
Experimental and theoretical studies on oxy-cracking of Quinolin-65 as a model molecule for residual feedstocks†
Abdallah D.
Manasrah
 a,
Amjad
El-Qanni
ab,
Ismail
Badran
ac,
Lante
Carbognani Ortega
a,
M. Josefina
Perez-Zurita
a and
Nashaat N.
Nassar
*a
a,
Amjad
El-Qanni
ab,
Ismail
Badran
ac,
Lante
Carbognani Ortega
a,
M. Josefina
Perez-Zurita
a and
Nashaat N.
Nassar
*a
aDepartment of Chemical and Petroleum Engineering, University of Calgary, 2500, University Drive NW, Calgary, Alberta, T2N 1N4 Canada. E-mail: nassar@ucalgary.ca; Fax: +1 403 210 3973; Tel: +1 403 210 9772
bDepartment of Chemical Engineering, An-Najah National University, P.O. Box 7, Nablus, Palestine
cDepartment of Chemistry, An-Najah National University, P.O. Box 7, Nablus, Palestine
First published on 3rd August 2017
Abstract
Oxy-cracking is a combination of oxidation and cracking reactions for converting heavy hydrocarbons into commodity products with minimal emission of CO2. This reaction takes place in basic aqueous media, at mild operation temperatures (200–230 °C) and pressures (500–750 psi). In this study, the main goal is to understand the oxy-cracking mechanism, involving oxidation and cracking reactions, of solid hydrocarbons represented by the model molecule Quinolin-65 (Q-65). In the experimental part, the oxy-cracking reaction was performed in a Parr batch reactor operated at an optimized oxygen partial pressure of 750 psi and temperatures between 200 °C and 230 °C. The reaction products were characterized by FTIR spectroscopy, TOC analysis, GC, NMR spectroscopy, and XPS. We found that the main products are composed of organic carboxylic, phenolic, and carbonyl-containing compounds, with small amounts of inorganic carbon (IC). In the theoretical part of the study, a comprehensive computational modeling of Q-65 reactivity was performed using high-level quantum theoretical calculations. The reaction studies indicated the attack of the hydroxyl radical (OH˙) and hydroxide anion (OH−) on the Q-65 molecule. The theoretical study employed the density functional theory (DFT) and the second-order Møller–Plesset perturbation theory (MP2) to study the reaction mechanisms under the same experimental conditions. Both the theoretical and the experimental studies confirmed the complexity of the reaction kinetics. The reaction kinetic results suggested that the Q-65 oxy-cracking reaction went through a parallel-consecutive reaction in which oxidative decomposition took place in the first step producing different aromatic intermediates. These intermediates were oxy-cracked consecutively into different organic acids and a small amount of CO2 gas.
1. Introduction
With the increased world population, modernization, and urbanization, the global demand for energy is projected to rise rapidly.1,2 Despite the variety of energy resources, the world still relies heavily on oil, which will inevitably deplete the world's supply of conventional crude oil.3,4 Therefore, there is an increasing demand to upgrade and recover unconventional crude oil, like bitumen and heavy oil, to meet current and future global energy demands. Typically, processing of conventional and unconventional oils to meet market or pipeline specifications involves “coking”, which is widely implemented in refineries and upgraders. This process generates a massive amount of solid-waste heavy hydrocarbon by-product known as petroleum coke (petcoke).5 Carbon is the most common species in these kinds of solid wastes with many other metals and heteroatoms like sulphur, nitrogen, and oxygen also present.6 As a result of increased production of unconventional oils, asphaltene and petcoke production will also increase in Canada and worldwide. Currently, there is no economically feasible use for these solid wastes and they are often disposed of in stockpiles. In Canada, the stockpiles are growing at a rate of about 4 million tonnes a year.7,8 These stockpiles pose long-term problems, requiring huge land space, potentially leaching heavy metals, particulate matter, and other contents into the environment.Thermal techniques have been proposed for treating these kinds of complex solid-waste heavy hydrocarbons and converting them into useful products. Gasification, a high-temperature process for converting carbonaceous solids into hydrogen and carbon monoxide gases, which subsequently could be used to produce liquid hydrocarbons through the Fischer–Tropsch process, is one common method.9–12 As of today, the best-known gasification process for heavy residue fraction upgrading is the Flexicoking process developed by Exxon Mobil.13 However, given the uncertainty of the optimum gasifier configuration, the lack of industrial scale gasifiers to confirm the feasibility and reliability of the gasification process, the significant coke/ash content generated from the gasifier that still requires stockpiling or landfilling, and the production of low-BTU waste gas, gasification is neither a green nor a reliable method for solid-waste conversion.10,14,15 Combustion is another potential technique for energy production. Again, this technique has limited prospects due to sulfur and CO2 emissions.9,16,17
Many researchers have investigated heavy hydrocarbon treatment processes, especially for asphaltene-type waste, such as carbon rejection and hydrogen addition.17,18 Carbon rejection processes include pyrolysis (visbreaking, coking and rapid thermal pyrolyzer),17–19 separation or extraction (solvent deasphalting process, SDA)20,21 and cracking (VGO and VR FCC).18,22 Hydrocracking and steam cracking gasification are conventional processes of hydrogen addition.9,23,24 These conventional hydroprocessing technologies still suffer from coke formation, high catalyst loading, and catalyst poisoning caused by metal deposition.23,24 Carbon rejection processes have simple configurations compared to hydrogen addition processes and do not require hydrogen injection and catalyst loading.18 Nevertheless, carbon rejection produces lower yields and generates significant coke waste. Therefore, these processes were deemed to be unreliable technologies for the treatment of solid-waste heavy hydrocarbons, specifically due to their high-energy intensity and CO2 emissions.10,14
Advanced oxidation processes (AOPs), based on the creation of reactive species like hydroxyl radicals (OH˙), are reliable and capable of breaking down a wide range of complex heavy hydrocarbon compounds quickly and non-selectively.25–27 One issue is the significant amount of gaseous CO2 produced, which requires further processing. Additionally, the solubilized organic species required further separation and treatment before safe disposal. Therefore, AOPs cannot be the sole method for conversion of heavy waste hydrocarbons.
A number of researchers investigated the conversion of solid-waste heavy hydrocarbons (lignites and coal) to humic substances in acidic media using oxidizing agents such as KMnO4 and HNO3.28,29 However, the process requires long reaction times for coal particles larger than 100 μm. It was also found that the acid used was more expensive compared to the obtained products. Moreover, the production of humic substances via coal oxidation was investigated industrially using a pre-oxidizing reaction.28–30 Using this mechanism, coal is converted to humic acids, then treated with formaldehyde and an alkali solution of bisulfate at 100 °C to form water-soluble products.31 Although coal pre-oxidation was proposed in several patents, this process is expensive and also has low conversion.32–34
Despite all the techniques that have been proposed for developing efficient and environmentally friendly technologies for treating solid-waste heavy hydrocarbons, there are no industrially feasible options. The challenges that face the oil industry create a need for new environmentally friendly and cost-effective technologies to convert solid-waste heavy hydrocarbons into valuable products. To overcome this challenge, our research group has recently introduced an alternative technique to produce light commodity hydrocarbons from asphaltenes. This method involves a combination of two types of reactions in aqueous alkaline media, namely oxidation and cracking, creating the name “oxy-cracking”.35 The oxy-cracking technique is inspired by asphaltene oxidation and ozonolysis studies reported in previous works.31,36–39 This new oxy-cracking technique offers a pathway that creates valuable material from heavy oil and refinery waste. Oxidized asphaltenes become soluble in water due to the polar functionalization of aromatic edges and paraffinic terminal carbons via oxygen incorporation. Changing the oxidation conditions will alter the mechanism and result in different product distributions. It is well known that the hydroxyl radical (OH˙) plays an essential role in the hydrocarbon wet oxidation process.40–42 Such a process is believed to involve the following primary initiation steps:43,44
| H2O + O2 → HO2˙ + OH˙ Ea = 68.6 kcal mol−1 |
| HO2˙ + H2O → H2O2 + OH˙ Ea = 32.8 kcal mol−1 |
| H2O + O˙ → OH˙ + OH˙ Ea = 17 kcal mol−1 |
These initiation steps are followed by different propagation and termination pathways. As reported by many studies,25,42,43,45 this indicated that a large range of intermediates and oxidized products may result from the above steps. The prevailing products in the liquid phase are alkylhydroperoxides, which are quickly converted into hydroxylated hydrocarbons and ketones/aldehydes.43 Hence, via the oxy-cracking process, waste heavy hydrocarbons could be oxidized in an aqueous alkaline medium at mild temperatures (170–230 °C) and pressures (500–750 psi).35,46,47 However, these heavy hydrocarbons do not oxidize completely to CO2, but instead form intermediate compounds that are soluble in water via oxygen incorporation. Thus, oxy-cracking enhances the tendency of large heavy hydrocarbon molecules to disaggregate, making them more accessible to subsequent hydrogenation and cracking at lower temperatures.48 Those intermediates (desired products) are likely analogs of humic acids, carboxylic acids, and their corresponding salts, or other products.35,46,47,49 Because of the selectivity and high solubility of such complex hydrocarbons in an alkaline medium, the process is considered environmentally friendly, as there is very little CO2 emitted.
Typically, these solid-waste heavy hydrocarbons, such as petcoke and asphaltenes, have no unique chemical identity and portray complex chemical structures that contain heteroatoms such as sulfur, nitrogen, and oxygen. However, some researchers have attempted to hypothesize model structures for asphaltenes, such as the Yen model or modified Yen model,50–58 based on physical and chemical methods. Understanding the reaction mechanism and the kinetics of such an innovative technique requires the chemical structure of the solid-waste heavy hydrocarbons to be known. Finding a model molecule with a well-known structure that mimics the real structure and properties of solid-waste heavy hydrocarbons would clarify the oxy-cracking reaction mechanism. Several researchers propose Quinolin-65 (Q-65) as a model molecule for solid-waste heavy hydrocarbons (like asphaltenes) because it has a large aromatic region in addition to side chains.59–61 Furthermore, Q-65 has been confirmed to resemble asphaltene molecules regarding physical and chemical properties, thermal cracking behavior, and adsorption on solid surfaces.62,63
Recently, we studied the complete dry oxidation of Q-65 as a heavy-oil model molecule.64 The research involved a comprehensive theoretical and thermogravimetric analysis of Q-65 in the absence of a catalyst. The study concluded that the thermo-oxidative decomposition of Q-65 is a complicated multi-step process involving different reaction pathways. The kinetic parameters from both the experimental and theoretical parts of the study strongly suggested catalyst incorporation.
Herein, we are interested in understanding the oxy-cracking of residual feedstocks using Q-65 as a model molecule. Therefore, the purposes of this study include: (1) carrying out an oxy-cracking reaction for Q-65 within a batch reactor under aqueous alkaline medium and mild operating conditions (i.e., temperatures varied between 200–230 °C and the operating pressure fixed at 750 psi) to keep the water in the liquid phase; (2) determining the experimental reaction kinetic parameters and potential mechanism for the oxy-cracking of Q-65 in the liquid phase using the generalized “triangular” lumped kinetic model for non-catalytic wet oxidation of organic compounds; (3) carrying out computational modeling to understand the oxy-cracking mechanism using state-of-the-art ab initio methods. The density functional theory (DFT) and the second-order Møller–Plesset perturbation theory (MP2) were implemented to study the oxy-cracking reaction mechanism under conditions similar to those in the experimental part. The theoretical study will not just validate the experimental findings, but it also serves twofold; first, it presents a new understanding for the oxy-cracking of Q-65 at the molecular level. Second, it provides vital kinetic parameters such as activation energies and Gibbs free energies of reaction for different possible oxidation pathways.
2. Experimental work
2.1 Materials
Quinolin-65 (Q-65) in powder form (C30H27NO2S, λmax = 565 nm, 80 wt% dye content) was purchased from Sigma-Aldrich (Ontario, Canada) and selected as a model compound mimicking the solid-waste hydrocarbons. The chemical structure of Q-65 is shown in Scheme 1.64 The Q-65 was used as-received without further purification as the impact of impurities on our results, from the application point of view, is minimal as real solid-waste hydrocarbons, when oxy-cracked, will contain unknown chemical structures.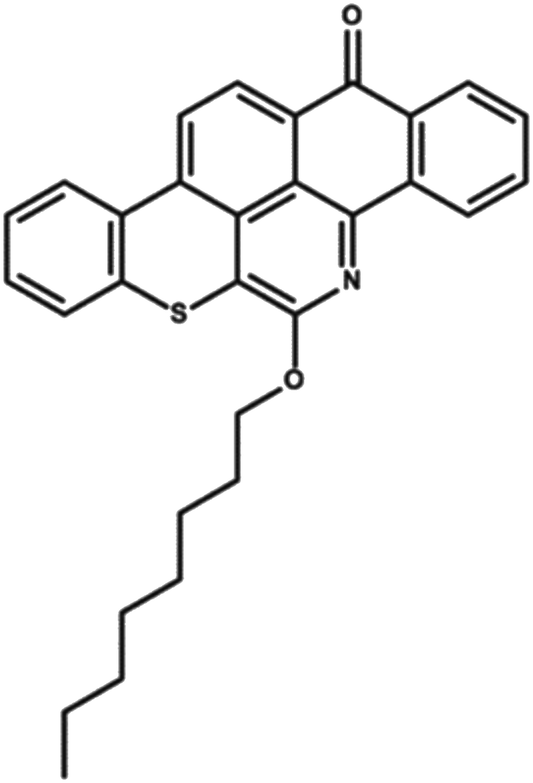 | ||
| Scheme 1 Q-65 molecular structure.64 | ||
Potassium hydroxide (KOH, ACS reagent, ≥85%, pellet form) purchased from Sigma-Aldrich (Ontario, Canada) was used to adjust the pH of the reaction medium (deionized water) and help in solubilizing Q-65 in solution. Oxygen (99.9% ultrahigh purity) purchased from Praxair (Calgary, Canada) was used as the oxidant gas.
2.2 Experimental setup and procedure
The oxy-cracking experiments were carried out in the setup shown schematically in Fig. 1. A 100 ml reactor vessel was used for the experiments (model number 4598, Parr Instrument Company, Moline, IL, USA). The reactor vessel was equipped with a heating band connected to a temperature control loop, a pressure gauge and a mechanical stirrer connected to a speed controller. The reactor is capable of handling pressures up to 1700 psi and temperatures up to 270 °C. In a typical experiment, 50 mg of Q-65 was charged into the reactor vessel containing 30 g of deionized water and a certain amount of KOH to keep the pH above 8.0 to help in solubilizing Q-65 and to avoid corrosion problems. The reaction was conducted at a stirring rate of 1000 rpm to minimize the interfacial mass resistance between the gas and liquid phases and to ensure uniform temperature and concentration profiles in the liquid phase. Leak tests were performed by pressurizing the reactor with O2 up to 1000 psi prior to fixing the operating pressure at 750 psi. The reactor was then heated to the desired temperature. Zero reaction time was set when the desired temperature was attained. The reaction was carried out at different times, namely 30, 60, 90 and 120 min. The predetermined operating temperatures were chosen to be 200, 215, and 230 °C. This temperature range was considered as an optimum range for favoring the Q-65 conversion with a minimum amount of CO2 produced. At the end of each experiment, the reactor was cooled down to room temperature. Then, the gas was analyzed using gas chromatography, GC (SRI 8610C, SRI Instruments), having thermal conductivity detection (TCD). The liquid layer containing the oxy-cracked Q-65 was carefully discharged from the reactor vessel for total organic carbon (TOC) analysis, and the reactor vessel and assembly were washed with soap and deionized water to remove any residue, therefore, the apparatus could be ready for another cycle of experiments. The oxy-cracked Q-65 was recovered using an evaporator for further analysis by FTIR and NMR spectroscopy (see the ESI,† S1.1) and XPS (see the ESI,† S1.2) techniques to investigate the nature of the formed compounds. It is worth noting here that a minimum amount of solid residue (i.e., insolubilized Q-65) could only be collected at low reaction conversion, which was recovered by using an evaporator for elemental analysis. The elemental analysis was performed by a combustion method using a PerkinElmer 2400 CHN (Waltham, Massachusetts, USA) analyzer to determine the amount of carbon, hydrogen, and nitrogen after the oxy-cracking reaction. All experiments were duplicated to confirm reproducibility. | ||
| Fig. 1 Schematic illustration of the experimental setup (not to scale). | ||
2.3 Characterization
3. Theoretical calculations
All theoretical calculations in this work were performed using the Gaussian 09 program.65 For the reactions involving the hydroxyl radical (OH˙), geometry optimization and frequency calculations were performed using the hybrid density functional, B3LYP,66,67 and the 6-31+G(d)68 basis set. For the reactions involving the hydroxide anion (OH−), the calculations were performed using the B97D3 functional with Grimme's dispersion.69 In addition, the OH− reactions were carried out in water by assigning (solvent water) in the scrf keyword in Gaussian. Zero-point energies (ZPE) were scaled by a factor of 0.9748, as suggested by Scott and Radom.70 In order to obtain accurate energy values, single point energies for all species, including transition states, were requested after complete optimization at the MP2/6-311+G(d,p) level of theory. Exact details on locating the transition states and the corresponding intrinsic reaction coordinates (IRCs) are described in detail in previous works.64,71,72 All transition states located in this work were confirmed to have one and only one imaginary frequency along the desired reaction coordinate, by plotting the corresponding IRCs in both directions.The activation and reaction parameters (enthalpy, entropy, and Gibbs free energy) were calculated as described in previous works.64,71,72 The rate constants for the unimolecular and bimolecular reactions were obtained using the two following equations, respectively:73
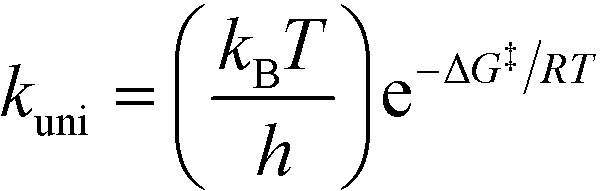 | (1) |
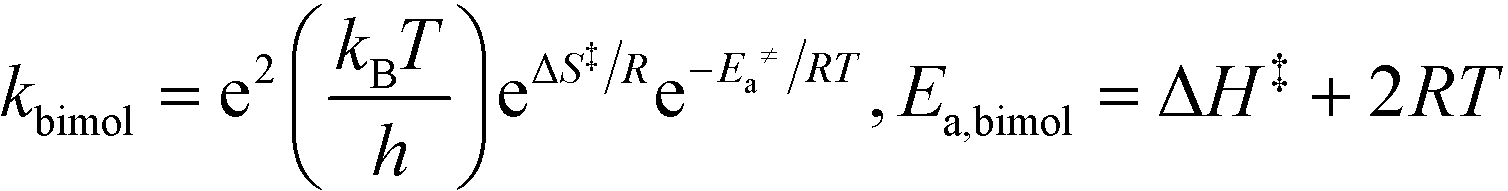 | (2) |
4. Results and discussion
4.1 Experimental determination of the oxy-cracking reaction kinetics
The effects of temperature and residence time on Q-65 oxy-cracking were investigated to understand and validate the reaction kinetic mechanism. Other important parameters, such as the operating partial pressure, the stoichiometric ratio of Q-65 with water and KOH, and the impeller speed, remained unchanged. Oxy-cracking is a gas–liquid reaction which includes various transport processes that might take place in series.35 For this reason, preliminary experiments were conducted to optimize the operating oxygen partial pressure and the impeller speed. The reaction was not significantly affected by pressures above 750 psi, which was thus used as the optimum operating partial pressure. Therefore, we ensured that oxygen in an excess amount is present. The rate of the oxy-cracking was found to be independent of the impeller speed between 500 and 1500 rpm, indicating the absence of interfacial mass transfer resistance in the liquid phase. This agrees excellently with other oxy-cracking and wet oxidation studies.26,35,46,47,74,75 Hence, all experiments were carried out at the impeller speed of 1000 rpm.After oxy-cracking of the Q-65, several soluble and insoluble intermediates were produced. Thus, the produced intermediates were considered as a lump component to describe the oxy-cracking reaction. Hence, the rate equations are developed based on the lumped concentrations of total organic carbon (TOC). A generalized “triangular” lumped kinetic model for non-catalytic wet oxidation of organic compounds in wastewater has been proposed by Li et al.43 It relies on the assumption that some of the organic compounds present in the wastewater are directly oxidized to CO2 and H2O, while the rest are converted to an intermediate product which is further oxidized. Recently, Ashtari et al.35 employed the “triangular” reaction pathway for asphaltene oxy-cracking, where oxygen was used to convert asphaltenes to water-soluble material which contains carboxylic, sulfonic, and phenolic functionalities in addition to a minimum amount of CO2 gas. The triangular reaction pathway was applied to the oxy-cracking of Q-65 while keeping the decomposition of alkylperoxo radicals under control to avoid the significant release of CO2 as shown in Fig. 2.
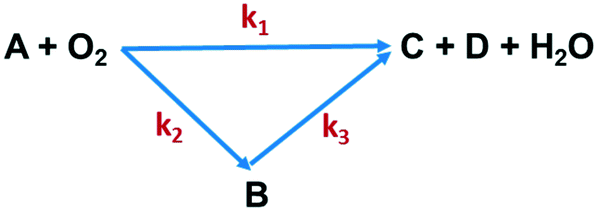 | ||
| Fig. 2 Triangular reaction scheme of Q-65 oxy-cracking. A is the Q-65, B is the soluble intermediate (TOC), C is CO2, and D is the inorganic carbon (IC). | ||
The mass balance was closed by conducting carbon-based balance before and after the oxy-cracking reaction of Q-65 as follows:
| Carbon content in the original Q-65 (CAo) = Carbon content in the liquid phase (CL) + Carbon content in the gas phase (CC) + Carbon content in the solid residue (CR) | (3) |
C Ao was calculated by multiplying the mass of Q-65 by the fraction of carbon in the Q-65 and CL was considered as TC. The overall carbon mass balance was closed to 98.21%, the leftover 1.79% was considered as a loss due to the experimental and instrumental errors. The latter percentage was added and presented alongside the TOC percentage error, which was less than 5% for all runs. The produced gas, most likely CO2, was analyzed online using GC. However, other gas contents were minimal, and thus, they were neglected. The ideal gas law was used to estimate the carbon content in the gas phase (CC) using the following equation:
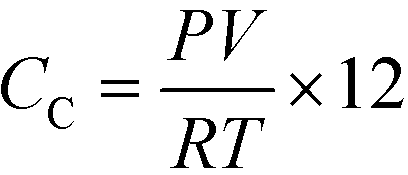 | (4) |
For the solid residue, the carbon content was estimated using elemental analysis: carbon content after reaction (insolubilized Q-65) = (mass of solid residue) × (carbon wt%). Moreover, the reaction conversion (X) based on carbon mass was calculated as follows:
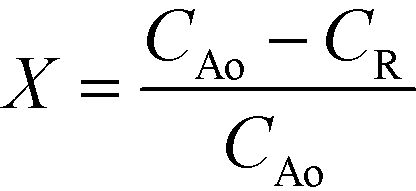 | (5) |
| CA = CAo(1 − X) | (6) |
The kinetic rate equations are presented by the set of the following three differential equations:
 | (7) |
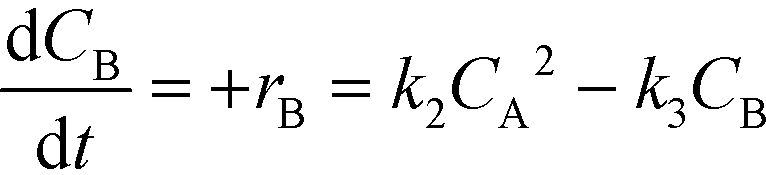 | (8) |
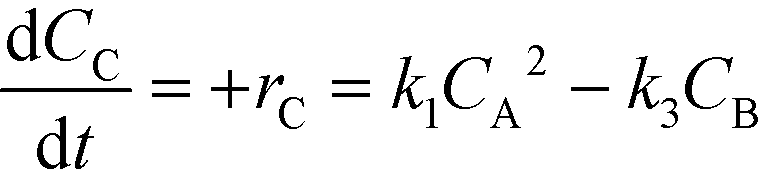 | (9) |
| ki = Aie−Eai/RT | (10) |
| Activation energy (kcal mol−1) | Frequency factor (unit) | |
|---|---|---|
| E a1 | 9.8 | 15.6 (L mmol−1 s−1) |
| E a2 | 17.7 | 9.3 × 104 (L mmol−1 s−1) |
| E a3 | 10.9 | 7.5 (s−1) |
Fig. 3 shows the experimental concentration profiles of Q-65, intermediate compounds (desired products), and CO2 expressed as a function of time during the oxy-cracking reaction at 200, 215, and 230 °C, together with the kinetic model fit. The second-order kinetic model fit well to the experimental results agreeing with a similar study on catalytic wet air oxidation of industrial effluents using the second-order lumped kinetic model.76,77 Moreover, this model described our reaction kinetics more accurately than the first-order kinetic model.35,43
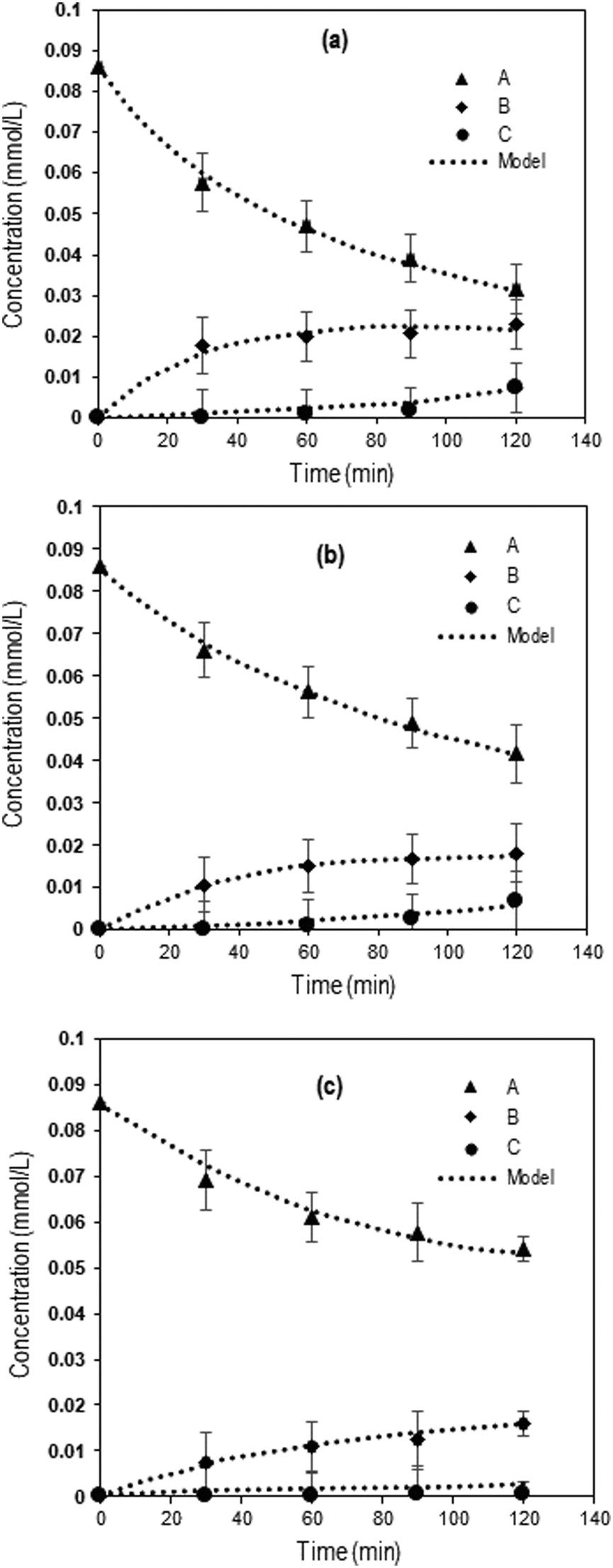 | ||
| Fig. 3 Concentrations as a function of time during Q-65 oxy-cracking at (a) 230 °C, (b) 215 °C, and (c) 200 °C. The symbols represent experimental data, and the dotted lines are from the kinetic model (eqn (7)–(9)). Experimental operating conditions: oxygen partial pressure: 750 psi, impeller speed: 1000 rpm, Q-65 amount: 0.05 g, KOH amount: 0.05 g, and water amount: 30 g. | ||
Clearly, the reaction temperature is a key parameter in the oxy-cracking reaction of Q-65. By increasing the temperature, organic functionalities are oxy-cracked in water due to the partial oxy-cracking of Q-65 into oxygenated intermediates, however, some of these intermediates are decomposed oxidatively into CO2 and H2O. Interestingly, the highest temperature of 230 °C appears to be the optimum temperature as the highest conversion, synchronized with low CO2 production, occurred at that temperature (Fig. 3a). This agrees well with similar studies reported for C7 asphaltene oxy-cracking35,46 and C7 asphaltene catalytic hydroprocessing.74
Fig. 4 shows the Arrhenius plot of the Q-65 oxy-cracking reaction for each step of the proposed triangular reaction mechanism. As seen, Arrhenius behavior was observed in the temperature range 200–230 °C. The values of activation energies and frequency factors are summarized in Table 1.
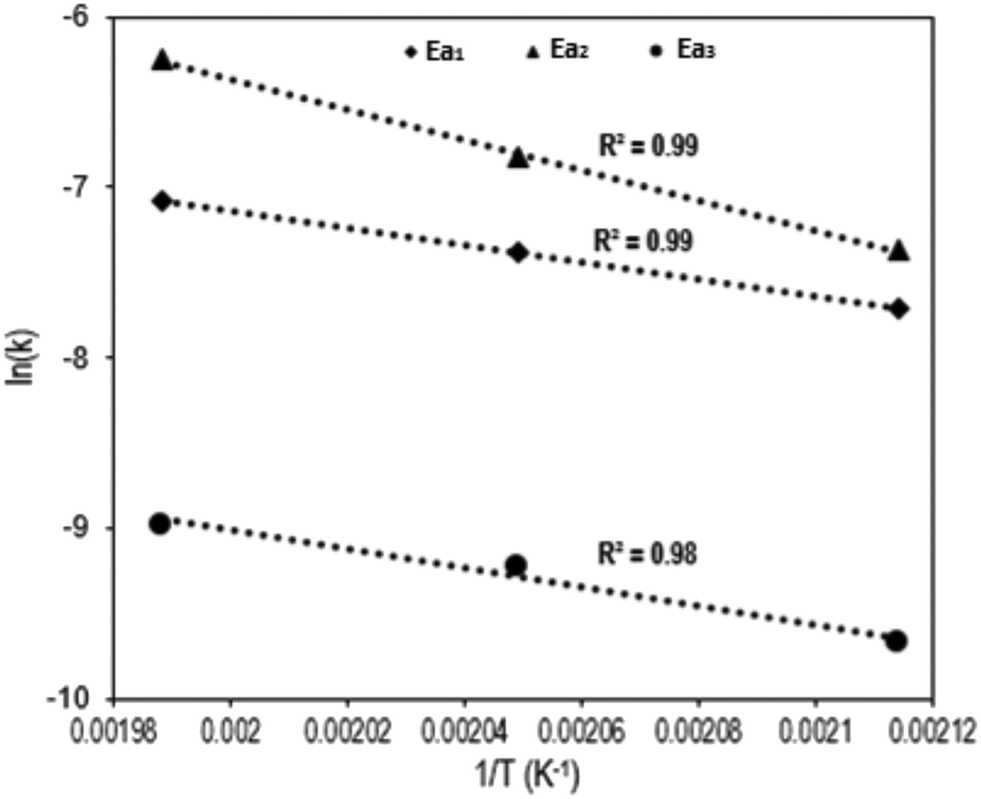 | ||
| Fig. 4 Arrhenius plots of Q-65 oxy-cracking for each reaction pathway. | ||
In our previous work regarding Q-65 thermo-oxidative decomposition, we proposed that the reaction begins with paraffinic chain loss, which was supported by both experimental and theoretical evidence.64 In this study, a similar behavior was observed. Initially during the oxy-cracking reaction, the alkyl chain presumably breaks up, followed by the dissociation of the weak carbons and sulfur bonds. Afterwards, complete cracking of the molecule takes place to produce analogs of humic substances and a small fraction of CO2, as reported in other works.78–81 A detailed insight into the reaction mechanism will be presented later in section 4.3.
The first reaction pathway (Ea1 = 9.8 kcal mol−1) involves a complete or deep oxidation reaction and might be favored to produce CO2 or demonstrates that Q-65 is partially soluble initially in the alkaline aqueous medium. In the second reaction pathway (Ea2 = 17.7 kcal mol−1), additional reactions are likely to occur due to the increased availability of aromatic moieties, forming heavier soluble and insoluble intermediates. This is also indicated by the higher value of the frequency factor (9.3 × 104 L mmol−1 s−1). However, CO2 could be produced in the third reaction pathway by total oxidation of some solubilized intermediates. Remarkably, the activation energies of the first and third reaction pathways are similar (Ea3 = 10.9 kcal mol−1). A similar trend was reported by Ashtari et al.35 for the oxy-cracking of C7 asphaltenes. However, the value of Ea2 obtained in the previous work35 was around 28.3 kcal mol−1. The difference in activation energies could be attributed to the structural complexity of asphaltene molecules and aggregation. Hence, asphaltene aggregates require more oxygen penetration during the oxy-cracking reaction to achieve the desired conversion and selectivity. However, Q-65 is more solubilized in water in comparison with asphaltene molecules.
4.2 FTIR spectroscopy of virgin and oxy-cracked Q-65
Fig. 5 shows the infrared spectra of the virgin and oxy-cracked Q-65 at 230 °C over a 2 h reaction period. The presence of oxygen in the aqueous phase causes the Q-65 molecules to oxy-crack into organic species bearing carboxylic and phenolic functional groups. A suggested mechanism of such a reaction will be further discussed (section 4.3). It is worth mentioning here that KOH aids the solubilization of Q-65 by neutralizing the acidic species forming water-soluble hydrocarbon salts. Many studies confirmed that the solubility of species containing oxygen increases in basic media.82,83 Patil et al.82 demonstrated that the solubility of lignin in water increased in the presence of NaOH.82 Likewise, n-C7 asphaltene oxy-cracking showed that the alkaline medium enhanced the cracking and subsequent solubilization of n-C7 asphaltenes in water.35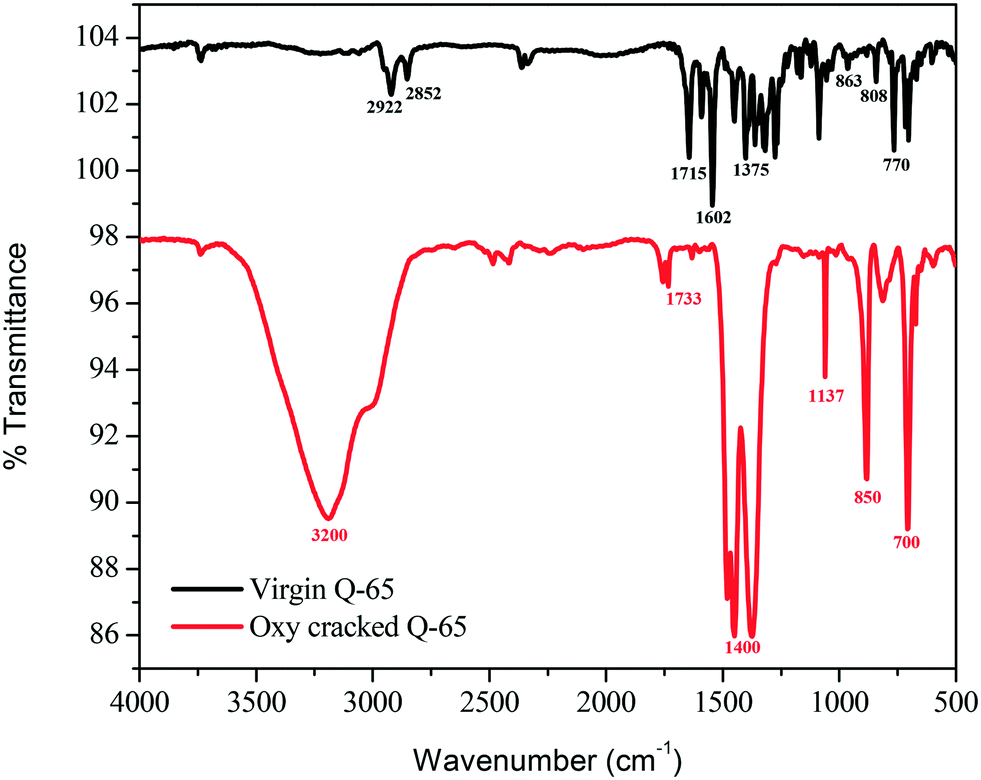 | ||
| Fig. 5 FTIR spectra of virgin and oxy-cracked Q-65 at 230 °C and 2 h residence time. | ||
As shown in Fig. 5, the featured bands of virgin Q-65 can be categorized into aliphatic and aromatic. For the aromatic bands, the C–H bending in 1,2-, 1,3-, and 1,4-disubstituted aromatics was detected at 770, 808, and 863 cm−1, respectively. These bands are associated with the out-of-plane bending C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration detected at 1602 cm−1 in the aromatic region. Furthermore, in the aliphatic region, the characteristic C–H stretching vibrations of alkyl groups were assigned at 2922 and 2852 cm−1. The bands at 1458 and 1375 cm−1 represent the C–H vibration of asymmetric and symmetric bending, respectively. The band corresponding to the CO group in Q-65 was represented at a frequency of 1715 cm−1. No additional bands which may imply the presence of impurities of a very different nature from Q-65 were observed.
C stretching vibration detected at 1602 cm−1 in the aromatic region. Furthermore, in the aliphatic region, the characteristic C–H stretching vibrations of alkyl groups were assigned at 2922 and 2852 cm−1. The bands at 1458 and 1375 cm−1 represent the C–H vibration of asymmetric and symmetric bending, respectively. The band corresponding to the CO group in Q-65 was represented at a frequency of 1715 cm−1. No additional bands which may imply the presence of impurities of a very different nature from Q-65 were observed.
The FTIR spectrum of the oxy-cracked Q-65 was dramatically different from that of virgin Q-65 (Fig. 5). The O–H band spanning between 3500–2500 cm−1 corresponds to –OH groups. This can originate from the presence of alcohols and/or H-bonded phenol groups. The presence of a carbonyl band at 1733 cm−1 was assigned to carboxylic groups. However, its contribution is negligible, indicating that these species are not important contributors to the spectrum, i.e., they are in salt forms. The most important doublet band centered at 1400 cm−1 was assigned to carboxylate anion stretching, suggesting that most carboxylic compounds are present as salts. The FTIR spectrum of oxy-cracked Q-65 shows the absence of the aromatic CC ring stretching band (1600 cm−1) and out-of-plane bands (950–750 cm−1) as compared to the virgin Q-65, indicating the disappearance of most aromatic moieties. Also, the alkyl groups are hardly visible in the range 3030–2800 cm−1. The IR band at 1137 cm−1 could be representative of sulfones (OSO) and those appearing at ∼700 and 850 cm−1 might correspond to sulphonic acid bonds. Overall, a similar water–asphaltene solubilization mechanism reported recently35,46 is also applicable to this analysis. It has been reported by Calemma et al.29 that the absorption bands between 1600 and 1800 cm−1 could belong to carboxyl groups, carbonyl groups, esters, aromatic esters, and ketones. This study showed that increasing the oxidation conditions activated the aromatic rings with hydroxyl groups. As a result, phenol groups are converted to quinones and subsequently transformed to carbonyl groups through the rupture of benzene rings.29 The above FTIR analysis of the virgin and oxy-cracked Q-65 demonstrates that phenolic and carboxylic functional groups are formed at 230 °C. More experimental evidence for the formation of such groups can be seen in the ESI† (S2.1 and S2.2).
4.3 Theoretical modeling
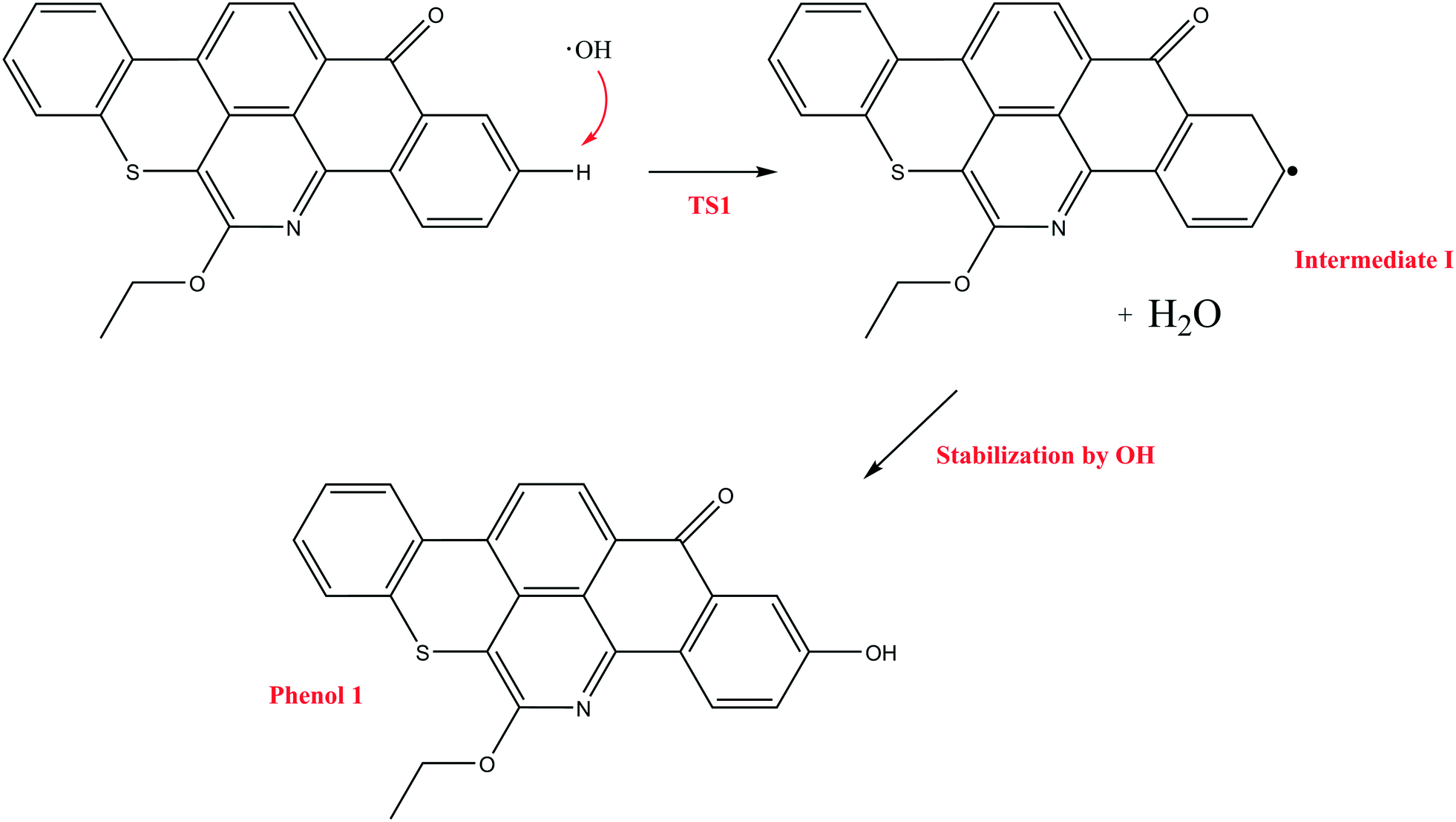 | ||
| Scheme 2 The reaction mechanism initiated by OH˙ attack on Q-65b to form H2O and phenol 1. | ||
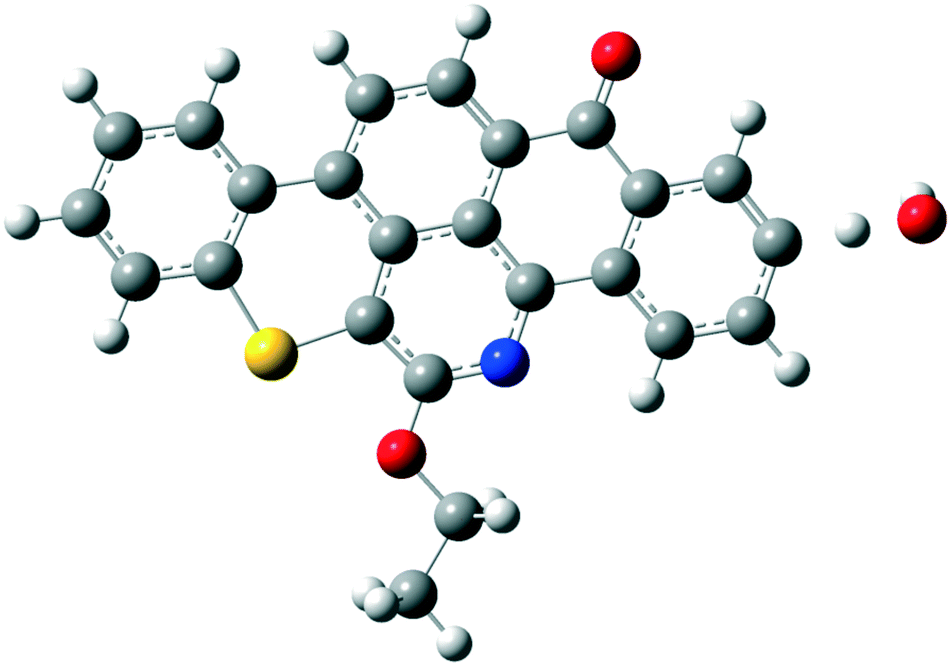 | ||
| Fig. 6 Optimized structure for the transition state of water formation (TS1). Grey atoms represent carbon, blue atoms represent nitrogen, white atoms represent hydrogen, yellow atoms represent sulfur and red atoms represent oxygen. | ||
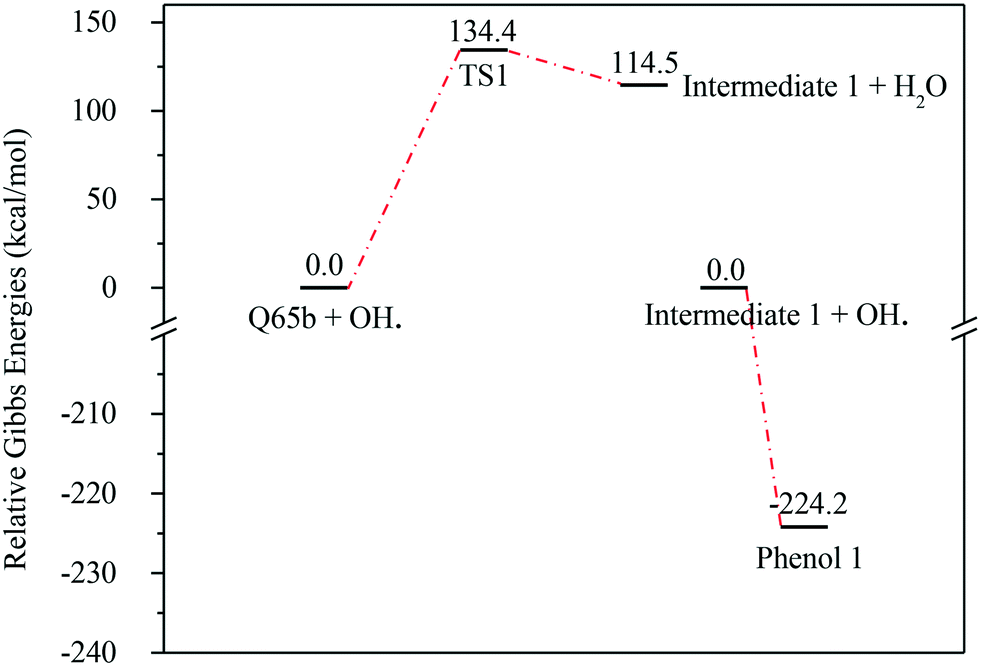 | ||
| Fig. 7 Energy level diagram for the water formation mechanism initiated by OH˙ attack on Q-65b. Energy values represent the relative Gibbs free energies at 298 K (ZPE corrections included). | ||
In contrast to the abstraction pathway, the direct attack of OH˙ on the aromatic carbon in Q-65b is more favourable. As depicted in Scheme 3, this reaction pathway proceeds first through the transition state TS2, leading to the formation of the radical intermediate II, followed by hydrogen loss via TS3 to form phenol I. The optimized structures for both transition states are shown in Fig. 8. The energy level diagram for this reaction pathway is plotted in Fig. 9. In addition to the slightly lower barrier than that in the first pathway (128.5 kcal mol−1), intermediate II is more stable than intermediate I, i.e., II lies 92.3 kcal mol−1 above Q-65b, whereas I lies 114.5 kcal mol−1 above Q-65b, as mentioned earlier.
 | ||
| Scheme 3 The formation mechanism of phenol I, initiated by OH˙ attack on Q-65b. | ||
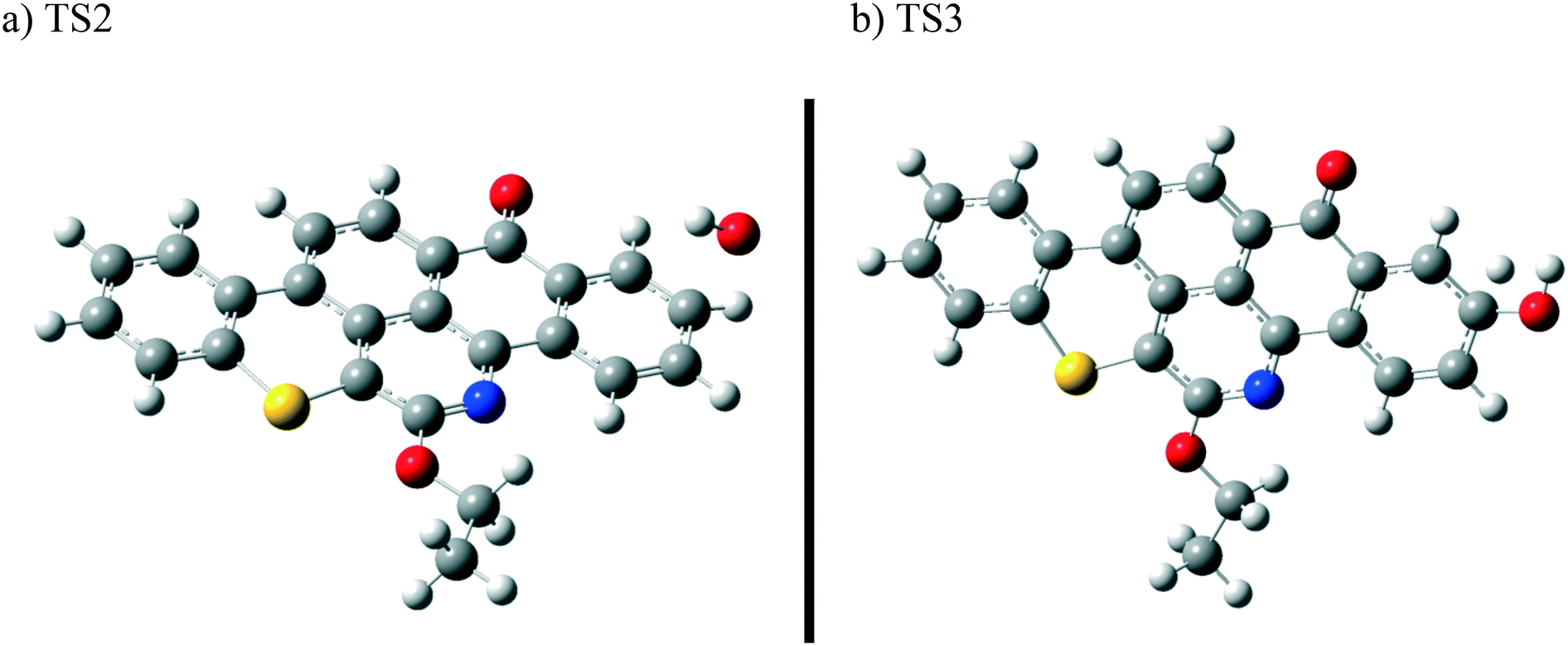 | ||
| Fig. 8 Optimized structures for the transition states involved in phenol I formation: a) TS2 and b) TS3. For the colour scheme, refer to Fig. 6. | ||
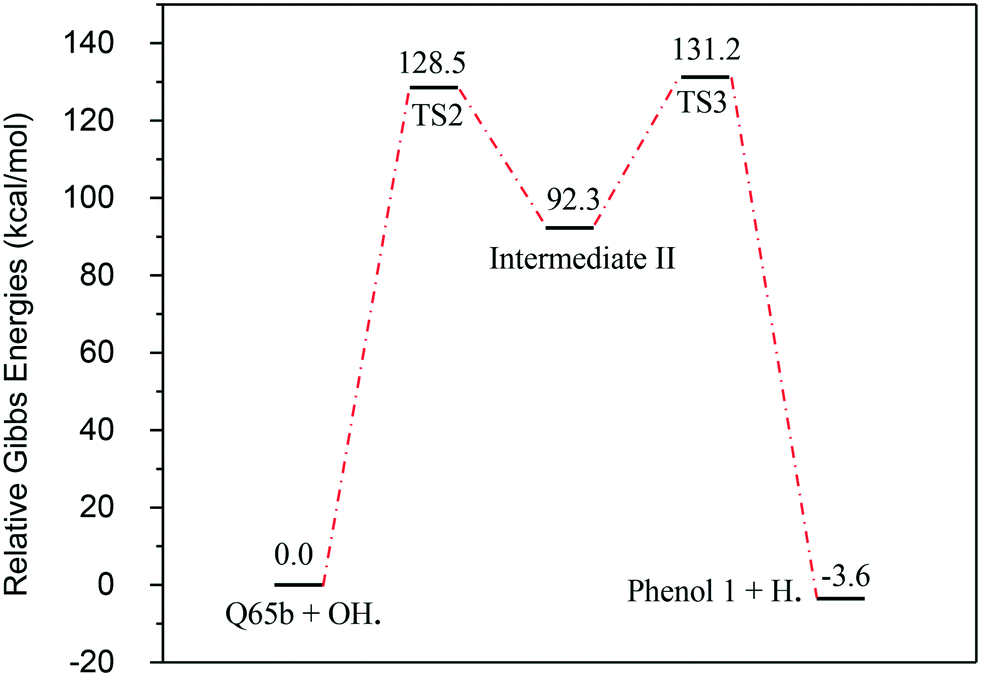 | ||
| Fig. 9 Energy level diagram for the phenol 1 formation mechanism initiated by OH˙ attack on Q-65b. Energy values represent the relative Gibbs free energies at 298 K (ZPE corrections included). | ||
It is worth noting that similar attack of OH˙ on aromatic carbon in small aromatic compounds, such as toluene86 and ethyl benzene,87 proceeds with very low barriers and have exothermic reaction enthalpies. Suh et al.86 calculated the reaction energies in the case of toluene to be −17.5 kcal mol−1 at the B3LYP/6-31G(d,p) level. The activation barrier was even negative at −3.2 kcal mol−1.86 The relatively high barrier and the large endothermic reaction energy of OH˙ attack on Q-65b can be explained by the high stability of the large aromatic system and the breakage of aromaticity caused by OH˙ attachment to the aromatic molecule.
The next reaction pathway involves the loss of the olefin chain in Q-65b; such loss can take place by a unimolecular 1,3-H shift mechanism, as explained in our previous study.64 Alternatively, the loss can occur by a primary attack of OH˙ on the carbon adjacent to nitrogen in Q-65b, where the lowest unoccupied molecular orbital (LUMO) is centred (details on the HOMO and LUMO of the Q-65b molecule were presented in our recent work64). This attack forms intermediate III through the transition state TS3. III can then decompose into phenol II and an ethoxy radical, as depicted in Scheme 4. The optimized structures of the two transition states involved in this mechanism are shown in Fig. 10. The proposed phenols suggested by this theoretical study (such as I and II) are in agreement with our FTIR observations, as shown earlier in section 4.2. The attack of OH˙ on the LUMO of Q-65b has an energy barrier and Gibbs free reaction energy close to those of the previous two attacks discussed earlier, as seen in Fig. 11. However, phenol II is more stable relative to phenol I, as clearly demonstrated from the two energy diagrams in Fig. 9 and 11.
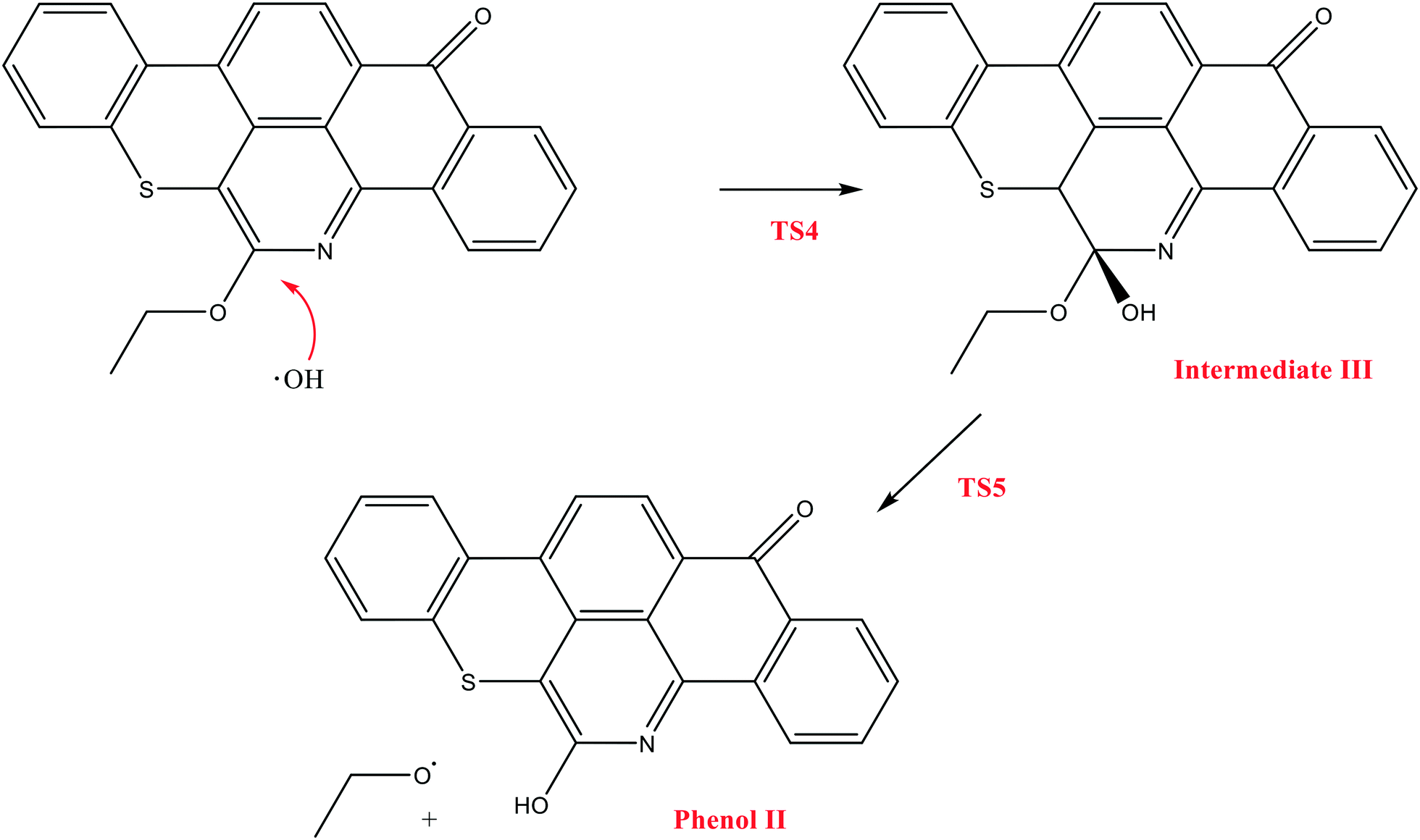 | ||
| Scheme 4 The formation mechanism of phenol II, initiated by OH˙ attack on the LUMO of Q-65b. | ||
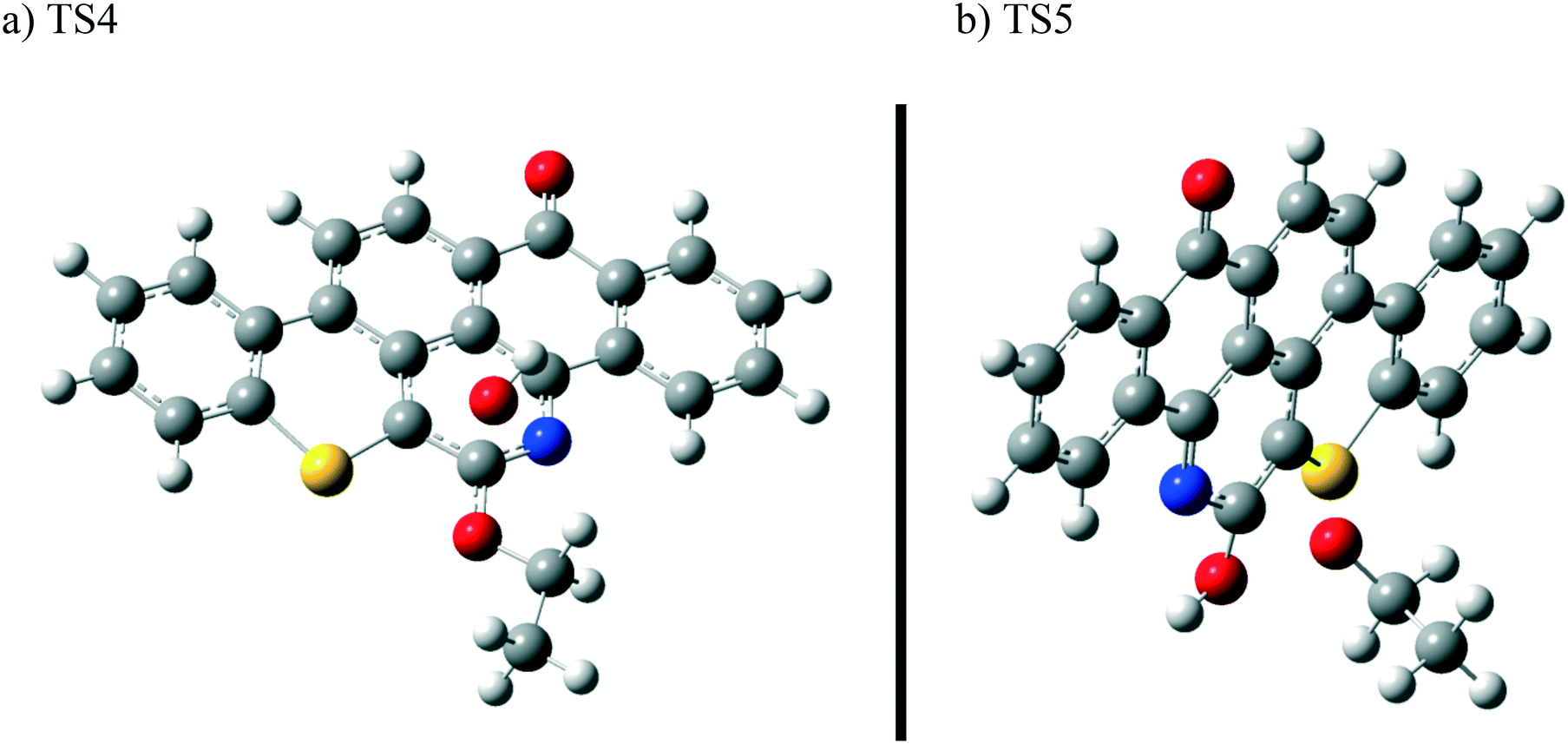 | ||
| Fig. 10 Optimized structures for the transition states involved in the formation of phenol 2: a) TS4 and b) TS5. For the colour scheme, refer to Fig. 6. | ||
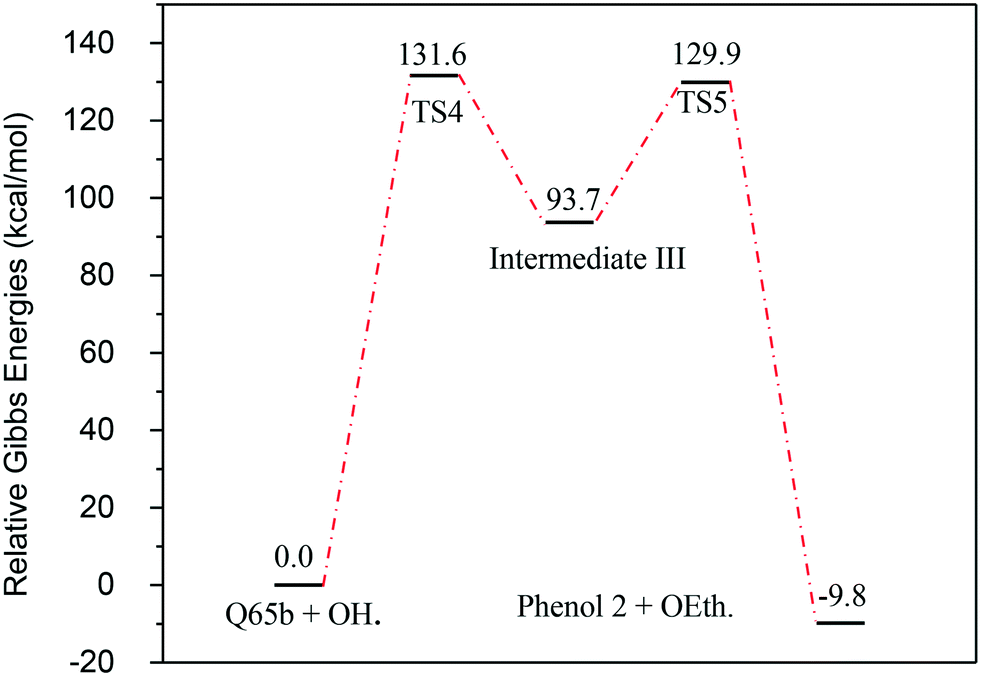 | ||
| Fig. 11 Energy level diagram for the phenol 2 formation mechanism initiated by OH˙ attack on Q-65b. Energy values represent the relative Gibbs free energies at 298 K (ZPE corrections included). | ||
The next reaction pathway of interest is the attack of OH˙ on the carbon adjacent to the carbonyl group in Q-65b, as illustrated in Scheme 5. In this mechanism, we chose to start from the product of the previous mechanism, phenol II, instead of the parent Q-65b molecule. This is based on the assumption that the course and the energies of the reactions would not be affected by this change. We have successfully located the transition state TS6, leading to the formation of the radical intermediate IV, which undergoes a quick hydrogen shift to form V through the transition state TS7, as shown in Scheme 5. Finally, intermediate V can be easily stabilized by an H radical to form phenol III, which also contains an aldehyde group.
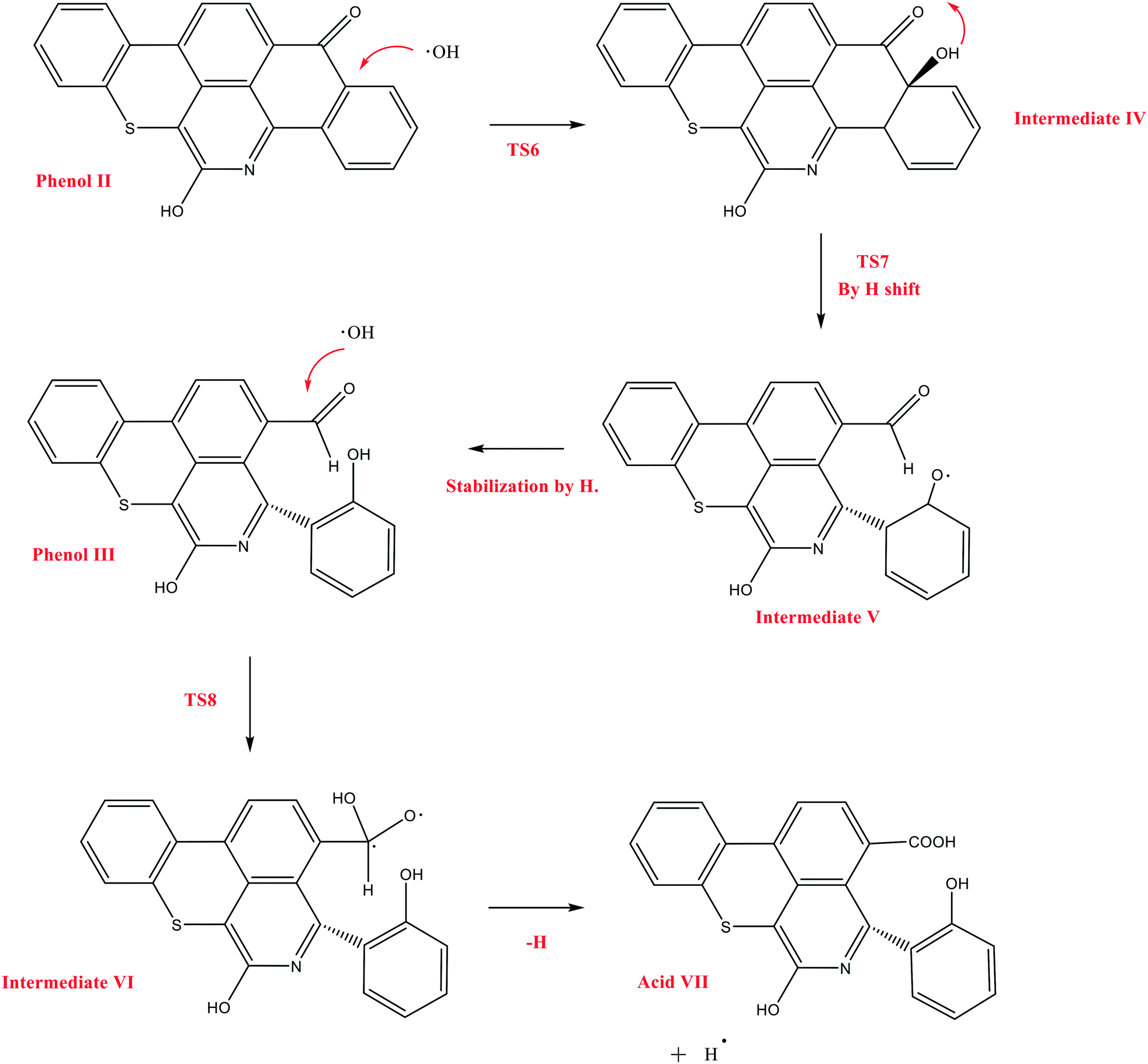 | ||
| Scheme 5 The decomposition mechanism of Q-65b, initiated by the attack of OH˙, and leading to the formation of acid VII. | ||
Other possible oxidations of phenol III to form a carboxylic acid have been explored. As seen from Scheme 5, this can take place by the attack of an OH˙ radical to phenol III, forming the intermediate VI, through a transition state TS8. Hydrogen loss converts intermediate VI into the carboxylic acid VII. The optimized structures of the transition states participating in this mechanism, along with acid VII, are shown in Fig. 12. The formation of the aldehyde group (phenol III) and the carboxylic acid shown in Scheme 5 is in agreement with the experimental results obtained in section 4.2.
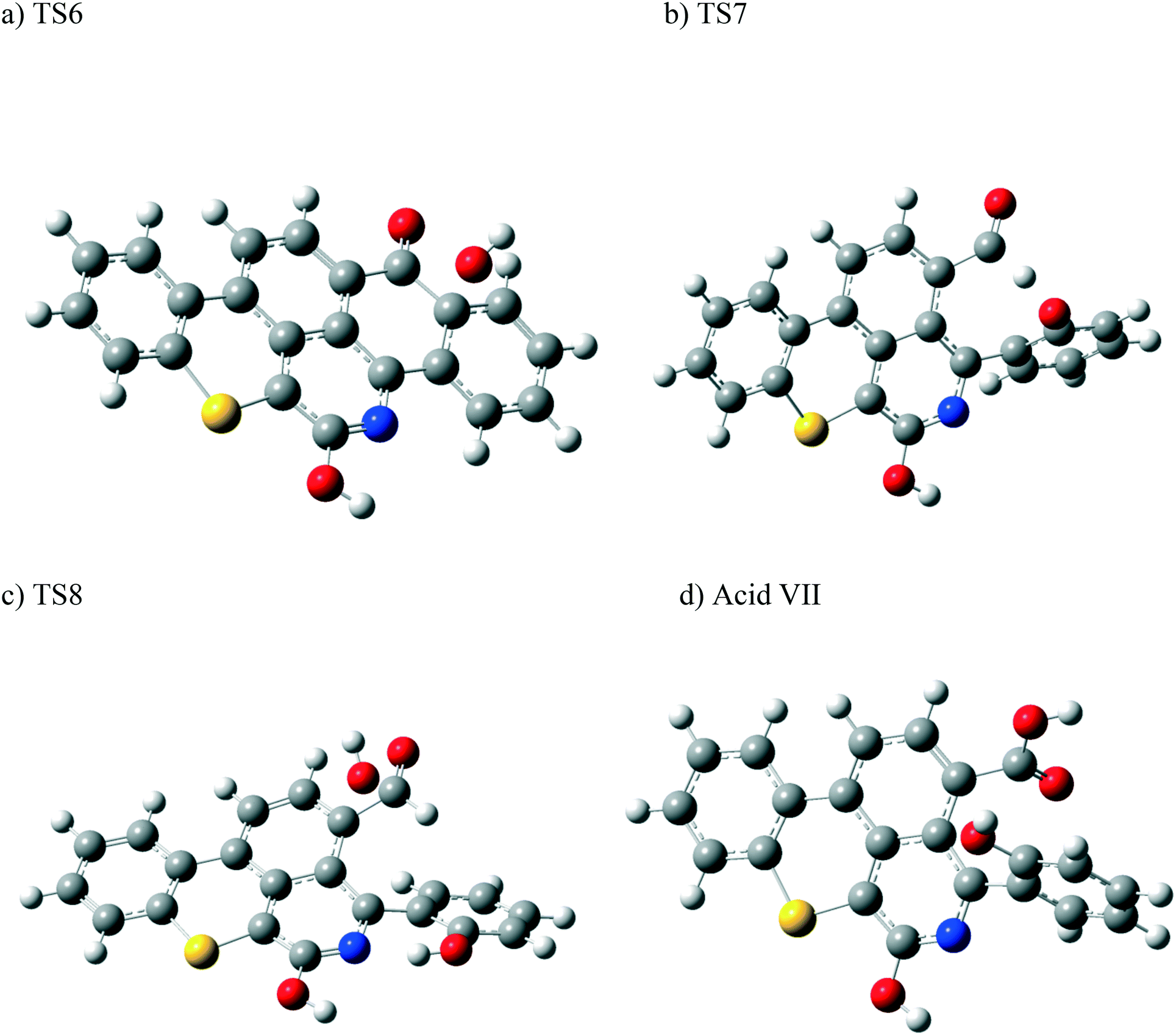 | ||
| Fig. 12 Optimized structures for the transition states involved in the formation of the carboxylic acid VII: a) TS6, b) TS7, and c) TS8. d) Optimized structure for the acid VII. For the colour scheme, refer to Fig. 6. | ||
Fig. 13 represents the energy level diagram for the reaction mechanism initiated by OH˙ radical attack on phenol III. The first activation barrier in this mechanism is high (127.8 kcal mol−1). This suggests that this reaction pathway is not kinetically favourable, similarly to the previous two mechanisms. However, once this barrier is reached, the next barrier from intermediate IV to V is relatively small (41.9 kcal mol−1). In addition, the stabilization of V into phenol III is very exothermic (−195.5 kcal mol−1). Despite the fact that this reaction pathway is kinetically unfavourable, it is thermodynamically preferred, due to the huge Gibbs free energy of reaction released by the formation of phenol III. This is similar to the previous case where phenol I is formed, as shown in Fig. 7.
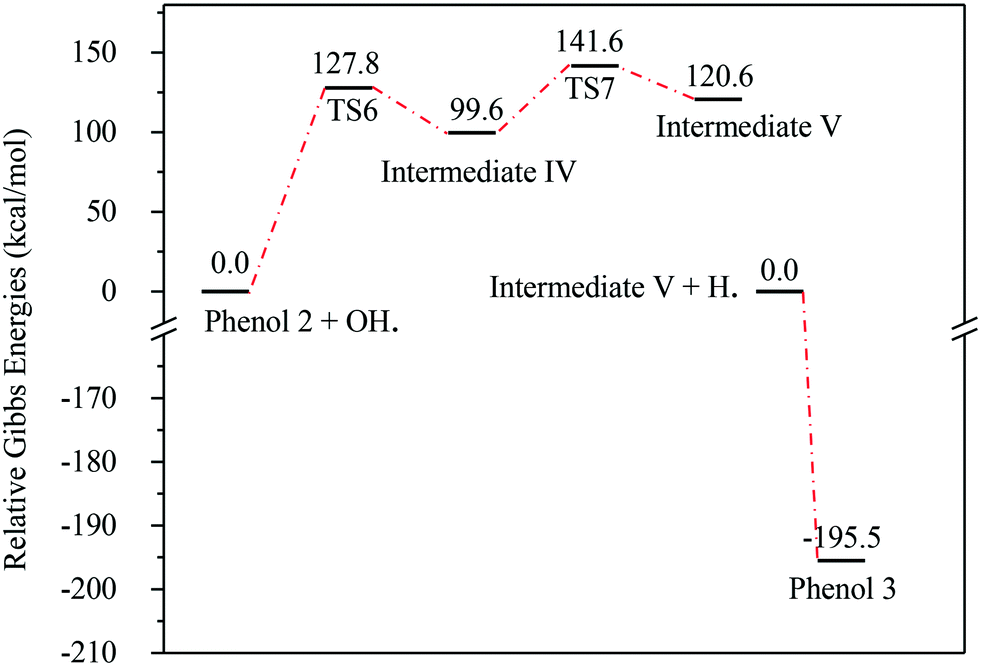 | ||
| Fig. 13 Energy level diagram for the phenol 3 formation mechanism initiated by OH˙ attack on phenol 2. Energy values represent the relative Gibbs free energies at 298 K (ZPE corrections included). | ||
The energies associated with the formation of the carboxylic acid VII are plotted in Fig. 14. Similar to the previous reaction pathways, the primary step for this reaction is also unfavourable due to the relatively high Gibbs free energy of activation (ΔG‡298) (134.3 kcal mol−1). Since the whole reaction is exergonic by −17.5 kcal mol−1, this reaction pathway is thermodynamically favourable.
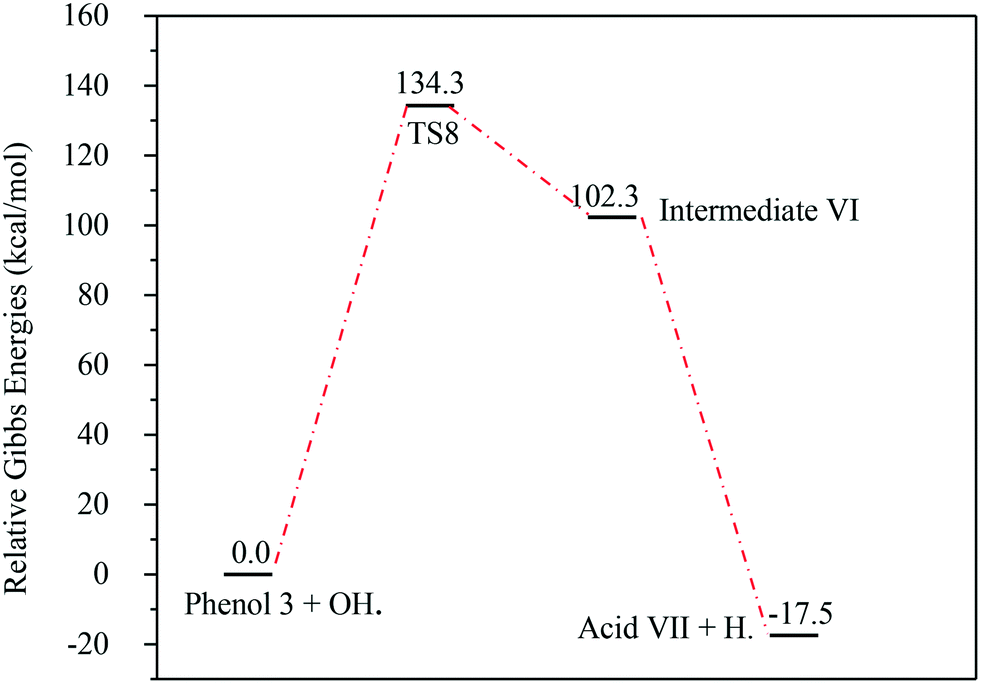 | ||
| Fig. 14 Energy level diagram for the acid VII formation mechanism initiated by OH˙ attack on phenol 3. Energy values represent the relative Gibbs free energies at 298 K (ZPE corrections included). | ||
Thus, different reaction pathways of the Q-65b molecule initiated by the OH˙ radical have been demonstrated. Although the role of the heteroatoms, S and N, in the reaction mechanism is not initially obvious, they help to create a location prone to free radical attack. It is known that direct attacks of free radicals on heteroatoms are not favourable.84,88 Thus, oxidation of heterocyclic compounds is more likely to occur from free radical attacks on carbon atoms adjacent to these heteroatoms. This prediction has been confirmed by performing calculations on the Q-65b molecule. It was discovered that attacks of OH˙ radicals on the heteroatoms N and S were highly energetic.
| Reactions with OH˙ | Reactions with OH− | |||
|---|---|---|---|---|
| ΔG‡298 (kcal mol−1) | k 298 (s−1) | ΔG‡298 (kcal mol−1) | k 298 (s−1) | |
| TS1 | 134.4 | 1.6 × 10−86 | ||
| TS2 | 128.5 | 3.3 × 10−82 | ||
| TS3 | 38.9 | 1.7 × 10−16 | ||
| TS4 | 131.6 | 1.9 × 10−84 | 16.3 | 7.1 |
| TS5 | 36.1 | 2.0 × 10−14 | 5.0 | 1.4 × 109 |
| TS6 | 127.8 | 1.8 × 10−82 | 23.8 | 2.2 × 10−5 |
| TS7 | 41.9 | 1.0 × 10−18 | 27.4 | 4.9 × 10−8 |
| TS8 | 134.3 | 1.9 × 10−86 | 18.0 | 0.41 |
Clearly, the activation barriers are much lower in the case of OH−. This leads to higher rate constants as Table 2 shows. Based on these findings, and because of the fact that the calculated rate constants obtained from the experimental part are closer to those obtained for OH− using DFT calculations, we propose that the oxy-cracking of Q-65 might also be initiated by OH− attacks on the aromatic molecule. This is supported by the fact that the reaction was performed under aqueous basic conditions (pH > 8). In addition, and since the activation barrier for TS4 is the lowest among all other reaction barriers involved in this work, we propose that the OH− attack on the LUMO of Q-65 is the most favourable reaction pathway. This observation is actually in agreement with our previous study on the thermal oxidation of Q-65.64 In addition, we cannot exclude the role of O and O2 species in initiating the primary steps of oxidizing the Q-65 molecule. In our previous work,64 the kinetic and thermodynamic parameters obtained from the attack of singlet atomic oxygen (O 1D) on Q-65 were also close to those obtained from the experiment.
In order to fully understand why the theoretical kinetic parameters for the OH˙ reactions with Q-65b obtained are different from the experimental ones, a thermal and pressure analysis over the temperature range of 25 to 500 °C and in the pressure range of 15 to 735 psi was conducted. Details on this procedure are explained elsewhere.64,71,72 Surprisingly, and according to the results of this analysis, increasing the temperature or the pressure did not decrease the calculated values of ΔG‡298 and ΔG0298. Because the reactions studied in this work are bimolecular and form a single product, the results of the temperature and pressure analysis can be explained using the entropy approach. It is also noteworthy to mention that increasing the pressure above 750 psi in this experimental setup did not alter the reaction kinetics of Q-65b.
5. Conclusions
The oxy-cracking process of the solid-waste hydrocarbon model molecule, Quinolin-65 (Q-65), was experimentally investigated in a batch reactor at different operating temperatures of 200, 215, and 230 °C and a constant oxygen partial pressure of 750 psi. The reaction residence times ranged between 0 and 2 h to establish the reaction kinetics. The concentration of the solubilized organic compounds in the liquid phase was monitored by total organic carbon (TOC) analysis, while the gas products were analyzed online using gas chromatography (GC). The Q-65 was oxy-cracked consecutively into different pathways in which oxidative decomposition took place in the first step producing different aromatic intermediates. The latter compounds were partially oxidized into different families of organic acids and a small amount of CO2. Furthermore, the oxy-cracked Q-65 has been characterized using FTIR, NMR and XPS techniques. The results showed that various types of oxygenated hydrocarbons were produced, such as carboxylic acids and phenolic compounds. To support our experimental findings, a theoretical study has been carried out by using the density functional theory (DFT) and the second-order Møller–Plesset perturbation theory (MP2) under the same proposed experimental conditions. The theoretical study succeeded in exploring many oxidation pathways of the Q-65 molecule initiated by the hydroxyl radical (OH˙). However, the Gibbs free energies of activation calculated from the OH˙ mechanisms were much higher than those obtained from the experiment. Since our experiments were carried out in a strong basic medium (pH = 13), and the fact that the pH dropped from 13 to 12 during the reaction, the theoretical calculations were performed on the hydroxide anion (OH−). The obtained activation energies were found to be much lower than those of the OH˙ radical and closer to the experimental ones. This suggests that the hydroxide anions play a role in the oxy-cracking reaction under our experimental conditions. In brief, the novelty of this study is considered as a vital backbone for the real oxy-cracking application of petroleum residue which will be addressed in the near future.Acknowledgements
The authors are grateful to the Natural Sciences and Engineering Research Council of Canada (NSERC), the Department of Chemical and Petroleum Engineering at the Schulich School of Engineering at the University of Calgary and the Western Canada Research Grid. The authors acknowledge Marianna Trujillo for helping in the NMR analysis.References
- M. Asif and T. Muneer, Energy supply, its demand and security issues for developed and emerging economies, Renewable Sustainable Energy Rev., 2007, 11(7), 1388–1413 CrossRef.
- B. Liddle, Impact of population, age structure, and urbanization on carbon emissions/energy consumption: evidence from macro-level, cross-country analyses, Popul. Environ., 2014, 35(3), 286–304 CrossRef.
- S. Brinklov, E. K. V. Kalko and A. Surlykke, Intense echolocation calls from two 'whispering' bats, Artibeus jamaicensis and Macrophyllum macrophyllum (Phyllostomidae), J. Exp. Biol., 2009, 212(1), 11–20 CrossRef PubMed.
- M. Anandan and S. Ramaswamy, Global oil market: Macro economic scenario, Glob. J. Res. Anal., 2016, 4(9), 53–57 Search PubMed.
- W. Zubot, M. D. MacKinnon, P. Chelme-Ayala, D. W. Smith and M. G. El-Din, Petroleum coke adsorption as a water management option for oil sands process-affected water, Sci. Total Environ., 2012, 427, 364–372 CrossRef PubMed.
- J.-H. Lv, X. Wei, Y.-H. Wang, T.-M. Wang, J. Liu, D.-D. Zhang and Z.-M. Zong, Characterization of condensed aromatics and heteroatomic species in Yanshan petroleum coke through ruthenium ion-catalyzed oxidation using three mass spectrometers, RSC Adv., 2016, 6, 61758–61770 RSC.
- P. Pourrezaei, A. Alpatova, P. Chelme-Ayala, L. Perez-Estrada, M. Jensen-Fontaine, X. Le and M. G. El-Din, Impact of petroleum coke characteristics on the adsorption of the organic fractions from oil sands process-affected water, Int. J. Environ. Sci. Technol., 2014, 11(7), 2037–2050 CrossRef CAS.
- J. M. Hill, A. Karimi and M. Malekshahian, Characterization, gasification, activation, and potential uses for the millions of tonnes of petroleum coke produced in Canada each year, Can. J. Chem. Eng., 2014, 92(9), 1618–1626 CrossRef CAS.
- N. N. Nassar, A. Hassan and P. Pereira-Almao, Application of nanotechnology for heavy oil upgrading: Catalytic steam gasification/cracking of asphaltenes, Energy Fuels, 2011, 25(4), 1566–1570 CrossRef CAS.
- M. Malekshahian and J. M. Hill, Kinetic analysis of CO2 gasification of petroleum coke at high pressures, Energy Fuels, 2011, 25(9), 4043–4048 CrossRef CAS.
- Y. Zhang, S. Nagamori, S. Hinchiranan, T. Vitidsant and N. Tsubaki, Promotional effects of Al2O3 addition to Co/SiO2 catalysts for Fischer−Tropsch synthesis, Energy Fuels, 2006, 20(2), 417–421 CrossRef CAS.
- A. P. Steynberg and H. G. Nel, Clean coal conversion options using Fischer–Tropsch technology, Fuel, 2004, 83(6), 765–770 CrossRef CAS.
- E. Mobil, FLEXICOKING™ Conversion Technology. http://www.exxonmobil.com/Apps/RefiningTechnologies/Files/sellsheet_06_flexicoking.pdf. (Accessed 6 February 2017).
- B. Group, BP statistical review of world energy June 2011, 2011. http://www.bp.com/content/dam/bp-country/de_de/PDFs/brochures/statistical_review_of_world_energy_full_report_2011.pdf. (Accessed October 15 2016).
- C. E. Baukal, The john zink hamworthy combustion handbook: Volume 1-Fundamentals, CRC press, 2012 Search PubMed.
- J. Wang, E. J. Anthony and J. C. Abanades, Clean and efficient use of petroleum coke for combustion and power generation, Fuel, 2004, 83(10), 1341–1348 CrossRef CAS.
- A. N. Sawarkar, A. B. Pandit, S. D. Samant and J. B. Joshi, Petroleum residue upgrading via delayed coking: A review, Can. J. Chem. Eng., 2007, 85(1), 1–24 CrossRef CAS.
- M. S. Rana, V. Samano, J. Ancheyta and J. Diaz, A review of recent advances on process technologies for upgrading of heavy oils and residua, Fuel, 2007, 86(9), 1216–1231 CrossRef CAS.
- F. M. Dautzenberg and J. C. De Deken, Reactor developments in hydrotreating and conversion of residues, Catal. Rev.: Sci. Eng., 1984, 26(3–4), 421–444 CAS.
- G. B. Brons, M. Siskin and K. O. Wrzeszczynski, Upgrading of bitumen asphaltenes by hot water treatment (C-2726), US Pat., 5326456, 1994 Search PubMed.
- O. R. Koseoglu and A. Bourane, System and process for integrated oxidative desulfurization, desalting and deasphalting of hydrocarbon feedstocks, US Pat., 2011/0226666, 2011 Search PubMed.
- J. G. Speight, New approaches to hydroprocessing, Catal. Today, 2004, 98(1), 55–60 CrossRef CAS.
- I. Mochida, X. Z. Zhao and K. Sakanishi, Catalyst deactivation during the hydrotreatment of asphaltene in an Australian brown coal liquid, Fuel, 1988, 67(8), 1101–1105 CrossRef CAS.
- S. Maity, V. Perez, J. Ancheyta, M. Rana and G. Centeno, Effect of asphaltene contained in feed on deactivation of Maya crude hydrotreating catalyst, Pet. Sci. Technol., 2007, 25(1–2), 241–249 CrossRef CAS.
- J. Levec and A. Pintar, Catalytic wet-air oxidation processes: a review, Catal. Today, 2007, 124(3), 172–184 CrossRef CAS.
- R. V. Shende and J. Levec, Kinetics of wet oxidation of propionic and 3-hydroxypropionic acids, Ind. Eng. Chem. Res., 1999, 38(7), 2557–2563 CrossRef CAS.
- F. Luck, Wet air oxidation: past, present and future, Catal. Today, 1999, 53(1), 81–91 CrossRef CAS.
- S. S. Fong, L. Seng, N. B. Majri and H. B. Mat, A comparative evaluation on the oxidative approaches for extraction of humic acids from low rank coal of Mukah, Sarawak, J. Braz. Chem. Soc., 2007, 18(1), 34–40 CAS.
- V. Calemma and R. Rausa, Process for the production of regenerated humic acids from coal, US Pat., 4788360, 1988 Search PubMed.
- E. M. Peña-Méndez, J. Havel and J. Patočka, Humic substances–compounds of still unknown structure: applications in agriculture, industry, environment, and biomedicine, J. Appl. Biomed, 2005, 3(1), 13–24 Search PubMed.
- S. E. Moschopedis, Sulfomethylation of humic acids, lignites, and coals and products thereof, US Pat., 3352902, 1967 Search PubMed.
- V. Calemma, R. Rausa, R. Margarit and E. Girardi, FT-ir study of coal oxidation at low temperature, Fuel, 1988, 67(6), 764–770 CrossRef CAS.
- B. Ignasiak, Production of metallurgical coke from oxidized caking coal, US Pat., 4259083, 1981 Search PubMed.
- E. Gorin and T. S. Robert, Size separation, preoxidation and fluidized low temperature carbonization of coal, US Pat., 3047472, 1962 Search PubMed.
- M. Ashtari, L. Carbognani Ortega, F. Lopez-Linares, A. Eldood and P. Pereira-Almao, New pathways for asphaltenes upgrading using the oxy-cracking process, Energy Fuels, 2016, 30(6), 4596–4608 CrossRef CAS.
- V. Platonov, A. Kudrya and S. Proskuryakov, Ozonolysis of asphaltenes from semicoking tar of G17 coal, Russ. J. Appl. Chem., 2003, 76(1), 148–152 CrossRef CAS.
- G. Escobar, P. Patiño, S. Acevedo, O. Escobar, M. A. Ranaudo and J. C. Pereira, Interfacial properties of the products of ozonolysis of hamaca crude oil, Pet. Sci. Technol., 2001, 19(1–2), 107–118 CrossRef CAS.
- S. E. Moschopedis and J. G. Speight, Chemical modification of bitumen heavy ends and their non-fuel uses, Shale Oil, Tar Sands, and Related Fuel Sources, Am. Chem. Soc., 1976, 144–152 CAS.
- S. E. Moschopedis and J. G. Speight, Oxidative degradation of Athabasca asphaltenes, Fuel, 1971, 50(2), 211–217 CrossRef CAS.
- F. J. Rivas, S. T. Kolaczkowski, F. J. Beltrán and D. B. McLurgh, Development of a model for the wet air oxidation of phenol based on a free radical mechanism, Chem. Eng. Sci., 1998, 53(14), 2575–2586 CrossRef CAS.
- M. N. Ingale, J. B. Joshi, V. V. Mahajani and M. K. Gada, Waste treatment of an aqueous waste stream from a cyclohexane oxidation unit: A case study, Process Saf. Environ. Prot., 1996, 74(4), 265–272 CrossRef CAS.
- S. K. Bhargava, J. Tardio, J. Prasad, K. Föger, D. B. Akolekar and S. C. Grocott, Wet oxidation and catalytic wet oxidation, Ind. Eng. Chem. Res., 2006, 45(4), 1221–1258 CrossRef CAS.
- L. Li, P. Chen and E. F. Gloyna, Generalized kinetic model for wet oxidation of organic compounds, AIChE J., 1991, 37(11), 1687–1697 CrossRef CAS.
- A. Lifshitz and J. V. Michael, Rate constants for the reaction, O+H2O→OH+OH, over the temperature range, 1500–2400 K, by the flash photolysis-shock tube technique: A further consideration of the back reaction, Symp. (Int.) Combust., [Proc.], 1991, 23(1), 59–67 CrossRef.
- S. Imamura, Catalytic and noncatalytic wet oxidation, Ind. Eng. Chem. Res., 1999, 38(5), 1743–1753 CrossRef CAS.
- M. Ashtari, L. Carbognani and P. Pereira-Almao, Asphaltenes aqueous conversion to humic and fulvic analogs via oxy-cracking, Energy Fuels, 2016, 30(7), 5470–5482 CrossRef CAS.
- P. Haghighat, L. C. Ortega and P. Pereira-Almao, Experimental study on catalytic hydroprocessing of solubilized asphaltene in water: A proof of concept to upgrade asphaltene in the aqueous phase, Energy Fuels, 2016, 30(4), 2904–2918 CrossRef CAS.
- P. Haghighat, L. Carbognani and P. Pereira-Almao, Experimental study on catalytic hydroprocessing of solubilized asphaltene in water: A proof of concept to upgrade asphaltene in the aqueous phase, Energy Fuels, 2016, 30(4), 2904–2918 CrossRef CAS.
- R. C. Silva, J. R. Radović, F. Ahmed, U. Ehrmann, M. Brown, L. Carbognani Ortega, S. Larter, P. Pereira-Almao and T. B. Oldenburg, Characterization of acid-soluble oxidized asphaltenes by Fourier transform ion cyclotron resonance mass spectrometry: Insights on oxycracking processes and asphaltene structural features, Energy Fuels, 2015, 30(1), 171–179 CrossRef.
- N. Rodriguez, H. Marsh, E. Heintz, R. Sherwood and R. Baker, Oxidation studies of various petroleum cokes, Carbon, 1987, 25(5), 629–635 CrossRef CAS.
- M. Legin-Kolar and D. Ugarković, Petroleum coke structure: Influence of feedstock composition, Carbon, 1993, 31(2), 383–390 CrossRef CAS.
- K. Akbarzadeh, H. Alboudwarej, W. Y. Svrcek and H. W. Yarranton, A generalized regular solution model for asphaltene precipitation from n-alkane diluted heavy oils and bitumens, Fluid Phase Equilib., 2005, 232(1), 159–170 CrossRef CAS.
- O. Castellano, R. Gimon and H. Soscun, Theoretical study of the σ–π and π–π interactions in heteroaromatic monocyclic molecular complexes of benzene, pyridine, and thiophene dimers: implications on the resin–asphaltene stability in crude oil, Energy Fuels, 2011, 25(6), 2526–2541 CrossRef CAS.
- P. G. Redelius, The structure of asphaltenes in bitumen, Road Mater. Pavement Des., 2006, 7(sup1), 143–162 CrossRef.
- J. J. Adams, Asphaltene adsorption, a literature review, Energy Fuels, 2014, 28(5), 2831–2856 CrossRef CAS.
- H. Wu and M. R. Kessler, Asphaltene: Structural characterization, molecular functionalization, and application as a low-cost filler in epoxy composites, RSC Adv., 2015, 5(31), 24264–24273 RSC.
- O. C. Mullins, The modified Yen model, Energy Fuels, 2010, 24(4), 2179–2207 CrossRef CAS.
- H. Groenzin and O. C. Mullins, Asphaltene molecular size and structure, J. Phys. Chem. A, 1999, 103(50), 11237–11245 CrossRef CAS.
- F. López-Linares, L. Carbognani, M. F. González, C. Sosa-Stull, M. Figueras and P. Pereira-Almao, Quinolin-65 and Violanthrone-79 as model molecules for the kinetics of the adsorption of C7 Athabasca asphaltene on macroporous solid surfaces, Energy Fuels, 2006, 20(6), 2748–2750 CrossRef.
- M. F. González, C. S. Stull, F. López-Linares and P. Pereira-Almao, Comparing asphaltene adsorption with model heavy molecules over macroporous solid surfaces, Energy Fuels, 2007, 21(1), 234–241 CrossRef.
- N. N. Marei, N. N. Nassar and G. Vitale, The effect of the nanosize on surface properties of NiO nanoparticles for the adsorption of Quinolin-65, Phys. Chem. Chem. Phys., 2016, 18(9), 6839–6849 RSC.
- A. Devard, R. Pujro, G. de la Puente and U. Sedran, Hydrocarbon yield structure in the conversion of heavy model molecules (Quinolin-65) on fluidized catalytic cracking catalysts, Energy Fuels, 2012, 26(8), 5015–5019 CrossRef CAS.
- N. N. Marei, N. N. Nassar, G. Vitale, A. Hassan and M. J. P. Zurita, Effects of the size of NiO nanoparticles on the catalytic oxidation of Quinolin-65 as an asphaltene model compound, Fuel, 2017, 207, 423–437 CrossRef CAS.
- I. Badran, N. N. Nassar, N. N. Marei and A. Hassan, Theoretical and thermogravimetric study on the thermo-oxidative decomposition of Quinolin-65 as an asphaltene model molecule, RSC Adv., 2016, 6(59), 54418–54430 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., R. A. Gaussian09, 1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38(6), 3098–3100 CrossRef CAS.
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33(12), 8822–8824 CrossRef.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements, J. Chem. Phys., 1988, 89(4), 2193–2218 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- A. P. Scott and L. Radom, Harmonic vibrational frequencies: An evaluation of Hartree−Fock, Møller−Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors, J. Chem. Phys., 1996, 100(41), 16502–16513 CrossRef CAS.
- I. Badran, Hot-wire chemical vapour deposition chemistry and kinetics of new precursors in the gas phase and on the wire surface, Department of Chemistry, University of Calgary, Calgary, Alberta, Canada, 2014, p. 310 Search PubMed.
- I. Badran, A. Rauk and Y. J. Shi, Theoretical study on the ring-opening of 1,3-disilacyclobutane and H2 elimination, J. Phys. Chem. A, 2012, 116(48), 11806–11816 CrossRef CAS PubMed.
- J. I. Steinfeld, J. S. Francisco and W. L. Hase, Chemical kinetics and dynamics, Prentice Hall, Upper Saddle River, N.J., 2nd edn, 1999 Search PubMed.
- P. Haghighat, L. Carbognani and P. Pereira-Almao, Kinetic study of preoxidized asphaltene hydroprocessing in aqueous phase, Energy Fuels, 2016, 30(7), 5617–5629 CrossRef CAS.
- J. H. Sinfelt and J. Wiley, Heterogeneous reactions: analysis, examples, and reactor design, Ann Arbor, 1984, vol. 1001, p. 48109 Search PubMed.
- A. Pintar, G. Berčič, M. Besson and P. Gallezot, Catalytic wet-air oxidation of industrial effluents: total mineralization of organics and lumped kinetic modelling, Appl. Catal., B, 2004, 47(3), 143–152 CrossRef CAS.
- G. Astarita, Lumping nonlinear kinetics: apparent overall order of reaction, AIChE J., 1989, 35(4), 529–532 CrossRef CAS.
- N. N. Nassar, C. A. Franco, T. Montoya, F. B. Cortés and A. Hassan, Effect of oxide support on Ni–Pd bimetallic nanocatalysts for steam gasification of nC 7 asphaltenes, Fuel, 2015, 156, 110–120 CrossRef CAS.
- N. N. Nassar, A. Hassan and P. Pereira-Almao, Thermogravimetric studies on catalytic effect of metal oxide nanoparticles on asphaltene pyrolysis under inert conditions, J. Therm. Anal. Calorim., 2011, 110(3), 1327–1332 CrossRef.
- Y. Hamedi Shokrlu and T. Babadagli, In-situ upgrading of heavy oil/bitumen during steam injection by use of metal nanoparticles: A study on in-situ catalysis and catalyst transportation, SPE Reservoir Eval. Eng., 2013, 16(03), 333–344 Search PubMed.
- M. N. Siddiqui, Catalytic pyrolysis of Arab heavy residue and effects on the chemistry of asphaltene, J. Anal. Appl. Pyrolysis, 2010, 89(2), 278–285 CrossRef CAS.
- P. T. Patil, U. Armbruster, M. Richter and A. Martin, Heterogeneously catalyzed hydroprocessing of organosolv lignin in sub-and supercritical solvents, Energy Fuels, 2011, 25(10), 4713–4722 CrossRef CAS.
- H. Konnerth, J. Zhang, D. Ma, M. H. Prechtl and N. Yan, Base promoted hydrogenolysis of lignin model compounds and organosolv lignin over metal catalysts in water, Chem. Eng. Sci., 2015, 123, 155–163 CrossRef CAS.
- C. Barckholtz, T. A. Barckholtz and C. M. Hadad, A mechanistic study of the reactions of H, O (3P), and OH with monocyclic aromatic hydrocarbons by density functional theory, J. Phys. Chem. A, 2001, 105(1), 140–152 CrossRef CAS.
- C.-Y. Lin and M. C. Lin, Thermal decomposition of methyl phenyl ether in shock waves: the kinetics of phenoxy radical reactions, J. Phys. Chem. A, 1986, 90(3), 425–431 CrossRef CAS.
- I. Suh, D. Zhang, R. Zhang, L. T. Molina and M. J. Molina, Theoretical study of OH addition reaction to toluene, Chem. Phys. Lett., 2002, 364(5–6), 454–462 CrossRef CAS.
- M. Huang, Z. Wang, L. Hao and W. Zhang, Theoretical investigation on the mechanism and kinetics of OH radical with ethylbenzene, Int. J. Quantum Chem., 2011, 111(12), 3125–3134 CrossRef CAS.
- A. Mellouki, G. Le Bras and H. Sidebottom, Kinetics and mechanisms of the oxidation of oxygenated organic compounds in the gas phase, Chem. Rev., 2003, 103(12), 5077–5096 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00048k |
| This journal is © The Royal Society of Chemistry 2017 |