part of the flow process.17–22
Although flow chemistry has been predominantly the domain of chemists and engineers working on the preparation of small molecular weight compounds it has recently started to gain more traction in the synthesis of macromolecular systems.23–28 Indeed, several types of polymerisation have already been shown to benefit from the application of various flow processing techniques which have been comprehensively summarised in a few recommended review articles.29–33 Considering all the potential polymerisation methods, free radical polymerisation is of particular interest from an industrial stand-point. A major virtue of free radical polymerisation is that it can be typically carried out under relatively undemanding conditions, allows for a wide range of monomers to be used and exhibits a high tolerance to stabilisers which are often present in the monomers.34 However, control over the molar mass distributions (MMD) in this type of polymerisation is harder to achieve requiring much more precise regulation of the reaction parameters. Therefore generating a new free radical derived polymer necessitates time intensive screening of the various reaction parameters, ensuring consistency of the polymerisation process and enabling the targeting of specific molecular weights. A solution to this problem may be found in the use of flow chemistry as this technique has repeatedly proven its strength in efficiently evaluating reaction parameters in a fast serial screening mode. Consequently the aim of this research was to screen and then define flow processing conditions to access various target molecular weight polymers which could then be reproducibly processed in a continuous mode to prepare larger quantities of material.
Poly(acrylic acid) was selected for study as it has been extensively researched and its behaviour is well known yet it presents several challenges in its synthesis.35–38 The acrylic acid monomer (1) is highly reactive,39 which raises two problems. First, due to the high polymerisation rate and exothermic nature of the process, heat evolution is an issue requiring careful regulation to control the reaction progress. This is necessary for a safe synthesis and to avoid gel formation. Secondly the final product inherently has a very high molar mass. Indeed, to obtain poly(acrylic acid) with a low molar mass, a low concentration of acrylic acid (1) is prepared which results in large batch volumes upon scale-up.40 Additionally, diffusion is an important phenomenon in free radical polymerisation.41 This aspect has different names in each mechanistic step, such as the cage effect for the initiation, glass effect for propagation and Tromsdorff or gel effect for termination, however at each stage good control over diffusion is necessary to achieve well defined polymers. It was anticipated that here again the advantages associated with flow processing relating to heat transfer, mixing and continuous operation would provide advantages creating an improved synthesis.
2 Experimental
2.1 Materials
Acrylic acid (1) (Alfa Aesar, 99%), 2,2′-azobis(2-methylpropionamidine) dihydrochloride (2) (Sigma Aldrich, 97%), sodium selenite (Alfa Aesar, 98%), 1,2-dimethoxyethane (DME, Alfa Aesar, 99% stabilized with BHT) and deionised D2O were used without further purification. The flow polymerisation was carried out on a FlowSyn (Uniqsis, Shepreth, United Kingdom), a reactor system available from Uniqsis Ltd.
2.2 Polymerisation
Stock solutions of acrylic acid (1) and initiator (2) were prepared in deionised water at known concentrations (Table 1). It is well known that, under free radical reaction conditions in concentrated homogeneous mixtures, gel formation can occur as soon as the conversion becomes high enough. It is also generally understood that the viscosity of reaction medium will constantly increase with increasing conversion.42 This can potentially become an issue using flow processing techniques where significant increases in viscosity followed by gel formation must be avoided to prevent clogging of the reactor. To limit the potential risks from clogging the maximum molecular weight and limiting monomer concentration were not researched. The maximum concentration of used monomer in this set-up was restricted to 1.0 mM.
Table 1 Screening parameters for acrylic acid (1) polymerisation
| Temperature (°C) |
Acrylic acid (mM) (1) |
Initiator (mol%) (2) |
Residence time (min) |
| 70 |
0.4, 0.7, 1.0 |
1.25, 2.50, 3.75 |
5, 10, 20, 30 |
| 80 |
0.4, 0.7, 1.0 |
1.25, 2.50, 3.75 |
5, 10, 20, 30 |
| 90 |
0.4, 0.7, 1.0 |
1.25, 2.50, 3.75 |
5, 10, 20, 30 |
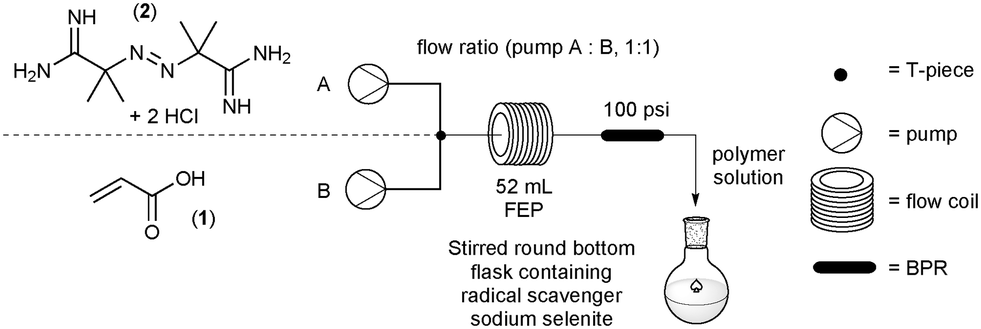
The two stock solutions were pumped through a FlowSyn reactor using the two independently controlled HPLC pumps, channel A and B respectively. The flow rates on each channel were always maintained at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and were adjusted to produce different residence times for the reactions. A PEEK cross assembly (1.30 mm thru hole and 22.8 μL swept volume fitted with a pressure transducer – obtained from Uniqsis Ltd., Shepreth, United Kingdom) was placed in-line to combine the two flows into a single homogeneous stream. The flow path was configured so that the channel A and B entered laterally and the mixed flow exited at right angles, which then passed into a 52 mL FEP coil which could be heated at different temperatures. A back pressure regulator (BPR, 100 psi) was placed at the exit to the coil reactor. The exiting solution of poly(acrylic acid) was collected in a stirred flask containing sodium selenite (0.005 mM) as a radical quencher to terminate any radical species and prevent further polymerisation (Fig. 1).
:1 ratio and were adjusted to produce different residence times for the reactions. A PEEK cross assembly (1.30 mm thru hole and 22.8 μL swept volume fitted with a pressure transducer – obtained from Uniqsis Ltd., Shepreth, United Kingdom) was placed in-line to combine the two flows into a single homogeneous stream. The flow path was configured so that the channel A and B entered laterally and the mixed flow exited at right angles, which then passed into a 52 mL FEP coil which could be heated at different temperatures. A back pressure regulator (BPR, 100 psi) was placed at the exit to the coil reactor. The exiting solution of poly(acrylic acid) was collected in a stirred flask containing sodium selenite (0.005 mM) as a radical quencher to terminate any radical species and prevent further polymerisation (Fig. 1).
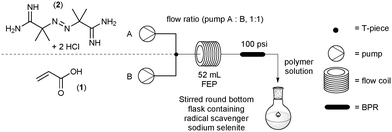 |
| | Fig. 1 Flow scheme for the polymerisation of acrylic acid (1). | |
2.3 Characterisation
For each transformation the monomer conversion was determined by 1H NMR, spectra were recorded using water suppression on either a Bruker-Avance 400 or Varian VNMRS-600 instrument. A 10% by volume aliquot of D2O was added in order to create a signal lock. Conversion was calculated based upon the determination of residual acrylic acid monomer in the sample at a known concentration. The technique used was based on the Watergate43 suppression technique as described by Morris et al.44,45 Although this technique is a powerful method to measure water rich samples it also has drawbacks. Bleaching of signals near water, saturation of exchangeable NH protons (for presaturation) and a tilted base line created by a large dispersive tail of the water signal need be taken into consideration when analysing the results. For these reasons an internal standard (DME) which was not affected by the water suppression was chosen to calibrate the conversion of acrylic acid (1). Gel permeation chromatography (GPC) analysis of the poly(acrylic acid) samples was performed using a Viscotek GPC max 2001 triple-detection system in aqueous solution (0.05 mol L−1 NaNO3, 2.81 mmol−1 NaOH and MeOH [ratio 4:1] using 2 × A6000M + guard column set (all purchased from Malvern)). The column and detector temperature was 50.00 °C, flow rate was 1.0000 mL min−1, injection volume was 50 μL and volume increment was 0.00333 mL.
2.4 Analysis
The conversion of monomer into polymer was determined by adding DME as an internal standard (either 5 mM or 10 mM) to the monomer stock solution. The standard used to assess the GPC system prior to each run was PEO (22 kDa, dn/dc (ml g−1) 0.1320, dispersity 1.045). For the GPC data outlying values were eliminated based upon the processed data (OmniSEC 4.7 software was used to build method and process data). To determine the average molecular weight and dispersity a minimum, of two measurements were used. Extreme outlaying data points relating to molecular weight and dispersity were removed from the data-set if these values deviated more than 10% from the mean value.
3 Results and discussion
Using the specified flow setup described above (Fig. 1) rapid screening of up to ten different test conditions could be conducted in a single working day (8 h), generating values for monomer conversion with associated dispersity and molecular weight range. The full data set represented by Table 1 was performed to generate a comprehensive profile of the polymerisation reaction in flow.
3.1 Initiator
At the conclusion of a conventional batch polymerisation of acrylic acid (1) it is common to add an extra dose of initiator to the reaction mixture to ensure any residual monomer is fully consumed. This avoids the post reaction requirement for expensive and time consuming sequestration of the monomer from the polymer solution but inevitably leads to a broadening of the dispersity. For this reason achieving full conversion is an important aim with it being most desirable to obtain full conversion of the monomer by exacting control over the individual reaction parameters. In free radical polymerisation it is assumed that the initiator activation follows first order kinetics. Therefore its half-life time is dependent on the reaction temperature. High temperature will initialise higher average radical concentrations. The higher the initiator content, the shorter the polymers that will be formed as the monomer concentration will not be sufficient to form long chains. In our study three initiator concentrations were selected; 1.25, 2.50 and 3.75 mol% of the monomer concentration. The upper limit was fixed at 3.75 mol% to avoid any potential problems resulting from clogging of the flow system through gel formation. Conversely the lower limit was also set as too low a value of initiator results in poor propagation due to the cage effect. This would negatively affect the polymerisation as the initiator would not be consumed optimally. This was considered particularly important for the short residence times we were targeting in this flow system.
3.2 Conversion
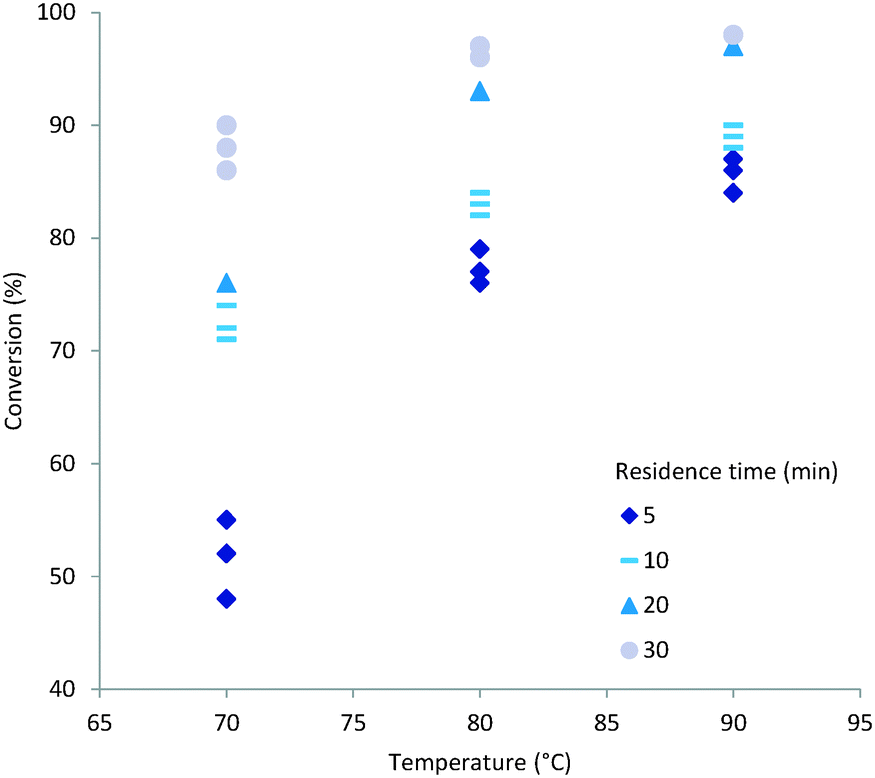
Both temperature and residence time contribute significantly to the overall conversion of acrylic acid (1). In general a high conversion could be reached at longer residence times (Fig. 2). To obtain good conversion at shorter residence times (i.e. 5 min) elevated temperatures were required. A drawback of elevated temperatures is however that extensive backbiting within the polymer will occur.46 As can be seen from the plots (Fig. 2) a secondary contributing effect on conversion was the amount of initiator (2) used. For low monomer concentration and high initiator ratio nearly full conversion was reached (Fig. 3). It was also shown that polymerisations at lower temperatures could also reach full conversion using high initiator input and low monomer concentrations. A direct correlation between the amount of initiator (2) and temperature could be rationalised via the half-life of the initiator. The half-life of initiator (2) is ten hours at 56 °C.47 The initiators fragmentation is expected to follow the Arrhenius equation and therefore the half-life at 80 °C would be in the order of 28 min. If the reaction residence time is below twice the half-life time of initiator (2) full conversion is hard to achieve.
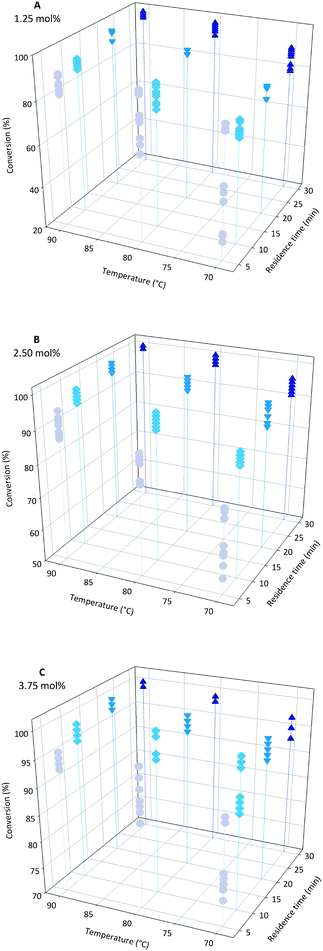 |
| | Fig. 2 A plot of conversion (%) against temperature (°C) and residence time (min) at various initiator (2) concentrations, A: 1.25 mol% of initiator (2), B: 2.50 mol% of initiator (2), C: 3.75 mol% of initiator (2). | |
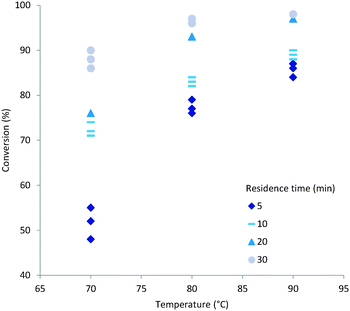 |
| | Fig. 3 Conversion (%) versus temperature (C) using high acrylic acid (1) concentration (1 mM) and low initiator (2) (1.25 mol%) input. | |
3.3 Molecular weight
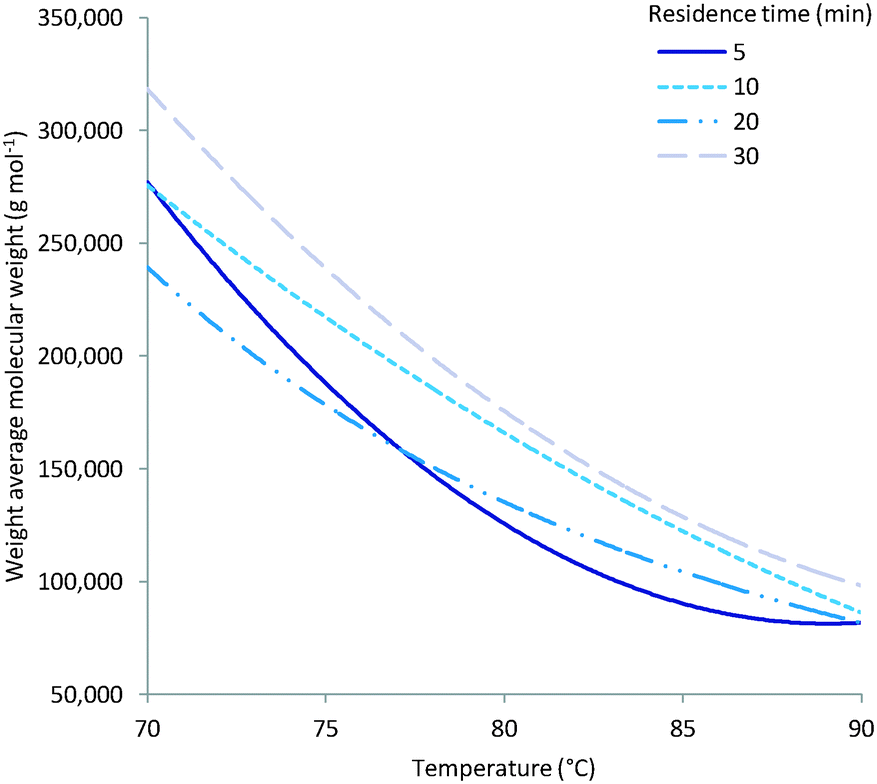
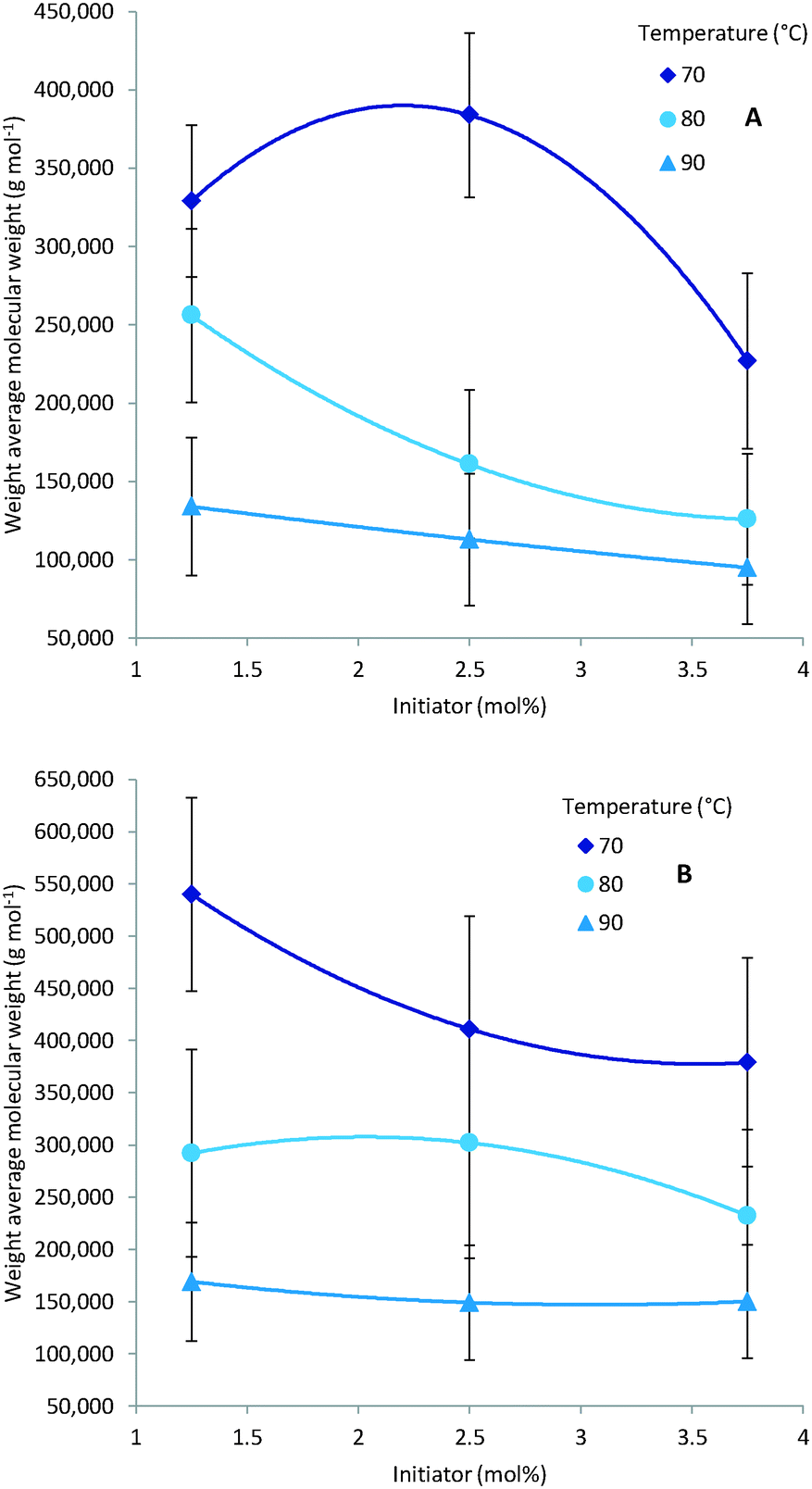
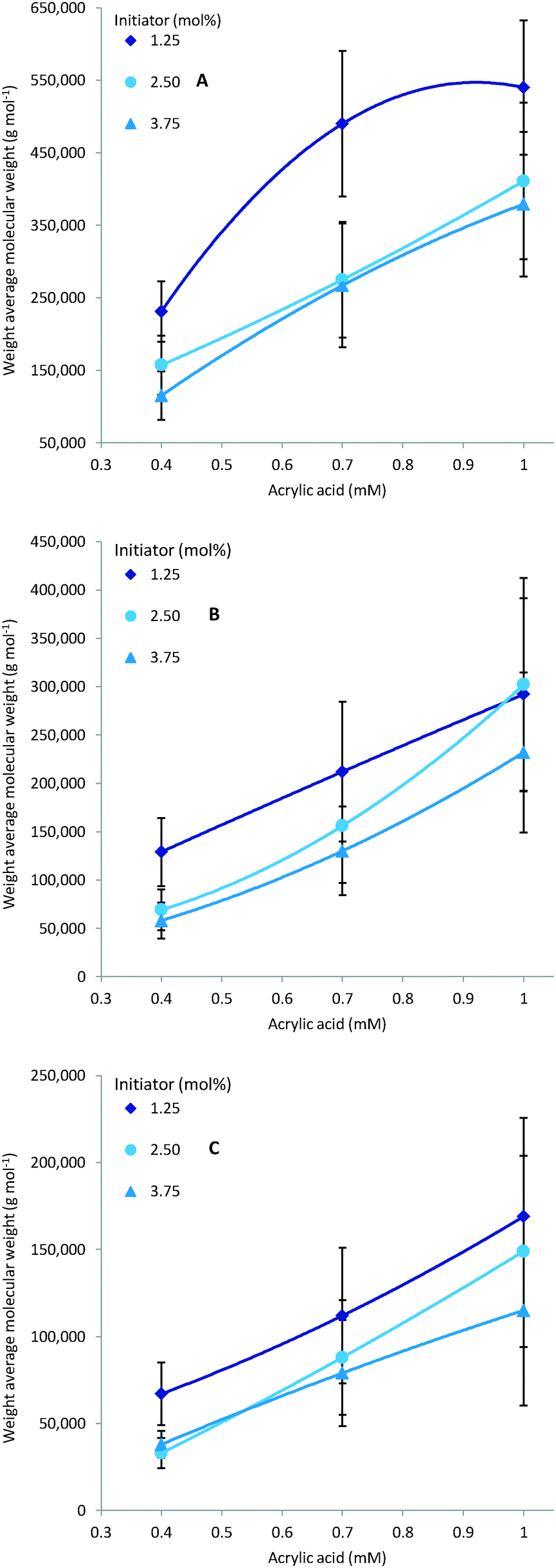
As expected multiple parameters influence the resulting molecular weight of the polymers synthesised. A general decrease in molecular weight was observed as the reaction temperature increased which can be ascribed to a corresponding increase in the concentration of radical species (Fig. 4). Consequently the amount of initiator added also greatly affects the molecular weight with a higher initial initiator concentration resulting in a higher concentration of initiator radicals and therefore multiple competing polymerisation events and a lower final molecular weight (Fig. 5).48 This effect is more pronounce at the higher temperatures evaluated as the initiator half-life time is shorter and so the systematic changes in starting initiator concentration are seen more significantly.
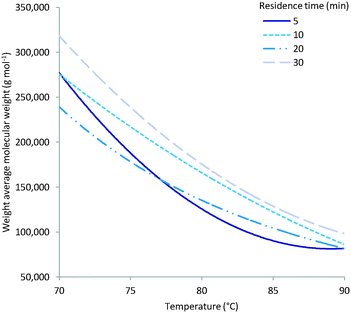 |
| | Fig. 4 Experimentally observed decrease of molecular weight over increase of temperature. | |
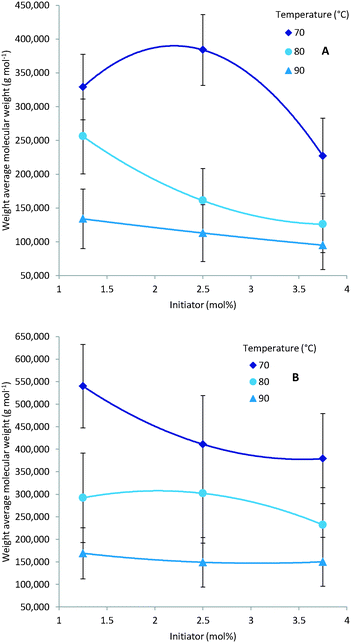 |
| | Fig. 5 GPC date: decrease of molecular weight for increase of initiator, the acrylic acid (1) concentration is kept constant at 1.0 mM with A: 5 min residence time and B: 30 min residence time. Other residence times and acrylic acid (1) concentrations show similar trends. | |
The final parameter that impacts the molecular weight is the monomer feed concentration. High concentrations results in longer chains, as there is sufficient monomer for extended propagation. The temperature has a similar effect on the molecular weight in relation to monomer concentration. The molecular weight will increase for low temperatures and high monomer concentration (Fig. 6A) but the relative difference is the same for the different monomer concentrations (Fig. 6).
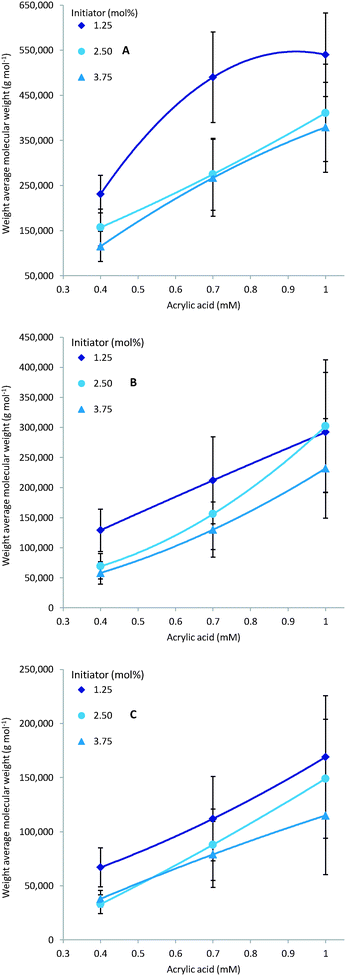 |
| | Fig. 6 Molecular weight increases for higher monomer feed. Lower temperature will increase the molecular weight. Residence time is 30 minutes, A: polymerisation at 70 °C, B: polymerisation at 80 °C and C: polymerisation at 90 °C. | |
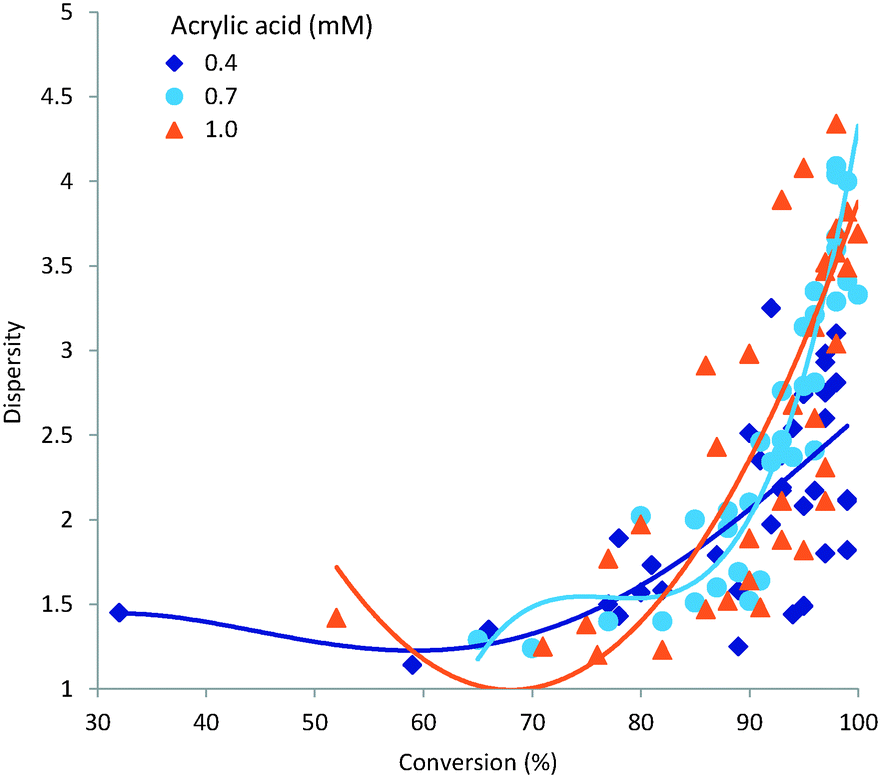
3.4 Dispersity
A narrow dispersity is generally desirable as it indicates a more homogeneous polymeric sample. The major factors influencing dispersity in our study were demonstrated to be residence time and processing temperature. These two variables also strongly influence the overall conversion; a plot of sample dispersity against conversion (%) is shown in Fig. 7. As can be seen obtaining a high monomer conversion whilst also maintaining a low dispersity is not readily achievable. This is understandable when acknowledging that steady state conditions for free radical polymerisation are arrived at very rapidly, orders of magnitude below the residence times used in this study. Therefore the rate of initiation will be equitable to that of termination during the entire reaction; assuming no limitation imposed by the initiator concentration or initiator half-life at the concentrations, residence times or temperatures investigated which was not the case. Consequently the dispersity of the polymer sample will steadily increase during the polymerisation process leading to a general broadening of dispersity following a classical Schulz–Flory distribution. In this respect a more progressed reaction – higher conversion of monomer – will generally give a larger dispersity.
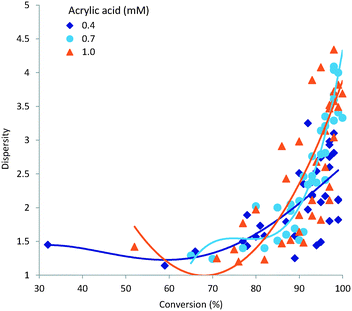 |
| | Fig. 7 Influence of conversion on dispersity. | |
3.5 Target molecular weight
Having performed a systematic evaluation of the parameters in flow we endeavoured to bring this knowledge together in a predictive fashion in order to target under optimal conditions a set of defined Mw polymers possessing narrow dispersity. Consequently, randomly selected target molecular weights from within three arbitrary groupings representing low (80000–200000 g mol−1), medium (210000–350000 g mol−1) and large (360000–500000 g mol−1) polymers were defined. The predicted conditions were derived from a 3 × 3 × 4 Full Factorial Design and Least Square Fit model using JMP Pro 12.1.0 software optimised for molecular weight. The randomly generated values of 120172, 311133 and 448542 g mol−1 along with the predicted parameters to synthesise the target polymers are given in Table 2. Upon performing the reactions under the defined reaction conditions analysis via GPC indicated a reasonable and reproducible correlation with a difference from predicted of 1.5% for low, 6.7% for medium and 5.9% for high molecular weight. Unfortunately, as anticipated the obtained dispersities were considerably higher than our basic molecular weight model predicted (Table 2).
Table 2 Parameters targeted molecular weights and obtained results
| Target Mw (g mol−1) |
Predicted conditions |
Obtained results |
| Temperature (°C) |
Residence time (min) |
Acrylic acid (1) (mM) |
Initiator (2) (mol%) |
M
w (g mol−1) |
Dispersity |
M
w (g mol−1) |
Dispersity |
| 120000 |
87 |
19.5 |
0.764 |
2.60 |
120000 |
3.22 |
118000 |
6.69 |
| 310000 |
73 |
10.0 |
0.785 |
1.26 |
311000 |
1.48 |
333000 |
2.43 |
| 450000 |
76 |
29.0 |
1.100 |
1.27 |
449000 |
2.10 |
477000 |
7.53 |
Throughout this investigation we have shown that this seemingly simple transformation is still a challenging synthetic endeavour. The difficulty of successfully predicting two desirable yet in-congruent outputs; molecular weight and dispersity, through an array of interlinked reaction parameters has not been achieved. However, through this work we have been able to identify from a standard DoE matrix (108 data points run in duplicate) the general trending characteristics for flow polymerisation of acrylic acid (sections 3.1–3.4). We now hope to use this data driven understanding to create a advanced model which incorporates additional Simplex algorithms to automatically suggest further optimization and refinement experiments leading to a closer match of the molecular weight and simultaneously an improvement in dispersity.
4 Conclusions
A range of conditions for the aqueous polymerisation of acrylic acid (1) under continuous flow conditions have been studied. The influence on polymer molecular weight and dispersity has been profiled against reaction parameters of temperature, initiator/monomer concentrations and residence time. The use of the flow reactor allowed the rapid iterative screening of multiple reactions generating large quantities of data on relatively small reaction samples; this would be of particular value when investigating more valuable monomers. As a proof of principle the data produced was analysed to identify potential reaction conditions aimed at delivering targeted molecular weight polymers. Although the basic model used gave reasonable accuracy with regards to molecular weight, the associated dispersities were far from ideal. It is, however, anticipated that further rounds of optimisation would allow improvement of the model and its predictive capabilities. We believe this approach therefore offers significant opportunities to research laboratories engaged in the discovery of new polymeric materials.
Acknowledgements
We are greatful for financial support by Unilever and EPSRC (RG64908 to LB) and the Royal Society (UF130576 to IRB).
References
- L. Malet-Sanz and F. Susanne, J. Med. Chem., 2012, 55, 4062–4098 CrossRef CAS PubMed.
- D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384–6389 CrossRef CAS PubMed.
- R. M. Myers, D. E. Fitzpatrick, R. M. Turner and S. V. Ley, Chem. – Eur. J., 2014, 20, 12348–12366 CrossRef CAS PubMed.
- S. Byrn, M. Futran, H. Thomas, E. Jayjock, N. Maron, R. F. Meyer, A. S. Myerson, M. P. Thien and B. L. Trout, J. Pharm. Sci., 2015, 104, 1520–6017 CrossRef PubMed.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef CAS PubMed.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS.
- T. Rohe, W. Becker, A. Krey, H. Nägele, S. Kölle and N. Eisenreich, J. Near Infrared Spectrosc., 1998, 6, 325–332 CrossRef CAS.
- C. Jamieson, M. S. Congreve, D. F. Emiabata-Smith and S. V. Ley, Synlett, 2000, 1603–1607 CAS.
- S. V. Ley and I. R. Baxendale, Nat. Rev. Drug Discovery, 2002, 1, 573–586 CrossRef CAS PubMed.
- H. Wikström, I. R. Lewis and L. S. Taylor, Appl. Spectrosc., 2005, 59, 934–941 CrossRef PubMed.
- C. F. Carter, H. Lange, S. V. Ley, I. R. Baxendale, B. Wittkamp, J. G. Goode and N. L. Gaunt, Org. Process Res. Dev., 2010, 14, 393–404 CrossRef CAS.
- T. Brodmann, P. Koos, A. Metzger, P. Knochel and S. V. Ley, Org. Process Res. Dev., 2012, 16, 1102–1113 CrossRef CAS.
- P. Filipponi, C. Ostacolo, E. Novellino, R. Pellicciari and A. Gioiello, Org. Process Res. Dev., 2014, 18, 1345–1353 CrossRef CAS.
- N. Holmes, G. R. Akien, R. J. D. Savage, C. Stanetty, I. R. Baxendale, A. J. Blacker, B. A. Taylor, R. L. Woodward, R. E. Meadows and R. A. Bourne, React. Chem. Eng., 2016, 1, 96–100 CAS.
- C. A. Hone, N. Holmes, G. R. Akien, R. A. Bourne and F. L. Muller, React. Chem. Eng., 2017, 2, 103–108 CAS.
- I. R. Baxendale, J. Chem. Technol. Biotechnol., 2013, 88, 519–552 CrossRef CAS.
- I. R. Baxendale, L. Brocken and C. J. Mallia, Green Process. Synth., 2013, 2, 211–230 CAS.
- D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680 RSC.
- L. F. Tietze and D. Liu, ARKIVOC, 2008, viii, 193–210 Search PubMed.
- T. Tsubogo, H. Oyamada and S. Kobayashi, Nature, 2015, 520, 329–332 CrossRef CAS PubMed.
-
Continuous-Flow Chemistry in the Research Laboratory, ed. T. Glasnov, Springer International Publishing, Cham, 1st edn, 2016, ISBN 978-3-319-32196-7 Search PubMed.
- E. Baeten, J. J. Haven and T. Junkers, Polym. Chem., 2017, 8, 3815–3824 RSC.
- N. Zhu, Y. Liu, W. Feng, W. Huang, Z. Zhang, X. Hu, Z. Fang, Z. Li and K. Guo, Eur. Polym. J., 2016, 80, 234–239 CrossRef CAS.
- X. Hu, N. Zhu, Z. Fang, Z. Li and K. Guo, Eur. Polym. J., 2016, 80, 177–185 CrossRef CAS.
- A. Chemtob, A. Rannée, L. Chalan, D. Fischer and S. Bistac, Eur. Polym. J., 2016, 80, 247–255 CrossRef CAS.
- S. Zhu, Y. Lu, K. Wang and G. Luo, Eur. Polym. J., 2016, 80, 219–226 CrossRef CAS.
- J. Gardiner, C. H. Hornung, J. Tsanaktsidis and D. Guthrie, Eur. Polym. J., 2016, 80, 200–207 CrossRef CAS.
- C. A. Serra and Z. Chang, Chem. Eng. Technol., 2008, 31, 1099–1115 CrossRef CAS.
- F. Bally, C. A. Serra, V. Hessel and G. Hadziioannou, Macromol. React. Eng., 2010, 4, 543–561 CrossRef CAS.
- F. Bally, C. A. Serra, V. Hessel and G. Hadziioannou, Chem. Eng. Sci., 2011, 66, 1449–1462 CrossRef CAS.
- C. Tonhauser, A. Natalello, H. Löwe and H. Frey, Macromolecules, 2012, 45, 9551–9570 CrossRef CAS.
- N. Chan, M. F. Cunningham and R. A. Hutchinson, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3081–3096 CrossRef CAS.
-
The Chemistry of Radical Polymerization, ed. G. Moad and D. H. Solomon, Elsevier Science Ltd, Amsterdam, 2nd edn, 2005 Search PubMed.
- L. Qiu, K. Wang, S. Zhu, Y. Lu and G. Luo, Chem. Eng. J., 2016, 284, 233–239 CrossRef CAS.
- J. Spěváček, M. Suchopárek and S. Al-Alawi, Polymer, 1995, 36, 4125–4130 CrossRef.
- Y. Hu, X. Jiang, Y. Ding, H. Ge, Y. Yuan and C. Yang, Biomaterials, 2002, 23, 3193–3201 CrossRef CAS PubMed.
- J. Loiseau, N. Doërr, J. M. Suau, J. B. Egraz, M. F. Llauro, C. Ladavière and J. Claverie, Macromolecules, 2003, 36, 3066–3077 CrossRef CAS.
- M. Buback, M. Egorov, R. G. Gilbert, V. Kaminsky, O. F. Olaj, G. T. Russell, P. Vana and G. Zifferer, Macromol. Chem. Phys., 2002, 203, 2570–2582 CrossRef CAS.
- S. S. Cutié, P. B. Smith, D. E. Henton, T. L. Staples and C. Powell, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 2029–2047 CrossRef.
- R. J. Minari, G. Caceres, P. Mandelli, M. M. Yossen, M. Gonzalez-Sierra, J. R. Vega and L. M. Gugliotta, Macromol. React. Eng., 2011, 5, 223–231 CrossRef CAS.
- G. A. O'Neil, M. B. Wisnudel and J. M. Torkelson, AIChE J., 1998, 44, 1226–1231 CrossRef.
- M. Liu, X. an Mao, C. Ye, H. Huang, J. K. Nicholson and J. C. Lindon, J. Magn. Reson., 1998, 132, 125–129 CrossRef CAS.
- R. W. Adams, C. M. Holroyd, J. A. Aguilar, M. Nilsson and G. A. Morris, Chem. Commun., 2013, 49, 358–360 RSC.
- J. A. Aguilar and S. J. Kenwright, Analyst, 2016, 141, 236–242 RSC.
- L. Pichavant, C. Guillermain, D. Harakat and X. Coqueret, Eur. Polym. J., 2016, 80, 99–116 CrossRef CAS.
-
J. Brandrup, Polymer handbook, Wiley New York, 1999, vol. 89 Search PubMed.
- P. M. de la Torre, G. Torrado and S. Torrado, J. Biomed. Mater. Res., Part B, 2005, 72, 191–197 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a