A direct strategy to synthesize hybrid benzothiazole–carbamate moieties via O-acylation of phenols under metal–organic framework catalysis†
Received
17th May 2017
, Accepted 13th July 2017
First published on 13th July 2017
Abstract
Carbamates containing a benzothiazole moiety were synthesized via cross-dehydrogenative coupling of phenols with N,N-disubstituted formamides utilizing the metal–organic framework Cu-CPO-27 as a productive and recyclable heterogeneous catalyst. These hybrid structures would combine the benefits from both carbamates and benzothiazoles in terms of biological and pharmaceutical activities. The Cu-CPO-27 framework exhibited better catalytic efficiency for the synthesis of carbamates than various MOFs. It was feasible to recover and reuse the catalyst without an appreciable deterioration in catalytic efficiency. To the best of our knowledge, this is the first heterogeneous catalytic protocol towards the generation of a hybrid carbamate–benzothiazole skeleton via direct coupling reaction.
1. Introduction
Organic carbamates have fascinated researchers and have been strikingly considered in indispensable functional and structural designs found in innumerable pharmaceuticals, bioactive natural chemicals, and agrochemicals, as well as valued intermediates in the generation of diverse functional materials.1–6 Different carbamates were prepared from non-environmentally favorable chemicals, and hence, the synthetic strategy for these structures must be modified.1–4 Kanai et al. developed a [Cu(NCMe)4]BF4-catalyzed C(sp3)–H bond modification concerning the facile synthesis of tertiary carbamates.5 Buchwald et al. reported the generation of aryl carbamates by adding alcohols into the reaction of Pd2(dba)3-catalyzed coupling between sodium cyanate and aryl halides.6 Jiao and Ren demonstrated a PdCl2-catalyzed approach using alcohols, CO, and organoazides to produce various carbamates.7 Skrydstrup et al. synthesized aryl acyl carbamates via a four-component Pd(cod)Cl2-catalyzed conversion of aryl halides in the presence of alcohols and potassium cyanate.8 Chung et al. pointed out that carbamates could be generated from N-tosylhydrazones, amines, and carbon dioxide, assisted by a base.3 Lately, Patel and et al. published the first demonstration of a synthetic approach to generate carbamates from phenols having benzothiazole directing substituents and dialkylformamides.9 These skeletons would combine the benefits from both benzothiazole and carbamate moieties, in terms of the biological and pharmaceutical efficiency. To adapt to greener trends in the production of carbamates containing a benzothiazole moiety, heterogeneous catalysts are definitely required.
Metal–organic frameworks (MOFs) have been developed as an advanced series of crystalline materials with established porosity, assembled by coupling metal-containing units with organic linkers.16–21 The characteristics of MOFs could be tuned by using appropriate organic bridging ligands and metal cations of various oxidation states as well as varied coordination geometries.22–27 Despite the fact that the commercialization of MOFs still demands more endeavors, the potential applications of these materials in various fields have been substantially investigated.10–15 MOFs have been explored by the catalysis community, and the striking aspects of MOFs have been one of the interesting topics in this field.16–20 Generally, it might be feasible to introduce the catalytically active site either at each metal cation or at each organic linker in the framework, accordingly offering advantages as compared to conventional solid catalysts.16,21–23 Throughout the last 10 years, a diversity of organic reactions using MOF-based catalysts have been reported in the literature.24–30 Among numerous universal MOFs, copper-based frameworks showed high efficiency in many organic transformations. In this paper, we would like to describe a direct strategy to synthesize carbamates containing benzothiazole proportions by coupling of phenols with N,N-disubstituted formamides utilizing the Cu-CPO-27 metal–organic framework as a robust heterogeneous catalyst. To the best of our awareness, the formation of a hybrid carbamate–benzothiazole skeleton has not been previously conducted with solid catalysts.
2. Experimental
2.1. Materials and instrumentation
Chemicals were commercially obtained from Acros Organics and Sigma-Aldrich and were used as received without any further purification. A D8 Advance Bruker powder diffractometer was used to record X-ray powder diffraction (XRD) patterns with a Cu Kα radiation source. Thermogravimetric analysis (TGA) was conducted on a Universal V4.5A TA instrument with a heating rate of 10 °C min−1 under argon. A Micromeritics 2020 volumetric adsorption analyzer system was utilized for nitrogen physisorption measurements. Samples were pretreated by heating at 150 °C for 3 h under vacuum prior to analysis. The samples were subsequently cooled with liquid nitrogen and analyzed by measuring the volume of nitrogen adsorbed at specific pressures. Micropore surface areas were calculated by using the Brunauer–Emmett–Teller (BET) model.31 The pore size distribution was estimated by using the non-local Density Functional Theory (DFT) method.32,33 Scanning electron microscopy investigation was performed using an S4800 scanning electron microscope (SEM). A JEOL JEM 1400 transmission electron microscope (TEM) was employed for transmission electron microscopy studies at 80 kV. The Cu-CPO-27 samples were dispersed on holey carbon grids for TEM observation. Elemental analysis with inductively coupled plasma (ICP) was implemented using a Shimadzu ICPE-9000. A Nicolet 6700 instrument was utilized to record Fourier transform infrared (FT-IR) spectra, with samples dispersed on potassium bromide pallets.
A Shimadzu GC 2010-Plus equipped with a flame ionization detector (FID) and an SPB-5 column (length = 30 m, inner diameter = 0.25 mm, and film thickness = 0.25 μm) was used for gas chromatographic (GC) analyses. The temperature program for GC analysis held the samples at 100 °C for 1 min, heated them from 100 to 280 °C at 40 °C min−1 and held them at 280 °C for 5.5 min. Inlet and detector temperatures were set constant at 280 °C. GC yields were calculated based on diphenyl ether as an internal standard. GC-MS analyses were implemented using a Shimadzu GCMS-QP2010 Ultra with a ZB-5MS column (length = 30 m, inner diameter = 0.25 mm, and film thickness = 0.25 μm). The temperature program for GC-MS analysis held the samples at 50 °C for 2 min, heated them from 50 to 280 °C at 10 °C min−1 and held them at 280 °C for 10 min. The inlet temperature was set constant at 280 °C. The MS spectra were compared with the spectra gathered in the NIST library. A Bruker AV 500 spectrometer was utilized to record 1H NMR and 13C NMR spectra with the residual solvent peak as a reference.
2.2. Synthesis of the metal–organic framework Cu-CPO-27
In a representative experiment, Cu(NO3)2·3H2O (1.18 g, 4.88 mmol) was added to a conical flask holding H2dhtp (H2dhtp = 2,5-dihydroxyterephthalic acid; 0.44 g, 2.29 mmol). A solution of isopropanol in N,N′-dimethylformamide (50 ml, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16 v/v) was then introduced to the flask. The mixture was magnetically stirred for 15 min at ambient temperature to dissolve the solid completely. The clear solution was then dispensed into seven pressurized vials. The vials were firmly covered, stabilized on a tray, put into an oven, and heated at 85 °C for 18 h. Crystals were generated on the vial wall during the course of the heating. Subsequent to this step, the vials were cooled down to ambient temperature. The crystals were collected by decantation and washed with N,N′-dimethylformamide (3 × 10 ml). The product was subsequently submerged in isopropanol (3 × 10 ml) at ambient temperature to replace the non-volatile N,N′-dimethylformamide with isopropanol. The framework was then dried under vacuum at 150 °C for 6 h on a Schlenk line, producing 0.62 g of Cu-CPO-27 as reddish black crystals (60% yield with respect to 2,5-dihydroxyterephthalic acid).
:16 v/v) was then introduced to the flask. The mixture was magnetically stirred for 15 min at ambient temperature to dissolve the solid completely. The clear solution was then dispensed into seven pressurized vials. The vials were firmly covered, stabilized on a tray, put into an oven, and heated at 85 °C for 18 h. Crystals were generated on the vial wall during the course of the heating. Subsequent to this step, the vials were cooled down to ambient temperature. The crystals were collected by decantation and washed with N,N′-dimethylformamide (3 × 10 ml). The product was subsequently submerged in isopropanol (3 × 10 ml) at ambient temperature to replace the non-volatile N,N′-dimethylformamide with isopropanol. The framework was then dried under vacuum at 150 °C for 6 h on a Schlenk line, producing 0.62 g of Cu-CPO-27 as reddish black crystals (60% yield with respect to 2,5-dihydroxyterephthalic acid).
2.3. Catalytic reaction
In a representative catalytic experiment, a solution of 2-(benzo[d]thiazol-2-yl)phenol (0.1136 g, 0.5 mmol) in N,N′-dimethylformamide (1.5 ml, 19 mmol) was added into an 8 ml pressurized vial holding the catalyst and tert-butyl hydroperoxide (tBuOOH, 70% wt in water; 0.291 ml, 2.0 mmol) as an oxidant. The catalyst amount employed was based on the copper/2-(benzo[d]thiazol-2-yl)phenol mole proportion. The vial was heated at 100 °C for 60 min under air on a magnetic stirrer. After the reaction was complete, the mixture was cooled down to ambient temperature, and right after that, diphenyl ether (0.0851 g, 0.5 mmol) in its role as an internal standard was introduced. Samples were extracted, dissolved in ethyl acetate (3 ml), shaken energetically with anhydrous Na2SO4, and then analyzed by GC with respect to the internal standard. The product of the reaction, 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate, was isolated by column chromatography. 1H NMR, 13C NMR, and GC-MS analyses were also performed to confirm the structure of the product. In the catalyst recycling studies, after the reaction, the catalyst was collected from the mixture, washed with a large amount of N,N′-dimethylformamide and methanol, dried at 150 °C under vacuum in 6 h in a Schlenk line, and reused for new experiments.
3. Results and discussion
3.1. Catalyst synthesis and characterization
Cu-CPO-27 was prepared in 60% yield via the reaction of 2,5-dihydroxyterephthalic acid and copper nitrate trihydrate in a mixture of N,N′-dimethylformamide and isopropanol following a solvothermal approach.34–36 The copper-based framework was then characterized by traditional analysis protocols (Fig. S1–S7†). X-ray powder diffraction analysis showed an exceptionally sharp peak at 2θ of 6.9, revealing that a crystalline framework was produced (Fig. S1†). The SEM image revealed the formation of well-shaped crystals with particle sizes of 10–20 μm (Fig. S2†). The TEM image showed that a porous structure was generated in the framework, though its pore patterns were complicated (Fig. S3†). A pore diameter of less than 10 Å was recorded for the framework, demonstrating that the material should be microporous (Fig. S4†). Nitrogen adsorption–desorption isotherms at 77 K exhibited typical microporous type I isotherms (Fig. S5†). BET surface areas of up to 1100 m2 g−1 were obtained for the framework, which were close to the value observed by Calleja et al. (1125 m2 g−1).36 Thermal degradation of solid materials is essential since several applications would depend on their thermal stability. TGA observations denoted that the second mass loss would occur at 100 °C, probably due to the physisorbed moisture in the framework. The most notable characteristic of the TGA results was that the material was thermally stable up to over 300 °C (Fig. S6†). FT-IR spectra of the Cu-MOF exposed a noticeable difference as compared to that of 2,5-dihydroxyterephthalic acid, confirming the deprotonation of –COOH groups in the linker upon the reaction with copper cations (Fig. S7†).
3.2. Catalytic studies
The framework was utilized as a heterogeneous catalyst in the reaction of 2-(benzo[d]thiazol-2-yl)phenol with N,N′-dimethylformamide to generate 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate as the dominant product (Scheme 1). With this approach, a hybrid carbamate–benzothiazole skeleton could be produced in a catalytic quantity of the framework. Preliminary studies focused on the influence of temperature on the yield of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate. The reaction was performed for 60 min at 5 mol% catalyst, with 4 equivalents of tBuOOH in water, using 13 equivalents of N,N′-dimethylformamide, at ambient temperature, 60 °C, 80 °C, 100 °C, and 120 °C, respectively. The reaction could not occur at ambient temperature, and no proof of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate was recorded after 60 min. Elevating the temperature led to a considerable improvement in the carbamate yield, affording yields of 23% and 70% after 60 min for the reaction conducted at 60 °C and 80 °C, respectively. It was noticed that the reaction performed at 100 °C generated 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate in 75% yield after 60 min. Boosting the reaction temperature to higher than 100 °C was observed to be pointless as the yield of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate was not enhanced significantly (Fig. 1).
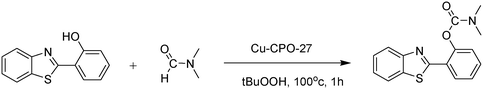 |
| | Scheme 1 The synthesis of carbamates containing a benzothiazole moiety utilizing the Cu-CPO-27 catalyst. | |
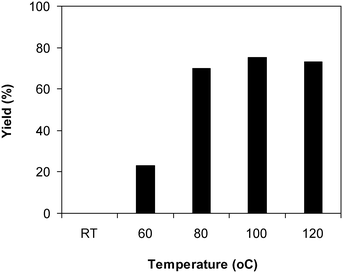 |
| | Fig. 1 Yields of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate at diverse temperatures. | |
One more feature that should be noted for the reaction between 2-(benzo[d]thiazol-2-yl)phenol and N,N′-dimethylformamide to produce 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate is the reactant mole proportion. In this reaction, an enormous excess of N,N′-dimethylformamide should be demanded in order to achieve high yields of the carbamate. The remaining N,N′-dimethylformamide also acted as medium for the reaction. In the first demonstration of the Cu(OAc)2-catalyzed production of carbamates from dialkylformamides and phenols, Patel et al. employed approximately 13 equivalents of N,N′-dimethylformamide for the conversion.9 The influence of the reactant mole proportion on the production of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate was accordingly assessed. The reaction was conducted at 100 °C for 60 min, in the presence of 5 mol% catalyst, with 4 equivalents of oxidant, using 3, 6.5, 13, 19, 25, 38, and 50 equivalents of N,N′-dimethylformamide, respectively. It was noticed that the reaction progressed with difficulty when 3 equivalents of N,N′-dimethylformamide were employed, though a yield of 41% 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate was recorded after 60 min. Increasing the quantity of N,N′-dimethylformamide led to a conspicuous improvement in the yield of the carbamate. Indeed, the reaction employing 13 equivalents of N,N′-dimethylformamide afforded a yield of 75% after 60 min. The yield of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate could be upgraded to 80% after 60 min for the reaction employing 19 equivalents of N,N′-dimethylformamide. Utilizing more than 19 equivalents of N,N′-dimethylformamide was noticed to be needless as the yield of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate was not enhanced considerably (Fig. 2).
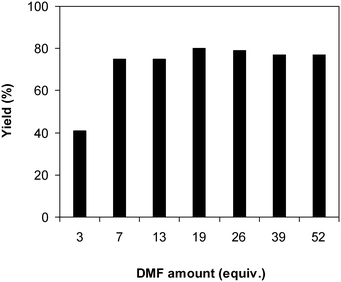 |
| | Fig. 2 Yields of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate at different reactant mole proportions. | |
Similar to other reactions via direct activation of C–H bonds, an oxidant must be used for the reaction of 2-(benzo[d]thiazol-2-yl)phenol with N,N′-dimethylformamide to generate 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate. In the first illustration of the Cu(OAc)2-catalyzed synthesis of carbamates from phenols and dialkylformamides possessing benzothiazole directing substituents, Patel and et al. explored several oxidants and pointed out that tBuOOH in water should be used for the conversion.9 We subsequently investigated the consequence of different oxidants on the generation of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate. The reaction was performed at 100 °C for 60 min, in the presence of 5 mol% catalyst, utilizing 19 equivalents of N,N′-dimethylformamide, with 4 equivalents of tBuOOH in water, tBuOOH in decane, tert-butyl perbenzoate, di-tert-butyl peroxide, cumyl hydroperoxide, dibenzoyl peroxide, potassium persulfate, respectively, as the oxidant. Potassium persulfate was observed to be entirely inert for the reaction, with no evidence of carbamates recorded after 60 min. Dibenzoyl peroxide and di-tert-butyl peroxide exhibited low performance, providing 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate in 16% and 30% yields, respectively, after 60 min. Among these oxidants, tBuOOH in water should be the oxidant of preference as the yield of the carbamate could be increased to 80% (Fig. 3). Additionally, the quantity of the oxidant also showed an impressive influence on the reaction, and the best yield of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate was recorded for the reaction utilizing 4 equivalents of tBuOOH in water. Furthermore, no product was observed after 60 min in the absence of aqueous tBuOOH, demonstrating the significance of the oxidant for the conversion (Fig. 4).
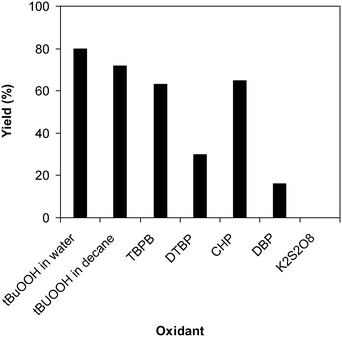 |
| | Fig. 3 Yields of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate with various oxidants. | |
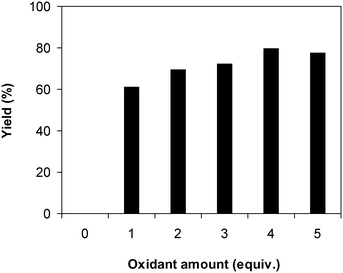 |
| | Fig. 4 Yields of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate at varied oxidant amounts. | |
Afterwards, we considered the consequence of catalyst concentration on the yield of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate in the reaction of 2-(benzo[d]thiazol-2-yl)phenol with N,N′-dimethylformamide. The reaction was conducted at 100 °C for 60 min, in the presence of 4 equivalents of oxidant, utilizing 19 equivalents of N,N′-dimethylformamide, at 0.5 mol%, 1 mol%, 3 mol%, 5 mol%, and 10 mol% catalyst, respectively. It must be mentioned that no evidence of a product was revealed in the absence of the catalyst, demonstrating the significance of the framework in the reaction. As anticipated, the presence of the catalyst generated a pronounced improvement in the production of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate, with a yield of 66% obtained after 60 min for the reaction utilizing 0.5 mol% catalyst. Increasing the catalyst concentration to 1 mol% resulted in a yield of 71% being recorded after 60 min. The yield of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate could be increased to 80% for the reaction employing 5 mol% catalyst. Utilizing more than 5 mol% catalyst was observed to be pointless as the yield of the carbamate was not increased any further (Fig. 5). Indeed, in the first illustration of the Cu(OAc)2-catalyzed synthesis of carbamates from phenols and dialkylformamide possessing benzothiazole directing substituents, Patel et al. screened the reaction conditions and demonstrated that the presence of 5 mol% Cu(OAc)2 led to the best yield being obtained for the conversion, and increasing the catalyst amount to 10 mol% did not considerably improve the yield of the product.9
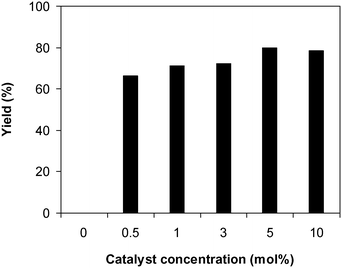 |
| | Fig. 5 Yields of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate at varied catalyst concentrations. | |
As the coupling of 2-(benzo[d]thiazol-2-yl)phenol with N,N′-dimethylformamide to form 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate using the framework catalyst was conducted in the liquid phase, the feasibility that some of the catalytically active sites on the solid catalyst could travel into the solution throughout the time of the experiment must be studied. In order to elucidate if copper species dissolved from the solid catalyst, if any, catalyzed the generation of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate, leaching assessment was then performed. Apparently, if 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate was still produced after the solid catalyst was isolated, the coupling reaction might not progress under actual heterogeneous catalysis conditions. The reaction was performed at 100 °C for 90 min, in the presence of 4 equivalents of oxidant, utilizing 13 equivalents of N,N′-dimethylformamide, at 5 mol% catalyst. The solid framework was separated from the reaction mixture after 5 min by centrifugation. The reaction solution was then transported to a new pressurized vial and magnetically stirred for an additional 85 min at 100 °C. The generation of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate, if any, was then monitored by GC as previously described. Experimental results revealed that no further 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate was generated in the absence of the solid catalyst (Fig. 6). It was accordingly suggested that the coupling of 2-(benzo[d]thiazol-2-yl)phenol with N,N′-dimethylformamide to generate 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate occurred via truly heterogeneous catalysis, and the donation of dissolved active copper species to the generation of the desired dimethylcarbamate product, if any, was insignificant.
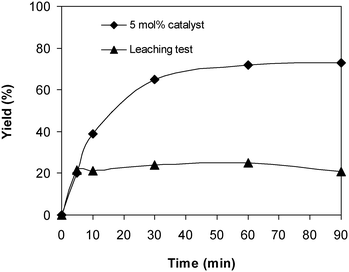 |
| | Fig. 6 Leaching assessment suggested that 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate was not formed in the absence of the catalyst. | |
To probe the reaction pathway of the reaction between 2-(benzo[d]thiazol-2-yl)phenol and N,N′-dimethylformamide to produce 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate using the framework catalyst, some mechanistic studies were conducted. In the first experiment, the reaction was conducted at 100 °C for 90 min, in the presence of 4 equivalents of oxidant, utilizing 13 equivalents of N,N′-dimethylformamide, at 5 mol% catalyst. After the first 5 min reaction period, the radical scavenger (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (conventionally called TEMPO) as the antioxidant was introduced to the reaction solution, and the resulting mixture was then heated at 100 °C with magnetic stirring for an additional 85 min. The experimental outcome revealed that the presence of TEMPO in the reaction mixture modified the transformation enormously, with no further 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate produced (Fig. 7). It must be mentioned that no 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate was recorded after 60 min in the absence of tert-butyl hydroperoxide but with the existence of 5 mol% catalyst. In the second experiment, the reaction was implemented under argon or under oxygen for 60 min. It was noticed that a similar yield of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate was obtained for the reaction performed either under air, argon, or oxygen.
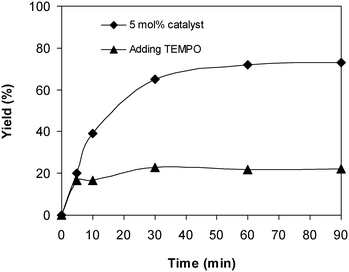 |
| | Fig. 7 Adding TEMPO to the reaction mixture after 5 min. | |
In the first illustration of the Cu(OAc)2-catalyzed production of carbamates from phenols possessing benzothiazole directing substituents and dialkylformamides, Patel and et al. proposed the reaction mechanism for this transformation.9 In the first step of the catalytic cycle, a six-membered copper complex was generated via the chelation of Cu(II) with 2-(benzo[d]thiazol-2-yl)phenol through N-binding of benzothiazole and O-binding of the phenolic group.9 We then explored further into the reaction pathway of the coupling of 2-(benzo[d]thiazol-2-yl)phenol with N,N′-dimethylformamide by using XRD observation. XRD patterns of the fresh catalyst were recorded prior to the experiment (Fig. 8a). 2-(Benzo[d]thiazol-2-yl)phenol and N,N′-dimethylformamide were added to the vial containing the catalyst, and the mixture was heated at 100 °C for 10 min. XRD patterns of this catalyst sample were then recorded. It was observed that the main peak positions shifted, suggesting a partial pore opening upon the chelation of copper sites with 2-(benzo[d]thiazol-2-yl)phenol (Fig. 8b).37,38 Right after that, tBuOOH was introduced into the mixture, allowing the reaction to carry on. After the 5 min reaction period at 100 °C, XRD patterns of the catalyst were then collected. It was noted that the main peak positions shifted closer back to those of the fresh catalyst (Fig. 8c). After 60 min reaction time with a yield of 80% 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate obtained, the XRD patterns of the catalyst were then recorded. The main peak positions of the recovered catalyst were noted to be identical to those of the fresh framework (Fig. 8d), suggesting that copper sites on the catalyst were liberated and prepared for a new catalytic cycle.37,38
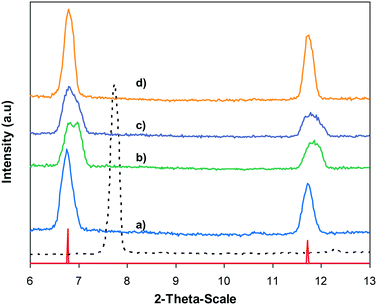 |
| | Fig. 8 XRD observations of the Cu-CPO-27 catalyst during the course of the reaction, including a fresh catalyst (a), a catalyst heated with 2-(benzo[d]thiazol-2-yl)phenol and N,N′-dimethylformamide for 10 min (b), a catalyst heated with 2-(benzo[d]thiazol-2-yl)phenol and N,N′-dimethylformamide for 10 min and then with tBuOOH for 5 min (c), and a catalyst at the end of the reaction (d). The broken line represents the XRD patterns of 2-(benzo[d]thiazol-2-yl)phenol. The red plot displays the simulated XRD patterns of Cu-CPO-27. | |
With these observations and based on previous reports,9,39,40 the mechanism of the reaction between 2-(benzo[d]thiazol-2-yl)phenol and N,N′-dimethylformamide to produce 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate using the framework catalyst was then suggested (Scheme 2). In activated Cu-CPO-27, each copper site was coordinated with five oxygen centers.34,41 The chelation of the copper site with 2-(benzo[d]thiazol-2-yl)phenol through N-binding of benzothiazole and O-binding of the phenolic group would proceed via the reversible displacement of two oxygen centers in the framework. It should be noted that the copper–organic framework was not collapsed during this linker displacement, as each copper site would still be connected to the framework via three other oxygen centers. Upon completion of the catalytic cycle, 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate would desorb from the framework. The 2,5-dihydroxyterephthalic acid linker could coordinate back to the copper center, ready for a new catalytic cycle. Indeed, Corma et al. previously reported a similar linker displacement and recoordination cycle in organic transformations using copper-framework catalysts.39 Kim et al. also demonstrated this phenomenon in several zinc-benzoate coordination polymers.40 However, further studies are needed to clarify the reaction mechanism.
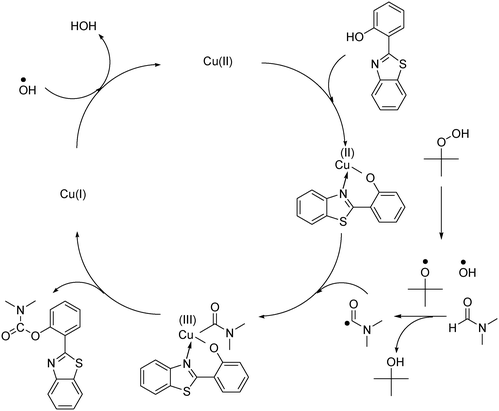 |
| | Scheme 2 Proposed reaction mechanism. | |
To additionally investigate where the catalytic reaction occurred in the coupling of 2-(benzo[d]thiazol-2-yl)phenol with N,N′-dimethylformamide to produce 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate, Cu-CPO-27 samples with different particle sizes were utilized under identical reaction conditions. Certainly, if the transformation proceeds on the external surface of the catalyst, smaller size particles generally offer higher catalytic activity owing to the increased external surface areas.42 The reaction was conducted at 100 °C for 60 min, in the presence of 4 equivalents of oxidant, deploying 19 equivalents of N,N′-dimethylformamide, at 5 mol% catalyst with mean sizes of 37 μm and 150 μm, respectively. It was noticed that the reaction using the Cu-MOF catalyst with a smaller particle size produced 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate in 80% yield after 60 min, while a yield of 69% was obtained for the experiment employing a larger particle catalyst. These observations would suggest that the coupling of 2-(benzo[d]thiazol-2-yl)phenol with N,N′-dimethylformamide would occur on the external surface as well as inside the cavities of the Cu-CPO-27 particles. However, further studies would be necessary to elucidate where the catalytic reaction proceeded.
To underline the important features of utilizing the Cu-CPO-27 catalyst for this conversion, the catalytic activity of this copper-based framework was compared with that of other MOFs. The reaction was conducted at 100 °C for 60 min, in the presence of 4 equivalents of oxidant, deploying 19 equivalents of N,N′-dimethylformamide, at 5 mol% catalyst. It was realized that only copper-based catalysts could be utilized for the synthesis of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate. Indeed, a yield of less than 5% was obtained after 60 min in the presence of Co-CPO-27, In(OH)(BDC), Fe3O(BDC)3, and Zr6O4(OH)4(BDC)6, while no trace quantities of the carbamate was observed after 60 min for the case of Zn-CPO-27 and Ni-CPO-27. The reaction employing the Cu2(OBA)2(BPY) catalyst could carry on to 59% yield of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate after 60 min. MOF-199, Cu(BDC), and Cu2(BDC)2(DABCO) revealed a similar catalytic activity for the reaction, with product yields of 60% and 58% obtained after 60 min. Cu2(BPDC)2(DABCO) was identified to be more active for the conversion, offering a yield of 71% after 60 min. In the midst of these heterogeneous catalysts, Cu-CPO-27 expressed the best efficiency, providing a carbamate yield of 80% after 60 min (Fig. 9).
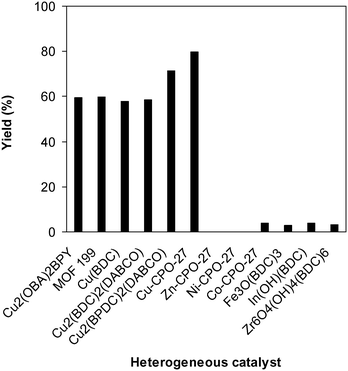 |
| | Fig. 9 Yields of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate with diverse MOF-based catalysts. | |
Under the view of green chemistry, one aspect that should be probed for the reaction of 2-(benzo[d]thiazol-2-yl)phenol with N,N′-dimethylformamide to generate 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate using the framework catalyst is the reusability of this Cu-MOF catalyst. For that reason, the catalyst was considered for reusability in the production of 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate over 9 consecutive runs. The reaction was performed at 100 °C for 60 min, in the presence of 4 equivalents of oxidant, deploying 19 equivalents of N,N′-dimethylformamide, at 5 mol% catalyst. After the 60 min reaction period, the catalyst was collected, washed with a large quantity of hot N,N′-dimethylformamide and methanol to remove any physisorbed reagents, and heated at 150 °C under vacuum in a Schlenk line for 6 h. The restored catalyst was then reused in new experiments. The experimental outcome identified that it was possible to reuse the catalyst in the reaction of 2-(benzo[d]thiazol-2-yl)phenol with N,N′-dimethylformamide to produce 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate. Certainly, a yield of 74% of the needed dimethylcarbamate product was still acquired in the 9th run (Fig. 10). Moreover, the structure of the catalyst could be preserved under the reaction condition, as confirmed by XRD (Fig. 11), FT-IR (Fig. 12), and nitrogen physisorption (Fig. 13) results.
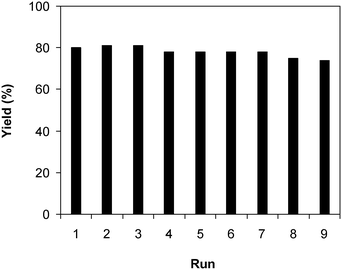 |
| | Fig. 10 Reutilization of the catalyst. | |
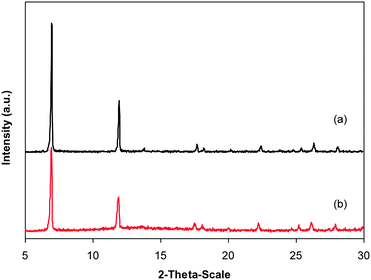 |
| | Fig. 11 XRD results of the new (a) and recovered (b) catalysts. | |
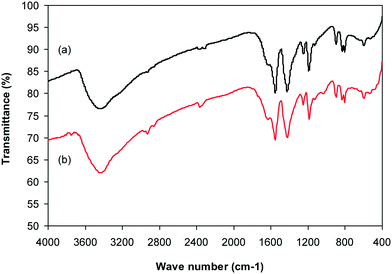 |
| | Fig. 12 FT-IR results of the new (a) and recovered (b) catalysts. | |
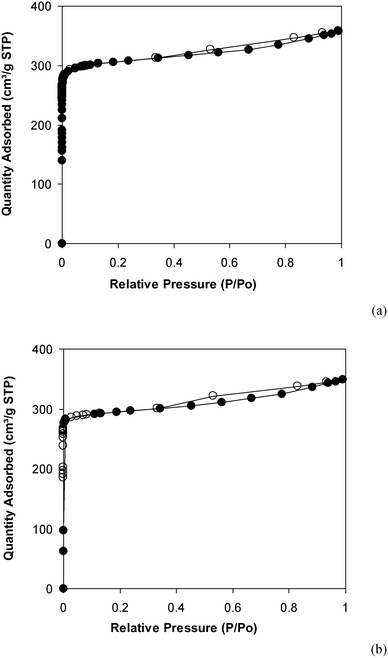 |
| | Fig. 13 Nitrogen adsorption/desorption isotherms of the new (a) and recovered (b) catalysts. Adsorption data are shown as closed circles and desorption data as open circles. | |
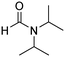
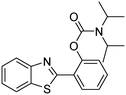
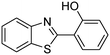
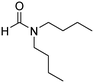
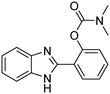
The scope of the research was then diversified into the synthesis of various carbamates containing a benzothiazole moiety. First, the coupling between 2-(benzo[d]thiazol-2-yl)phenol and different formamides was conducted. The reaction was performed at 100 °C for 60 min, in the presence of 4 equivalents of oxidant, deploying 19 equivalents of formamide, at 5 mol% catalyst. Utilizing this procedure, 2-(benzo[d]thiazol-2-yl)phenyl dimethylcarbamate was produced in 77% isolated yield. More bulky formamides were observed to be less reactive towards the coupling reaction. Moving to N,N′-diethylformamide, the reaction generated 2-(benzo[d]thiazol-2-yl)phenyl diethylcarbamate in 72% yield, while a yield of 67% 2-(benzo[d]thiazol-2-yl)phenyl diisopropylcarbamate was obtained for the case of N,N-diisopropylformamide. As expected, the coupling reaction between N,N-dibutylformamide and 2-(benzo[d]thiazol-2-yl)phenol proceeded with more difficulty, producing 2-(benzo[d]thiazol-2-yl)phenyl dibutylcarbamate in 61% yield. Piperidine-1-carbaldehyde was able to react with 2-(benzo[d]thiazol-2-yl)phenol in the presence of the framework catalyst, with a yield of 66% 2-(benzo[d]thiazol-2-yl)phenyl piperidine-1-carboxylate recorded. Similarly, a yield of 64% 2-(benzo[d]thiazol-2-yl)phenyl morpholine-4-carboxylate was achieved for the reaction of morpholine-4-carbaldehyde with 2-(benzo[d]thiazol-2-yl)phenol, while a yield of 62% 2-(benzo[d]thiazol-2-yl)phenyl cyclohexylcarbamate was observed for the case of N-cyclohexylformamide. The coupling reaction of N,N′-dimethylformamide with 2-(benzo[d]oxazol-2-yl)phenol or 2-(1H-benzo[d]imidazol-2-yl)phenol was also conducted utilizing the framework catalyst. Under this condition, 2-(benzo[d]oxazol-2-yl)phenyl dimethylcarbamate was generated in 66% yield, while a yield of 70% was observed for 2-(1H-benzo[d]imidazol-2-yl)phenyl dimethylcarbamate (Table 1).
Table 1 Synthesis of various carbamates containing a benzothiazole moiety
| Entry |
Phenols |
Formamides |
Products |
Isolated yield (%) |
| 1 |
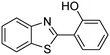
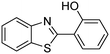
|
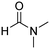
|
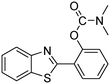
|
77 |
| 2 |
|
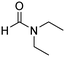
|
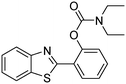
|
72 |
| 3 |
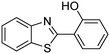
|
|
|
67 |
| 4 |
|
|
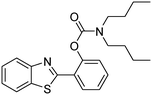
|
61 |
| 5 |
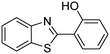
|
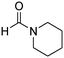
|
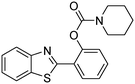
|
66 |
| 6 |
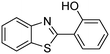
|
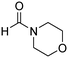
|
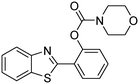
|
64 |
| 7 |
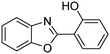
|
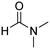
|
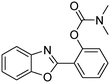
|
66 |
| 8 |
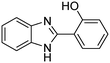
|
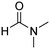
|
|
70 |
4. Conclusions
The metal–organic framework Cu-CPO-27 was synthesized from dihydroxyterephthalic acid and copper nitrate trihydrate by a solvothermal method. The framework was used as a recyclable catalyst for the synthesis of carbamates containing a benzothiazole moiety via cross-dehydrogenative coupling of phenols with N,N-disubstituted formamides. These hybrid benzothiazole–carbamate structures would combine the benefits from both carbamate and benzothiazole moieties in terms of biological and pharmaceutical activities. Using this protocol, a benzothiazole–carbamate moiety could be generated by utilizing a catalytic quantity of the framework. The conversion occurred via truly heterogeneous catalysis, and the donation of dissolved active species to the generation of the desired dimethylcarbamate product, if any, was unimportant. It was feasible to recover and reuse the catalyst for the synthesis of carbamates without an appreciable deterioration in catalytic efficiency. The feature that carbamates containing a benzothiazole moiety could be generated by the cross-dehydrogenative coupling of phenols with N,N-disubstituted formamides using a recyclable catalyst should be of benefit, and would attract consideration from the chemical industry.
Acknowledgements
We would like to thank the Vietnam National University – Ho Chi Minh City (VNU-HCM) for financial support via contract No. B2017-20-08.
References
- R. Srivastava, M. D. Manju, D. Srinivas and P. Ratnasamy, Catal. Lett., 2004, 97, 41–47 CrossRef CAS.
- P. Wang, Y. Ma, S. Liu, F. Zhou, B. Yang and Y. Deng, Green Chem., 2015, 17, 3964–3971 RSC.
- J. Y. Hong, U. R. Seo and Y. K. Chung, Org. Chem. Front., 2016, 3, 764–767 RSC.
- A. R. Sardarian and I. D. Inaloo, RSC Adv., 2015, 5, 76626–76641 RSC.
- P. K. Chikkade, Y. Kuninobu and M. Kanai, Chem. Sci., 2015, 6, 3195–3200 RSC.
- E. V. Vinogradova, N. H. Park, B. P. Fors and S. L. Buchwald, Org. Lett., 2013, 15, 1394–1397 CrossRef CAS PubMed.
- L. Ren and N. Jiao, Chem. Commun., 2014, 50, 3706–3709 RSC.
- H. Yin, A. M. D. Almeida, M. V. D. Almeida, A. T. Lindhardt and T. Skrydstrup, Org. Lett., 2015, 17, 1248–1251 CrossRef CAS PubMed.
- W. Ali, S. K. Rout, S. Guin, A. Modi, A. Banerjee and B. K. Patel, Adv. Synth. Catal., 2015, 357, 515–522 CrossRef CAS.
- I.-H. Park, R. Medishetty, H.-H. Lee, C. E. Mulijanto, H. S. Quah, S. S. Lee and J. J. Vittal, Angew. Chem., Int. Ed., 2015, 54, 7313–7317 CrossRef CAS PubMed.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- A. Torres-Knoop and D. Dubbeldam, ChemPhysChem, 2015, 16, 2046–2067 CrossRef CAS PubMed.
- D. E. Williams and N. B. Shustova, Chem. – Eur. J., 2015, 21, 15474–15479 CrossRef CAS PubMed.
- D. Andirova, Y. Lei, X. Zhao and S. Choi, ChemSusChem, 2015, 8, 3405–3409 CrossRef CAS PubMed.
- H. J. Lee, J. We, J. O. Kim, D. Kim, W. Cha, E. Lee, J. Sohn and M. Oh, Angew. Chem., Int. Ed., 2015, 54, 10564–10568 CrossRef CAS PubMed.
- Z.-R. Jiang, H. Wang, Y. Hu, J. Lu and H.-L. Jiang, ChemSusChem, 2015, 8, 878–885 CrossRef CAS PubMed.
- A. Rapeyko, M. J. Climent, A. Corma, P. Concepción and S. Iborra, ChemSusChem, 2015, 8, 3270–3282 CrossRef CAS PubMed.
- R. M. Abdelhameed, M. M. Q. Simões, A. M. S. Silva and J. Rocha, Chem. – Eur. J., 2015, 21, 11072–11081 CrossRef CAS PubMed.
- Y. Yang, W. Zhang, X. Ma, H. Zhao and X. Zhang, ChemCatChem, 2015, 7, 3454–3459 CrossRef CAS.
- R. Sen, D. Saha, S. Koner, D. Mal, P. Brandão and Z. Lin, ChemPlusChem, 2015, 80, 591–598 CrossRef CAS.
- Y. Chen, X. Feng, X. Huang, Z. Lin, X. Pei, S. Li, J. Li, S. Wang, R. Li and B. Wang, Chem. – Eur. J., 2015, 21, 13894–13899 CrossRef CAS PubMed.
- M. A. Gotthardt, R. Schoch, T. S. Brunner, M. Bauer and W. Kleist, ChemPlusChem, 2015, 80, 188–195 CrossRef CAS.
- J. Li, J. Yang, Y.-Y. Liu and J.-F. Ma, Chem. – Eur. J., 2015, 21, 4413–4421 CrossRef CAS PubMed.
- B. Bueken, F. Vermoortele, D. E. P. Vanpoucke, H. Reinsch, C.-C. Tsou, P. Valvekens, T. D. Baerdemaeker, R. Ameloot, C. E. A. Kirschhock, V. V. Speybroeck, J. M. Mayer and D. D. Vos, Angew. Chem., Int. Ed., 2015, 54, 13912–13917 CrossRef CAS PubMed.
- D. Ruano, M. Díaz-García, A. Alfayate and M. Sánchez-Sánchez, ChemCatChem, 2015, 7, 674–681 CrossRef CAS.
- G. Huang, L. Yang, X. Ma, J. Jiang, S.-H. Yu and H.-L. Jiang, Chem. – Eur. J., 2016, 22, 3470–3477 CrossRef CAS PubMed.
- L. M. Aguirre-Díaz, M. Iglesias, N. Snejko, E. Gutiérrez-Puebla and Á. Monge, Chem. – Eur. J., 2016, 22, 6654–6665 CrossRef PubMed.
- S. Sartipi, M. J. V. Romero, E. Rozhko, Z. Que, H. A. Stil, J. D. With, F. Kapteijn and J. Gascon, ChemCatChem, 2015, 7, 3243–3247 CrossRef CAS.
- Y.-J. Tang, M.-R. Gao, C.-H. Liu, S.-L. Li, H.-L. Jiang, Y.-Q. Lan, M. Han and S.-H. Yu, Angew. Chem., Int. Ed., 2015, 54, 12928–12932 CrossRef CAS PubMed.
- W. Xi, Y. Liu, Q. Xia, Z. Li and Y. Cui, Chem. – Eur. J., 2015, 21, 12581–12585 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- C. Lastoskie, K. E. Gubbins and N. Quirke, J. Phys. Chem., 1993, 97, 4786–4796 CrossRef CAS.
- C. Lastoskie, K. E. Gubbins and N. Quirke, Langmuir, 1993, 9, 2693–2702 CrossRef CAS.
- M. J. Katz, A. J. Howarth, P. Z. Moghadam, J. B. DeCoste, R. Q. Snurr, J. T. Hupp and O. K. Farha, Dalton Trans., 2016, 45, 4150–4153 RSC.
- R. Sanz, F. Martínez, G. Orcajo, L. Wojtas and D. Briones, Dalton Trans., 2013, 42, 2392–2398 RSC.
- G. Calleja, R. Sanz, G. Orcajo, D. Briones, P. Leo and F. Martínez, Catal. Today, 2014, 227, 130–137 CrossRef CAS.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. A. Vallet-Regí, M. Sebban, F. Taulelle and G. R. Férey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS PubMed.
- S. Henke, A. Schneemann, A. Wütscher and R. A. Fischer, J. Am. Chem. Soc., 2012, 134, 9464–9474 CrossRef CAS PubMed.
- I. Luz, A. León, M. Boronat, F. X. L. s. i. Xamena and A. Corma, Catal. Sci. Technol., 2013, 3, 371–379 CAS.
- H. Kwak, S. H. Lee, S. H. Kim, Y. M. Lee, B. K. Park, Y. J. Lee, J. Y. Jun, C. Kim, S.-J. Kim and Y. Kim, Polyhedron, 2009, 28, 553–561 CrossRef CAS.
- L. J. Wang, H. Deng, H. Furukawa, F. Gándara, K. E. Cordova, D. Peri and O. M. Yaghi, Inorg. Chem., 2005, 53, 5881–5883 CrossRef PubMed.
- R. Selvin, H.-L. Hsu and T.-M. Her, Catal. Commun., 2008, 10, 169–172 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00067g |
|
| This journal is © The Royal Society of Chemistry 2017 |

 *
*