
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew findings about human dipeptidyl peptidase III based on mutations found in cancer†
M. Matovina a,
D. Agićb,
M. Abramića,
S. Matića,
Z. Karačića and
S. Tomić*a
a,
D. Agićb,
M. Abramića,
S. Matića,
Z. Karačića and
S. Tomić*a
aDivision of Organic Chemistry and Biochemistry, Ruđer Bošković Institute, Bijenička cesta 54, Zagreb, Croatia. E-mail: sanja.tomic@irb.hr
bJosip Juraj Strossmayer University of Osijek, Faculty of Agriculture in Osijek, Vladimira Preloga 1, Osijek, Croatia
First published on 21st July 2017
Abstract
Dipeptidyl peptidase III (DPP III) is a cytosolic enzyme belonging to the metallopeptidase family M49, involved in the final steps of protein catabolism. More than a hundred missense mutations can be found in the coding region of the human DPP3 gene when searching cBioPortal for Cancer Genomics. The role of two highly conserved residues in the family M49, whose mutations G313W and R510W were detected in human cancer, was investigated using combined experimental and computational approaches (substrate docking and MD simulations). Several mutants of human DPP III were expressed and purified as recombinant proteins, and their biochemical properties were determined. The conservative substitution of Arg510 with lysine mildly decreased enzyme activity activity for Arg-Arg-2-naphtylamide substrate, while the substitutions of Arg510 with glutamine and Gly313 with alanine substantially decreased enzyme activity, and tryptophan substitutions found in cancer, G313W and R510W, almost abolished enzyme activity. MD simulations showed that substitution of Gly313, and especially Arg510 with tryptophan, significantly increases the enzyme flexibility, particularly that of the binding site including the H450ELLGH455 motif, and influences the substrate interactions with the catalytic His568. The results clearly indicate that, besides the enzyme structure, its dynamics properties also significantly influence the human DPP III activity.
Introduction
The dipeptidyl-peptidase III (DPP III; EC 3.4.14.4) is a monozinc exopeptidase from the metallopeptidase family M49 (according to the MEROPS database, http://www.merops.ac.uk), found to be broadly distributed in mammalian tissues, and thought to contribute to the final steps of normal intracellular protein catabolism. There are strong indications of its role in the endogenous pain-modulation system,1 as well as in the endogenous defence against oxidative stress.2 Pathophysiological roles of human DPP III (h.DPP III) are indicated in malignant growth,3,4 and influenza virus infection.5 Recently, it was proven that DPP III contributes to the activation of transcription factor Nrf2, a constituent of the Nrf2-Keap1 signalling pathway, the main defence mechanism against many environmentally toxic agents and carcinogens in cells.6,7 However, (patho)physiological roles of DPP III are still not elucidated, and better understanding of its broad substrate specificity would be a step toward understanding its biological function.From a structural point of view, the DPP III is a two-domain protein with the motifs HEXXGH and EE*XR(K)AE(D) considered as its trademark.8,9 Histidines from the first motif and glutamate* from the second one take part in the zinc ion coordination, while the glutamate from the first motif is crucial for the catalytic activity of DPP III, namely the hydrolysis of dipeptides from the N-terminus of its substrates consisting of three or more amino acids.10
Until now several 3D structures of h.DPP III have been determined, ligand free and in complex with natural peptides. The structure of the ligand-free enzyme (PDB codes: 3FVY and 5EGY) was determined in the so-called open and closed form, while that of the liganded enzyme only in a more compact, closed, form (PDB codes: 3T6B, 3T6J, 5E33, 5E3A, 5E2Q, 5EHH, 5E3C).11 The open, ligand-free enzyme structure is characterized with the wide inter-domain cleft (∼40 Å wide and ∼40 Å high) with the catalytic zinc ion bound at its “roof”. Using computational approaches we have traced the protein closure,12,13 gained better insight into enzyme broad substrate specificity and determined the reaction mechanism of h.DPP III.14–16
More than hundred missense mutations can be found in the coding region of h.DPP III gene by searching cBioPortal for Cancer Genomics (http://www.cbioportal.org/; accessed September 2014).17,18 Most of those mutations were predicted to have low or medium impact on protein function, however, two of them, G313W and R510W, which reside in the conserved regions 1 and 4 of DPP III,9 were predicted to have high influence on protein functionality. Those mutations were found in the tumour samples from lung adenocarcinoma, and colorectal adenocarcinoma samples, respectively. Mutation G313W was found in the provisional TCGA (The Cancer Genome Atlas) study, and was removed from newer releases of cBioPortal (http://www.cbioportal.org/; accessed October 2015), however, this study was aimed at finding the cause of the DPP III inactivation by this mutation, and not at studying the potential role of these mutations in the development of cancer so we decided to proceed with the research of this mutant regardless of that. We investigated the influence of those mutations on the enzyme biochemical and physicochemical properties. Those mutants exhibited almost complete loss of activity towards synthetic substrate, Arg-Arg-2-naphthylamide (Arg2-2NA; RRNA) compared to the wild type (wt) h.DPP III, and we set out to determine the cause of this activity loss by combining experimental approaches with computational simulations. In order to determine the root of the activity loss in these two mutants, we investigated three other amino acid substitutions, in which the native amino acid residues were substituted by an amino acid that differs less from the original than mutants found in tumour cells. In this way we were able to gain better understanding of how amino acid substitutions at particular position influence enzyme structure and activity. Altogether, we analysed 5 different mutants, G313W and R510W that were found in cancer, and the more similar (conserved) amino acid substitutions at the same positions, G313A, R510K and R510Q.
Results and discussion
Biochemical properties of h.DPP III mutants
We purified and biochemically characterized two recombinant protein mutants of h.DPP III, G313W and R510W, that were found in lung adenocarcinoma, and colorectal adenocarcinoma samples respectively (http://www.cbioportal.org/; accessed September 2014), in order to test their enzymatic activities in comparison to wt h.DPPIII. Both residues are located in conserved regions of the family M49 (DPP III family), with R510 being the part of EECRAE motif of the active site, and G313 being the part of the first conserved region (amino acids 313 to 332).9 Both Gly313 and Arg510 are completely conserved among M49 family members (Fig. S1†). Since these amino acid substitutions, G313W and R510W, almost abolished h.DPP III activity towards its preferred Arg2-2NA substrate (Table 1), we purified and biochemically characterized three more mutants with more conservative amino acid substitutions at the same positions, G313A, R510K, and R510Q.| Protein h.DPP III | Specific activity/nmol min−1 mg−1 | RA Ala-Arg-2NA/% | RA Phe-Arg-2NA/% |
|---|---|---|---|
| a RA Relative activity towards substrates (normalized to that for Arg2-2NA). | |||
| WT | 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 900 |
26 | 27 |
| G313W | 2 | 40 | 40 |
| G313A | 76 | 6 | 20 |
| R510W | 31 | 18 | 23 |
| R510K | 21515 |
25 | 19 |
| R510Q | 3284 | 5 | 5 |
Measurements of specific activity of the wt h.DPP III and the mutants towards Arg2-2NA showed that tryptophan mutants, G313W and R510W, have approximately 24000, and 1600 fold lower specific activity than the wt h.DPP III, respectively. Substitution of alanine at amino acid residue 313 increased the activity of the protein compared to the G313W mutant, however, it was still considerably lower than the wt h.DPP III, showing that glycine at position 313 is essential for the h.DPP III activity. On the other hand, substitution by lysine instead of tryptophan at position 510 returned the specific activity of the mutant to approximately half of the activity of the wt h.DPP III, while substitution by glutamine raised enzyme activity comparing to the R510W mutant, but the mutant R510Q still had approximately 14 fold lower specific activity than the wt h.DPP III (Table 1). That result showed that for the efficient enzymatic activity h.DPP III requires a positively charged amino acid at position 510. The pH optimum for Arg2-2NA hydrolysis was around 8.6 for R510 mutants, while G313W, and G313A had the optimum at pH 8.0, and 7.4, respectively. The studied substitutions slightly change the relative preferences for different dipeptidyl 2-naphthylamides (Arg2-2NA, Ala-Arg-2NA and Phe-Arg-2NA) compared to the wt, however, Arg2-2NA was still the preferred substrate for all mutants (Table 1).
The catalytic properties of the wt h.DPP III and all mutated forms were determined for the hydrolysis of characteristic synthetic substrate Arg2-2NA from two or three independent measurements at pH 8.6 (Table 2), except for the G313A whose catalytic properties were determined from one measurement at pH 7.4. We did not determine the G313W kinetic parameters due to extremely low enzymatic activity of that mutant. All tested proteins showed similar KM as the wt h.DPP III, except G313A that had more than 20 times higher KM, while kcat differed by 2–3 orders of magnitude for mutants G313A and R510W, respectively. The kcat value of R510Q was 26 times lower than the wt, and R510K had similar kcat as the wt.
| Protein h.DPP III | KM/μM | kcat/s−1 | kcat/KM × 106 M−1 s−1 |
|---|---|---|---|
| a The values are the average ± st. dev. of 2 (WT and R510W) or 3 (R510K and R510Q) independent measurements at pH 8.6, except G313A for which we conducted 1 measurement at pH 7.4. | |||
| WT | 5.45 ± 0.53 | 35.400 ± 6.364 | 6.475 ± 0.538 |
| G313W | — | — | — |
| G313A | 114.57 | 0.224 | 0.002 |
| R510W | 4.48 ± 1.49 | 0.012 ± 0.003 | 0.003 ± 0.000 |
| R510K | 4.06 ± 0.87 | 27.533 ± 9.905 | 7.066 ± 3.269 |
| R510Q | 2.10 ± 1.32 | 1.350 ± 0.365 | 0.864 ± 0.533 |
We have revealed or confirmed the importance of several conserved amino acid residues situated outside of the two active-site motifs H450ELLGH455 and E507ECRAE512, in human DPP III (Tyr318, Trp300, His568, Asp496), so far.19–22 Among previously characterized h.DPP III mutants, H568N showed the lowest kcat value for the hydrolysis of Arg2-2NA, three orders of magnitude lower than the wt h.DPP III,21 without significant change in KM value. According to steady-state kinetic parameters, this protein variant is similar to R510W characterized in our present study.
Molecular simulations
We simulated 6 different variants of h.DPP III, wild type and its mutants, G313A, G313W, R510K, R510Q and R510W. All variants were simulated without substrate and in complex with the substrate Arg2-2NA (RRNA), see Table 3. For each variant we performed four MD simulations (100 ns long), two for the ligand free enzyme variant and two for its complex with Arg2-2NA, see Table S1.† Structure of the wt h.DPP III–RRNA complex is shown in Fig. 1 and positions of the amino acid residues mutated in this study are indicated. Influence of the mutations to the protein structure and flexibility was studied using the structure of the ligand free enzyme in its open form, while the influence of the point mutations on the ligand binding was studied using the closed form of the enzyme complexed with the preferred synthetic substrate Arg2-2NA. Our earlier studies13,14,16 showed that such a closed enzyme form is the active one, i.e. it ensures environment suitable for the peptide hydrolysis. Orientation of Arg2-2NA in the active site of the closed form closely resembles the position of Leu-enkephalin in the human DPP III binding site, for which the reaction mechanism was determined using QM/MM approach.16| h.DPP III variant | Zn2+ coordination | Rgyr | |||
|---|---|---|---|---|---|
| H450 | H455 | E451 | E508 | Range (Å) | |
| a In the case of two simulation of variants, R510K and R510Q, His455 reoriented and during about 4.5% of the total simulation time (200 ns) have not coordinated the zinc ion. | |||||
| WT | + | + | m | m | 26.9–28.4 |
| G313W | + | + | m | m/b | 26.2–28.6 |
| G313A | + | + | m | m/b | 26.4–28.5 |
| R510K | + | ±a | m | m/b | 26.3–28.2 |
| R510W | + | + | m | m | 26.2–28.3 |
| R510Q | + | ±a | m | m | 26.4–28.4 |
| WT-closed | + | + | m | m/b | 25.0–25.5 |
| WT-RRNA | + | + | m | b | 24.9–25.5 |
| G313W-RRNA | + | + | m/b | b | 24.8–25.6 |
| G313A-RRNA | + | + | m/b | b | 24.9–25.5 |
| R510W-RRNA | + | + | m | b | 24.9–25.7 |
| R510Q-RRNA | + | + | m/b | m/b | 24.8–25.5 |
| R510K-RRNA | + | + | m | m/b | 24.8–25.7 |
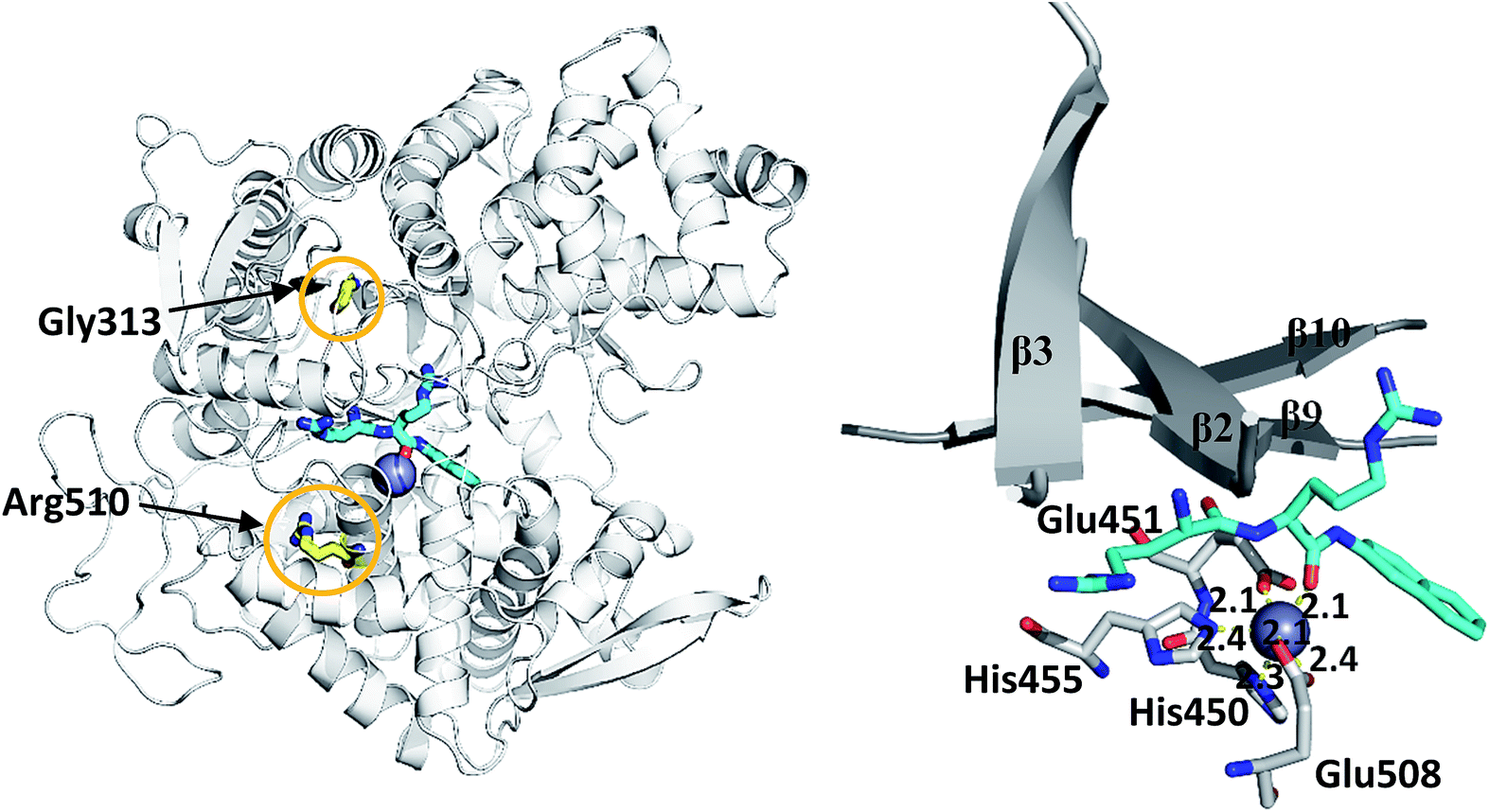 | ||
| Fig. 1 (left) Structure of wild type DPP III (light grey) in complex with preferred synthetic substrate Arg2-2NA (cyan), with marked positions of mutated residues (yellow) and the active site Zn2+ (grey sphere), (right) part of the beta-sheet core of the enzyme and the coordination of Zn2+ obtained after 100 ns of MD simulations. The distances (in Å) between coordinating atoms and Zn2+ are presented. | ||
For the G313W variant, two different orientation of the indole ring were considered. In one, the Trp side chain at position 313 points towards the active site and in the other it points toward the lower domain interior. However, since the orientation toward the lower domain interior turned out to be unfavourable, in this work we consider only results obtained with Trp313 pointing towards the interdomain cleft active site. According to the results of MD simulations, the point mutations at positions 313 and 510, G313A, G313W, R510K, R510Q, and R510W do not influence the metal ion coordination (Table 3). The simulations showed that, like in the wt protein,25 the Zn2+ ion is mostly hexacoordinated in the simulated mutants, as well. In the ligand free variants the metal dication was coordinated by two histidines, H450 and H455, two glutamates, E451 and E508, and one or two water molecules, depending on the position of E451 and E508 carboxyl groups, during the simulations. The water molecules coordinating Zn2+ often exchanged with bulk water during the simulations and they are not shown in Fig. 1. In the case of two simulation (variants R510K and R510Q) His455 reoriented, and during about 4.5% of the total simulation time have not coordinated the zinc ion. However, during the simulations of the other two replicas of the same variants the zinc coordination has been preserved during entire simulation time. Apparently, the zinc ion coordination is mostly preserved in all simulations and rarely occurring reorientation of H455 can be attributed to the stochastic nature of the simulations.
MD-simulations of the ligand free h.DPP III variants
Although the mutations at positions 313 and 510 do not significantly influence the overall protein fold and the Zn2+ ion coordination, they induce local structural changes which distort the active site conformation, and in this way hinder substrate positioning in the orientation that is the most suitable for the enzymatic reaction. Gly313 is located in the β6 sheet of the h.DPP III lower domain. At the end of the sheet there is a Glu316 residue, which upon protein closure approaches the substrate and interacts electrostatically with the N-terminal amino group of its first amino acid residue (Fig. S2†). The residue conservation within M49 family and large decrease of the G313W mutant enzyme activity, suggest that Gly residue at position 313 is essential for proper positioning of the substrate upon closing of the domains.MD simulations revealed that, although Gly313 does not interact with the substrate, it dictates the position of Glu316.
Indeed, Trp at position 313 distorts the β6 sheet conformation, changes the Glu316 position and prevents proper substrate positioning in the active site (Fig. 2 and S2†). The kinetic parameters of G313W mutant could not be determined due to its low activity and KM of the more conservative mutant G313A was significantly increased compared to the wt (Table 2). This supports the finding that the mutation in G313 in general does not prevent the substrate binding, but disturbs its positioning in the active site in the catalytically productive orientation.
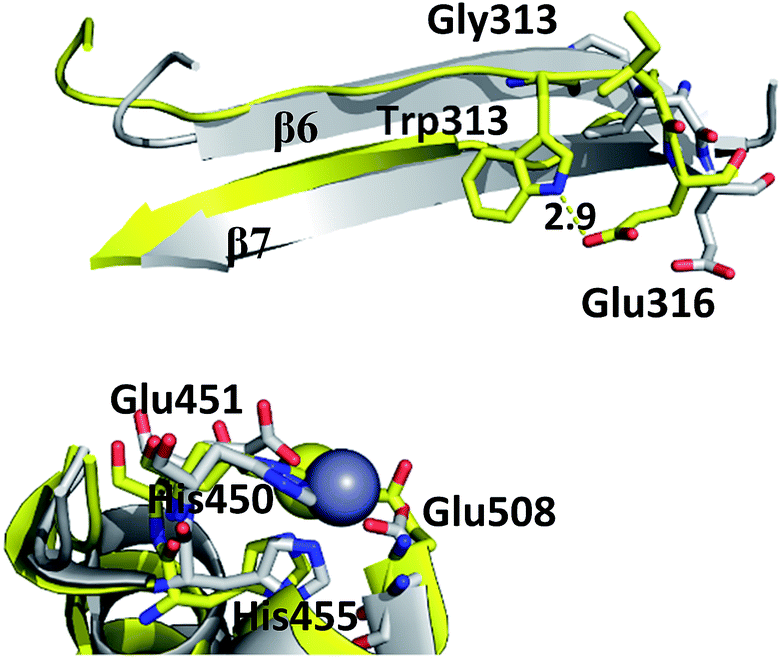 | ||
| Fig. 2 Position of residues 313 (Gly and Trp, respectively) and Glu316 in β-sheets β6–β7 in wt protein (grey) and mutant variant (yellow), in representative structures of 100 ns simulations. The distance (in Å) between Trp313 and Glu316 of mutant structure is labeled. Zn2+ is represented as a grey sphere and the residues of active site are shown as sticks. | ||
In order to get an insight into the overall protein flexibility, and to find out how the point mutations influence this flexibility, we computed the average Root Mean Square Fluctuations (RMSF) for the wild type protein and its mutants (Fig. 3). The results show that the lower (satellite) domain, especially its N and C terminal segments (residues 4 to 300 and 680 to 726, respectively) are the most flexible parts of the enzyme, followed by the substrate binding β-sheet region, consisting of two β-strands, β9 and β10. Analysis of the RMSF clearly indicated that mutations, either at position 313 or in position 510, increase protein flexibility, especially of the lower domain β-core (β-sheets β2–β3 and β9–β10). The R510K flexibility closely resembles that of the wild type protein.
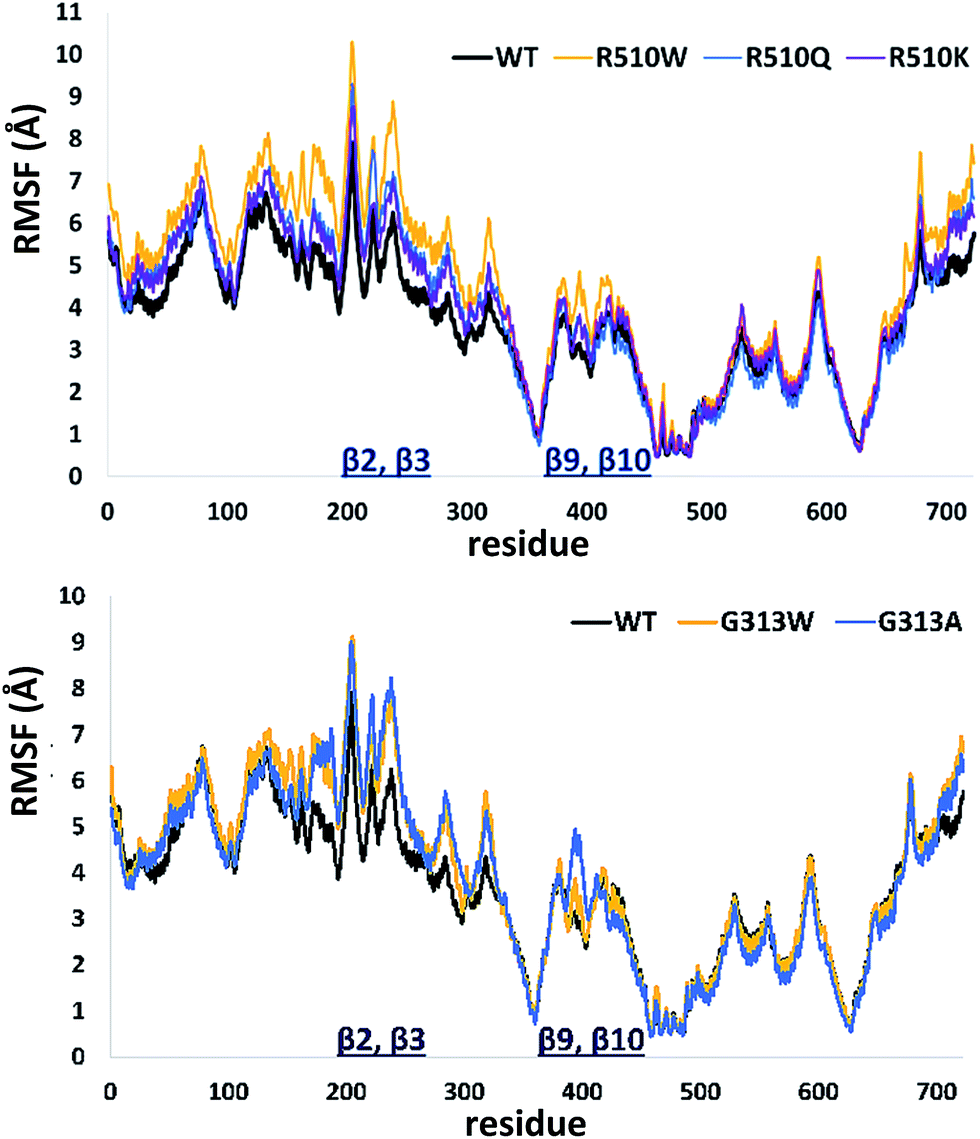 | ||
| Fig. 3 Root Mean Square Fluctuations (RMSF) profiles of the human DPP III variants obtained by averaging over two 100 ns long simulations (see Table S1†). | ||
Apparently, increased flexibility of the lower domain β-core, especially of β-sheets β2–β3 and β9–β10, is negatively correlated with the enzyme activity. The mutants with the highest fluctuations determined during MD simulations were also showing the lowest activity. On the other hand, both the activity and flexibility of R510K are very similar to those of the wild type protein. Further on, the RMSF profile revealed that substitution of Arg510 by Trp significantly increases fluctuations of the H450ELLGH455 motif and its surrounding (Fig. 3 and S3†).
Consequently, the interactions between the amino acid residues from this region with the rest of the protein are weaker in mutants than in the wt h.DPP III. In the wt protein, Arg510 interacts electrostatically with carbonyl groups of Gly454, Leu453 and His455 (Fig. 4). The Arg510 to Trp and Gln mutations resulted with decrease of these interactions and as a consequence flexibility of this motif increased in accord with the RMSF results. The highest fluctuation of the H450ELLGH455 motif has been found in the R510W mutant, followed by R510Q, while the fluctuations in the R510K mutant and the wild type protein are similar (Fig. S3†). It seems that destabilization of the H450ELLGH455 motif does not disturb the Zn2+ coordination in the R510W and R510Q mutants but it significantly influences the enzymatic activity. This is not surprising if we know that Glu451 directly participates in the peptide bond hydrolysis, acting as a general base during the deprotonation of water molecule that attacks the scissile amide bond.16 The increased fluctuations of this motif in the Arg510 mutants has been observed in both open and closed protein forms. Apparently, the H450ELLGH455 instability can be accounted for the decreased peptidase activity of h.DPP III mutants.
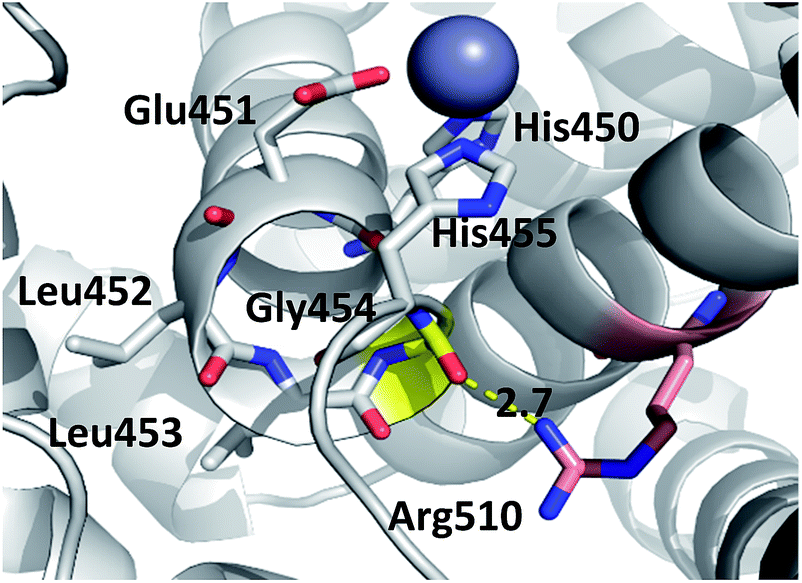 | ||
| Fig. 4 Position of highly conserved residue R510 (pink) in the structure of h.DPP3 (PDB 3FVY) interacting with Gly454 (yellow, distance in Å) from the conserved H450ELLGH455 motif (shown as sticks) responsible for coordinating Zn2+ (grey sphere). | ||
Additionally, the hydrogen bond between residue 510 and Ser457, present in the wild type enzyme, is significantly less stable in Arg510 mutants, while the Arg510–Gln626 hydrogen bond is preserved in the R510Q mutant, but not in R510W and R510K mutants (Table 4). Further on, the hydrogen bond between fully conserved Tyr318 and Glu327 is preserved in the R510Q but not in R510K and R510W mutants.
| Residues | % H-bond | |||||
|---|---|---|---|---|---|---|
| Variant | ||||||
| wt | G313W | G313A | R510W | R510Q | R510K | |
| Y318–E327 | 66 | 87 | 52 | 31 | 78 | 34 |
| H450–H455 | 22 | 22 | 19 | 17 | 19 | 14 |
| E451–N406 | 59 | 41 | 11 | 49 | 52 | 55 |
| H455–S457 | 2 | 3 | 3 | 2 | 3 | 16 |
| H455–E507 | 84 | 89 | 86 | 63 | 77 | 73 |
| E508–S504 | 8 | 11 | 6 | 5 | 4 | 8 |
| E512–H568 | 68 | 74 | 32 | 73 | 70 | 61 |
| X510–G454 | 78 | 80 | 74 | 35 | 12 | 13 |
| X510–S457 | 62 | 50 | 72 | 6 | 1 | 4 |
| X510–Q626 | 73 | 71 | 79 | 2 | 78 | 7 |
MD-simulations of the h.DPP III–Arg2-2NA complexes
In the studied complexes two arginine residues of the substrate (Arg2-2NA) form a β-strand that in the antiparallel fashion binds to the five-stranded β-core of DPP III (as shown in Fig. 1). Such binding closely resembles the tynorphin binding mode in the experimentally determined structure of the inactive DPP III mutant–tynorphin complex.26 Binding of the substrate Arg2-2NA into the active site of human DPP III stabilized the protein structure, especially the lower domain β-core (Fig. 3 and 5) and slightly changed the flexibility profile pattern. Apparently, binding of the synthetic substrate Arg2-2NA to the β9 and β10 sheets decreases their mobility in the wt protein. However, despite the striking similarity of the substrate orientation in the simulated variants, the protein structure stabilization induced by the substrate binding is not the same in all variants (Fig. S4 and S5†), and the complex with the R510W mutant is significantly more flexible than the other simulated complexes (Fig. S3†).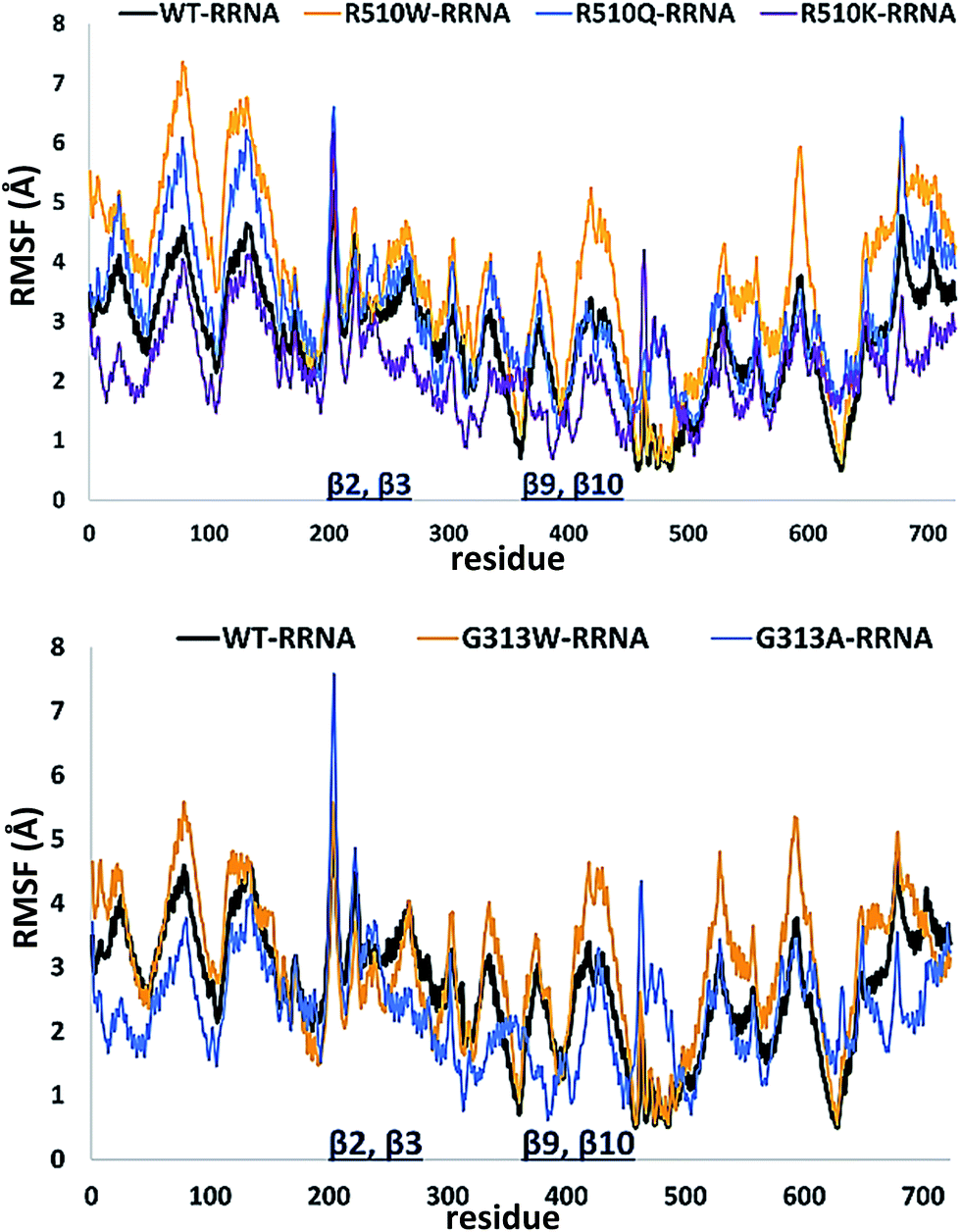 | ||
| Fig. 5 Root Mean Square Fluctuations (RMSF) calculated for the 100 ns of MD simulations of the complexes between different human DPP III variants and Arg2-2NA obtained by averaging over two 100 ns long simulations. | ||
The hydrogen bond analysis revealed that, similarly to the ligand free protein, interaction between residues at position 510 and Gln626 is completely abolished in the R510W–Arg2-2NA mutant complex, while the hydrogen bond between Arg510 and Gly454 has not been detected during the simulation of the R510Q complex (Table 5). Oppositely, the Tyr318 stabilization by Asp496 and His568 is the most stable in the complex with the G313A and the R510Q mutant, respectively (Fig. S7†) as confirmed by the hydrogen bond analysis (Table 5). We have showed that the 100% conserved Tyr318 is important for substrate stabilization in the active site.16 Its reorientation into direction of either Asp496 or His568 might be one of the reasons for the enzymatic inactivation of the R510Q mutant, also. Analysis of the substrate–protein hydrogen bonds revealed that in general there are no significant differences among the complexes (Fig. 6, S4 and S5†). However, the stability of the hydrogen bond between the substrate and highly conserved residue G385 as well as the catalytic histidine H568 is the most stable in the complex with wt enzyme (Table 5 and Fig. 6 and S6†). It has been shown that His568 plays a significant role in the transition state stabilisation and the enzymatic reaction. By forming a stable hydrogen bond with the substrate carbonyl oxygen, His568 is dragging the negative charge away from the peptide C atom and in this way facilitates nucleophile attack of the activated water.16 So the lack of this hydrogen bond in the complexes with mutated protein may explain the enzymatic inactivity of these variants.
| H-bonds | WT-RRNA | R510W-RRNA | R510Q-RRNA | G313W-RRNA | G313A-RRNA |
|---|---|---|---|---|---|
| Y318–D496 | <1 | <1 | 40 | 3 | 97 |
| Y318–E508 | 76 | 44 | 1 | 85 | <1 |
| Y318–H568 | <1 | <1 | 21 | <1 | 1 |
| X510–G454 | 57 | 34 | <1 | 74 | 41 |
| X510–Q626 | 68 | <1 | 71 | 76 | 63 |
| E512–H450 | 88 | 73 | 71 | 80 | 90 |
| E316–RRNA | 100 | 99 | 100 | 100 | 56 |
| E329–RRNA | 21/63 | 87 | 100 | 100 | 95 |
| G385–RRNA | 49 | <1 | 5 | <1 | <1 |
| G389–RRNA | 54 | 77 | 74 | 55 | <1 |
| N391–RRNA | 51 | 100 | 88 | 100 | 77 |
| N394–RRNA | 89 | 64 | 90 | 88 | 83 |
| D396–RRNA | 50 | 23 | 45 | 62 | 84 |
| D496–RRNA | 41 | 85 | 95 | 100 | 96 |
| H568–RRNA | 47 | 7 | 8 | <1 | <1 |
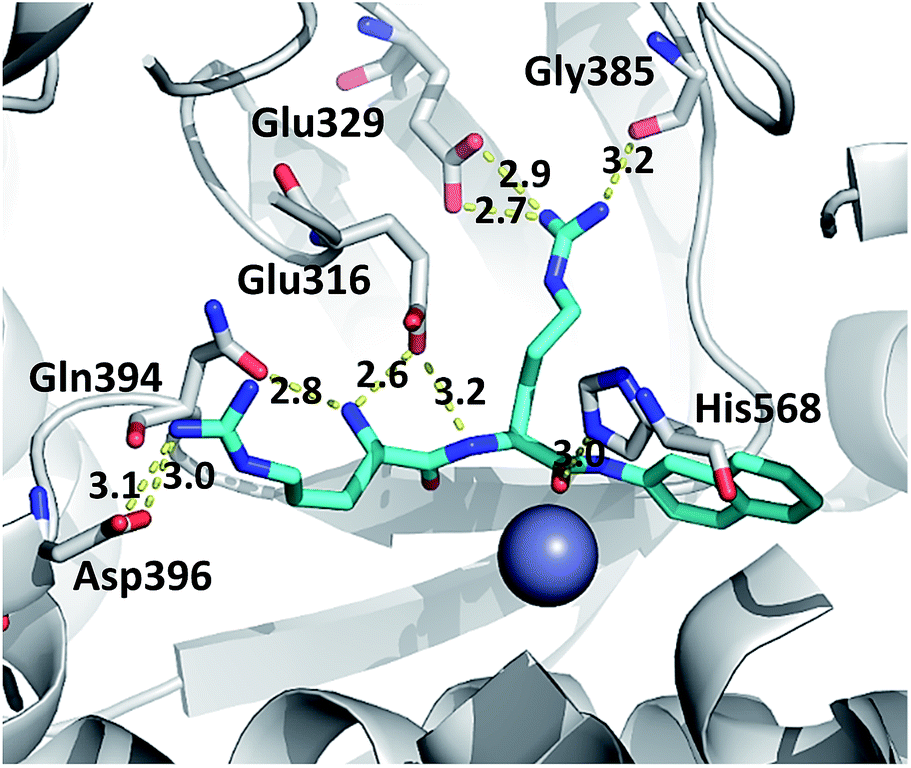 | ||
| Fig. 6 The representative structure of the h.DPP III–Arg2-2NA complex. The residues interacting with the substrate are presented as grey and the substrate as cyan sticks. The distances are given in Å. | ||
Conclusions
We expressed and biochemically characterized two mutant h.DPP III proteins, G313W and R510W, with mutations in the highly conserved regions of DPP III, to analyse their biochemical and physicochemical properties in comparison to the wild type h.DPP III. Aforementioned mutants exhibited dramatically lower activity towards the synthetic DPP III substrate, Arg2-2NA, which prompted us to investigate the origins of that activity loss by combining experimental and computational approach. We also analysed 3 additional mutants with more conservative amino acid substitution at the same positions, G313A, R510K and R510Q. The results of these investigations gave new insight into biochemical and structural importance of conserved residues Gly313 and Arg510 for the enzyme activity. Experimental approach indicated that Gly313 to Ala mutation significantly influences the binding of the Arg2-2NA substrate in contrast to the mutations of Arg510 that did not result in the significant change of KM. In agreement with biochemical results, MD simulations revealed that Gly313 is essential for the proper positioning of the substrate, although there is no direct contact between them. However Gly313 dictates position of Glu316, conserved residue that interacts electrostatically with the N-terminal amino group of the substrate. The efficiency of Arg2-2NA hydrolysis (kcat/KM) was significantly decreased in all mutants except R510K. One possible reason for the enzymatic inactivity of the mutants is increased flexibility of their active site in comparison with the wild type enzyme, and the other is lack of the hydrogen bond between the substrate and catalytic His568, which is known to be important for the transition state stabilization.16Apparently, the disrupted hydrogen bond between Arg2-2NA and catalytic His568 in combination with the increased active site fluctuations, especially of the H450ELLGH455 motif provide possible explanation for inactivity of the G313W and R510W mutants.
This is the first report on negative correlation of DPP III activity with increased flexibility of the lower domain β-core.
Experimental
Protein expression and purification
Mutants of h.DPP III, G313W, G313A, R510W, R510K and R510Q were prepared by site-specific mutagenesis of wt h.DPP III His-tagged expression construct cloned in pET21b vector with QuikChange II XL Site-Directed Mutagenesis kit (Agilent technologies, USA). Proteins were expressed in E. coli strain BL21-CodonPlus(DE3)-RIL, and purified by Ni-NTA affinity chromatography, as described by Špoljarić et al. (2011).23Protein concentrations were determined using the protein-dye binding assay, with bovine serum albumin as a standard.24
Enzyme activity assay and determination of kinetic parameters
The enzymatic activities of wt and mutant h.DPP IIIs were determined by a standard assay at 37 °C with Arg2-2NA as a substrate, using the colorimetric method as previously described.25 Kinetic parameters for hydrolysis of Arg2-2NA were determined at 25 °C and at pH 8.6 for mutants R510W, R510K and R510Q, and at pH 7.4 for G313A, in the presence of 50 μM CoCl2 by initial rate measurements, as previously described.25 The kinetic parameters were calculated using Hanes–Woolf plot, with Arg2-2NA concentrations from 2.5 to 40 μM for all mutants except G313A for which concentrations range from 20 to 240 μM was used. Kinetic parameters for mutant G313W were not determined due to low enzyme activity.Molecular simulations
The enzyme variants, either ligand free or its complexes with the synthetic substrate Arg2-2NA, were neutralized by adding Na+ ions and solvated in an octahedron box filled with TIP3P28 water molecules ensuring an at least 11 Å thick water molecules buffer around the protein. The systems were parametrized by the AMBERTools14 modules antechamber and tleap, using GAFF29 and ff14SB30 force fields to parameterize the substrate and the protein, respectively. For the zinc cation, Zn2+, parameters were derived from our previous work12 and modified according to the PDB survey.31,32
000 (ligand free) and ∼64000 (complexes) atoms (∼27500 and ∼17500 molecules of water, respectively), were simulated using periodic boundary conditions. The electrostatic interactions were calculated using the particle-mesh Ewald method.33 The PMEMD module running at GPU was used to conduct the molecular dynamics (MD) simulation. Before MD simulations, three rounds of geometry optimization, with different degree of the protein restraints, were performed by the module sander. In the first cycle 1500 optimization steps were performed, where the first 450 steps were of the steepest descent method, and the rest was the conjugate gradient. Both, the protein atoms and the metal ion, were constrained using a harmonic potential of 32 kcal (mol Å2)−1, in order to equilibrate water molecules. In the second cycle 2500 steps of optimization was done. The metal ion and the protein backbone were constrained with 12 kcal (mol Å2)−1. Finally, in the third cycle the same number of optimization steps was used as in the first cycle, and the protein backbone and the metal ion were constrained with 1 kcal (mol Å2)−1 and 12 kcal (mol Å2)−1, respectively. The minimized system was heated from 0 to 300 K during 30 ps of MD simulation using a canonical ensemble (NVT), followed by equilibration stage of 30 and 100 ps (for the ligand free enzyme and complexes), during which the water density was adjusted. The equilibrated system was then subjected to 100 ns of the productive MD simulations, 50 ns of conventional MD (cMD) followed by 50 ns of the dual boost accelerated MD (aMD) simulations. One boost was applied to torsional terms and the other to total potential. The latter facilitates diffusion of the explicit solvent molecules around the biomolecule and thus reduces hindrance that water induce on the large amplitude displacements of a solute. In this way more efficient conformational sampling is enabled.Type and duration of the performed simulations is given in Table S1.†34 The constant temperature (300 K) and pressure (1 atm) were ensured using Langevin dynamics with a collision frequency of 1 ps−1, and Berendsen barostat, respectively.35,36 The time step during the periods of heating and cooling was 1 fs, and for the rest of the simulation 2 fs (to restrain the motion of hydrogens the SHAKE algorithm was used).
Conflict of interest
There are no conflicts of interest to declare.Acknowledgements
The work has been supported by Croatian Science Foundation under the project “7235 Flexibility, activity and structure correlations in the dipeptidyl peptidase III family” and by UKF grant agreement 21/15.Notes and references
- T. Chiba, Y.-H. Li, T. Yamane, O. Ogikubo, M. Fukuoka, R. Arai, S. Takahashi, T. Ohtsuka, I. Ohkubo and N. Matsui, Peptides, 2003, 24, 773 CrossRef CAS.
- Y. Liu, J. T. Kern, J. R. Walker, J. A. Johnson, P. G. Schultz and H. Luesch, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5205 CrossRef CAS PubMed.
- Š. Šimaga, D. Babić, M. Osmak, M. Šprem and M. Abramić, Gynecol. Oncol., 2003, 91, 194 CrossRef.
- M. He, D. P. Mangiameli, S. Kachala, K. Hunter, J. Gillespie, X. Bian, H.-C. J. Shen and S. K. Libutti, Clin. Cancer Res., 2010, 16, 249 CrossRef CAS PubMed.
- V. A. Meliopoulos, L. E. Andersen, P. Brooks, X. Yan, A. Bakre, J. K. Coleman, S. M. Tompkins and R. A. Tripp, PLoS One, 2012, 7, e37169 CAS.
- B. E. Hast, D. Goldfarb, K. M. Mulvaney, M. A. Hast, P. F. Siesser, F. Yan, D. N. Hayes and M. B. Major, Cancer Res., 2013, 73, 2199 CrossRef CAS PubMed.
- B. E. Hast, E. W. Cloer, D. Goldfarb, H. Li, P. F. Siesser, F. Yan, V. Walter, N. Zheng, D. N. Hayes and M. B. Major, Cancer Res., 2014, 74, 808 CrossRef CAS PubMed.
- K. Fukasawa, K. M. Fukasawa, M. Kanai, S. Fujii, J. Hirose and M. Harada, Biochem. J., 1998, 329, 275 CrossRef CAS PubMed.
- M. Abramić, J. Špoljarić and Š. Šimaga, Period. Biol., 2004, 2, 161 Search PubMed.
- J. M. Chen and A. J. Barrett, Dipeptidyl–Peptidase III, in Handbook of Proteolytic Enzymes, ed. A. J. Barrett, N. D. Rawlings and J. F. Woessner, Elsevier, Academic Press, London, 2004, pp. 809–812 Search PubMed.
- P. Kumar, V. Reithofer, M. Reisinger, S. Wallner, T. Pavkov-Keller, P. Macheroux and K. Gruber, Sci. Rep., 2016, 6, 23787 CrossRef CAS PubMed.
- A. Tomić, M. González and S. Tomić, J. Chem. Inf. Model., 2012, 52, 1583 CrossRef PubMed.
- A. Tomić, M. Berynskyy, R. C. Wade and S. Tomić, Mol. BioSyst., 2015, 11, 3068 RSC.
- A. Tomić and S. Tomić, Dalton Trans., 2014, 43, 15503 RSC.
- J. Matić, F. Šupljika, N. Tir, P. Piotrowski, C. Schmuck, M. Abramić, I. Piantanida and S. Tomić, RSC Adv., 2016, 6, 83044 RSC.
- A. Tomić, B. Kovačević and S. Tomić, Phys. Chem. Chem. Phys., 2016, 18, 83044 RSC.
- E. Cerami, J. Gao, U. Dogrusoz, B. E. Gross, S. O. Sumer, B. A. Aksoy, A. Jacobsen, C. J. Byrne, M. L. Heuer, E. Larsson, Y. Antipin, B. Reva, A. P. Goldberg, C. Sander and N. Schultz, Cancer Discovery, 2012, 2, 401 CrossRef PubMed.
- J. Gao, B. A. Aksoy, U. Dogrusoz, G. Dresdner, B. Gross, S. O. Sumer, Y. Sun, A. Jacobsen, R. Sinha, E. Larsson, E. Cerami, C. Sander and N. Schultz, Sci. Signaling, 2013, 6, pl1 CrossRef PubMed.
- B. Salopek-Sondi, B. Vukelić, J. Špoljarić, Š. Šimaga, D. Vujaklija, J. Makarević, N. Jajčanin and M. Abramić, Biol. Chem., 2008, 389, 163 CrossRef CAS PubMed.
- J. Špoljarić, B. Salopek-Sondi, J. Makarević, B. Vukelić, D. Agić, Š. Šimaga, N. Jajčanin-Jozić and M. Abramić, Bioorg. Chem., 2009, 37, 70 CrossRef PubMed.
- A. Tomić, M. Abramić, J. Špoljarić, D. Agić, D. M. Smith and S. Tomić, J. Mol. Recognit., 2011, 24, 804 CrossRef PubMed.
- M. Abramić, Z. Karačić, M. Šemanjski, B. Vukelić and N. Jajčanin-Jozić, Biol. Chem., 2015, 396, 359 CrossRef PubMed.
- J. Špoljarić, A. Tomić, B. Vukelić, B. Salopek-Sondi, D. Agić, S. Tomić and M. Abramić, Croat. Chem. Acta, 2011, 84, 259 CrossRef.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248 CrossRef CAS PubMed.
- M. Abramić, Š. Šimaga, M. Osmak, L. Čičin-Šain, B. Vukelić, K. Vlahoviček and L. J. Dolovčak, Int. J. Biochem. Cell Biol., 2004, 36, 434 CrossRef.
- G. A. Bezerra, E. Dobrovetsky, R. Viertlmayr, A. Dong, A. Binter, M. Abramic, P. Macheroux, S. Dhe-Paganon and K. Gruber, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6525 CrossRef CAS PubMed.
- D. A. Case, V. Babin, J. T. Berryman, R. M. Betz, Q. Cai, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, H. Gohlke, A. W. Goetz, S. Gusarov, N. Homeyer, P. Janowski, J. Kaus, I. Kolossváry, A. Kovalenko, T. S. Lee, S. LeGrand, T. Luchko, R. Luo, B. Madej, K. M. Merz, F. Paesani, D. R. Roe, A. Roitberg, C. Sagui, R. Salomon-Ferrer, G. Seabra, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu and P. A. Kollman, AMBER 14, University of California, San Francisco, 2014 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696 CrossRef CAS PubMed.
- I. Dokmanić, M. Šikić and S. Tomić, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64, 257 Search PubMed.
- A. Tus, A. Rakipović, G. Peretin, S. Tomić and M. Šikić, Nucleic Acids Res., 2012, 40, W352 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS.
- D. Hamelberg, J. Mongan and J. A. McCammon, J. Chem. Phys., 2004, 120, 11919 CrossRef CAS PubMed.
- G. Grest and K. Kremer, Phys. Rev. A: At., Mol., Opt. Phys., 1986, 33, 3628 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02642k |
| This journal is © The Royal Society of Chemistry 2017 |