
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced photocatalytic oxygen evolution over Mo-doped Ca2NiWO6 perovskite photocatalyst under visible light irradiation
Ying Luoa,
Jiawei Xuea,
Xiaodi Zhua,
Jose Daniela,
Xiang Gaoa,
Song Sun*a,
Chen Gao*ab and
Jun Bao *a
*a
aNational Synchrotron Radiation Laboratory & Collaborative Innovation Center of Chemistry for Energy Materials, University of Science and Technology of China, Hefei, Anhui 230029, China. E-mail: baoj@ustc.edu.cn; suns@ustc.edu.cn
bCAS Key Laboratory of Materials for Energy Conversion, Department of Materials Science and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: cgao@ustc.edu.cn
First published on 17th January 2017
Abstract
A series of Mo-doped Ca2NiWO6 (Ca2NiW1−xMoxO6, x = 0–0.05) samples were synthesized by a solid-state reaction. The physical and optical properties of these photocatalysts were characterized by X-ray diffraction, X-ray absorption fine structure, UV-visible diffuse reflectance spectra, and the band structure along with density of states were calculated by the plane-wave-based density functional theory. The photocatalytic activity of oxygen evolution from water was evaluated under visible light irradiation. The Mo doping significantly increased the photocatalytic activity of Ca2NiWO6. The optimal Ca2NiW0.97Mo0.03O6 showed an oxygen evolution rate approximately 2 times higher than that of the pure Ca2NiWO6. The characterization results indicated that the Mo-doped Ca2NiWO6 maintained the double perovskite structure. The Mo6+ ions were substituted into the W6+ sites in the lattice, causing a certain amount of lattice distortion and promoting the formation of oxygen vacancies. Furthermore, the Mo doping introduced impurity energy levels into the band structure of Ca2NiWO6. Consequently, the conduction band changed from discrete to continuous and the bottom of the conduction band was shifted to more positive potential, compared to that of pure Ca2NiWO6, leading to a lower band gap and higher absorbance in the wider visible light region. These characteristics are responsible for the high photocatalytic activity of the oxygen evolution reaction.
Introduction
With increasing demands for clean energy, photocatalytic water splitting to generate hydrogen and oxygen has attracted significant attention because of the low energy consumption, simple operation process and mild reaction conditions.1–5 An ideal photocatalyst for overall water splitting should have proper band position, i.e., the conduction band minimum (CBM) should be more negative than the hydrogen evolution potential (H+/H2 0.0 eV vs. normal hydrogen electrode (NHE)), meanwhile, the valence band maximum (VBM) should be more positive than the oxygen evolution potential (O2/H2O +1.23 eV vs. NHE). Despite several decades of intensive studies, to date, only a few photocatalysts have been discovered to be active for overall water splitting into hydrogen and oxygen under visible light irradiation.6–9 Many attempts have been made to increase the activity of the half reaction of water splitting, the oxidation of water to form oxygen or the reduction of protons to produce hydrogen.10 For the oxygen evolution reaction (OER), high energy is required to undertake the O–H bond breaking and the attendant O–O bond formation, thus requiring a more positive VB level of the efficient photocatalyst.11–13 In recent years, notable progress has been achieved in the search for highly active oxygen evolution photocatalysts, including simple oxides (e.g. TiO2 (ref. 14) and CoOx (ref. 15)), complex oxides (e.g. SrTiO3 (ref. 16) and BiVO4 (ref. 17)), nitrides (e.g. C3N4 (ref. 18)), and sulfides (e.g. Cu2ZnSnS4 (ref. 19)).Perovskite structured compounds have attracted significant attention as photocatalysts due to their unique physicochemical properties, demonstrating the potential for application in water splitting and decontamination.20–23 It was found that the band structure, optical properties, and chemical stability of perovskite photocatalysts can be modified and the photocatalytic activity can be improved by doping or partial replacement, on the basis of maintaining the basic structure. Some researchers have reported that doping with transition metal ions such as rhodium, chromium, vanadium and tungsten results in the red shift of the light absorption of perovskite-type compounds to the visible light region.24–27 For instance, Kim et al.28 have found that the W-doped Sr2FeNbO6 showed a photocatalytic quantum yield two times higher than that of the pristine sample under visible light irradiation. A similar conclusion was arrived at from La2Ti2O7 perovskite nanosheets with a narrow band gap after Ni doping.29 Cheng et al.30 reported that the La1−xSrxCoO3 perovskites have good photocatalytic activity for OER. The experimental and theoretical investigation confirmed that Sr3+ substitutions straightened the octahedral cage, aligned atoms along the Co–O–Co axis, and increased the average oxidation state of the Co ions. Consequently, the overlap between the occupied O 2p VB and the unoccupied Co 3d CB increased and thus improved the OER activity. Recently, the perovskite-type (Ln0.5Ba0.5)CoO3−x (Ln = Pr, Sm, Gd and Ho) have also been reported as a family of efficient photocatalysts for the OER in alkaline solution.31
The double-perovskite-type compound Ca2NiWO6 consists of a layered structure of corner-sharing regular octahedrons, WO6 and NiO6, with a unique chain of the –Ni–W–O–. The VB of Ca2NiWO6 is controlled by the hybrid O 2p and Ni t2g orbitals, and the CB is composed of two discrete energy levels, the W 5d and Ni eg levels.32 The excited electrons from the VB to the W 5d CB are responsible for the absorption in the UV region, and the other from the VB to the Ni eg CB corresponds to the absorption in the visible light region. Consequently, the Ca2NiWO6 perovskite showed photocatalytic activity for oxygen evolution under visible light irradiation. However, the band gap (2.61 eV) is still relatively large, limiting its further application in the visible light region. It is expected that the doping modification may expand the spectral response of the photocatalyst and increase the photocatalytic activity by introducing the appropriate impurity level into the band gap. To the best of our knowledge, the study on the modification of Ca2NiWO6 by metal doping has not been reported so far.
In this study, the role of the Mo dopant in the structural features of Ca2NiWO6 and the photocatalytic activity for the OER were investigated. A series of Mo-doped Ca2NiWO6 with different doping levels was synthesized by a conventional solid-state reaction. The Mo-doped samples showed much higher OER activity than that of the pristine sample under visible light irradiation. With a combination of experimental and theoretical investigations, the effects of Mo6+ ion doping on the crystal structure, band structure and optical properties of double-perovskite Ca2NiWO6 were studied in detail and the structure–performance relationship was discussed.
Experimental
Ca2NiW1−xMoxO6 (x = 0, 0.01, 0.02, 0.03, 0.04, 0.05) was prepared by a conventional solid-state reaction. The starting materials consisted of CaCO3, WO3, NiO and MoO3, which were mixed in the molar ratio Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni:W:Mo = 2:1:1 − x:x (x = 0.0.01, 0.02, 0.03, 0.04, 0.05). The solid mixture was ground thoroughly and then calcined in air at 1273 K for 20 h. After cooling to room temperature, the mixture was reground and calcined once again at 1553 K for 10 h.
:Ni:W:Mo = 2:1:1 − x:x (x = 0.0.01, 0.02, 0.03, 0.04, 0.05). The solid mixture was ground thoroughly and then calcined in air at 1273 K for 20 h. After cooling to room temperature, the mixture was reground and calcined once again at 1553 K for 10 h.
The as-prepared samples were characterized by various analytical techniques. X-ray diffraction (XRD) patterns were recorded on a Rigaku (D/MAX-A) X-ray diffractometer using a Cu Kα radiation source. UV-visible diffuse reflectance spectra (UV-vis DRS) were measured on a UV-visible spectrophotometer (Solidspec DUC-3700) using BaSO4 as a reference. The Mo K-edge X-ray absorption fine structure (XAFS) measurements were performed at the endstation of 1W1B of the Beijing Synchrotron Radiation Facility (BSRF, China). The storage ring energy was 2.5 GeV with an average current of 200 mA. Spectra were collected in the fluorescence mode using a Si (111) double crystal monochromator. The energy calibration of the Mo K-edge was performed using a Mo foil. Data processing was carried out using the ATHENA software package.33 The electronic band structures, along with densities of states were calculated by the plane-wave-based density functional theory (DFT), using the CASTEP program.34,35 The exchange correlation potential and ultrasoft pseudopotentials for electron–ion interactions were modeled using the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE). The kinetic energy cut-off was taken to be 380 eV for Ca2NiWO6. A supercell consisting of a (2 × 2 × 1) unit cell was used in the calculations. The lattice parameter of Ca2NiW1−xMoxO6 was based on the structure of Ca2NiWO6: a = 5.4048 Å, b = 5.5369 Å, c = 7.6889 Å, which formed a layered crystal structure with the space group P21/n.
The photocatalytic O2 evolution of the as-prepared samples were evaluated on commercial water splitting equipment (LABSOLAR-IIAGA, Beijing PERFECTLIGHT Technology Co. Ltd.). In a typical test, 0.185 g of photocatalyst powder were dispersed into 100 ml of distilled water in a quartz reaction cell with an external cooling jacket. AgNO3 (1.85 mmol) was used as the sacrificial electron acceptor. A 300 W xenon arc lamp (PLS-SXE300 UV, Beijing Perfectlight Technology Co. Ltd) with a 400 nm cut-off filter was used as the light source. Prior to irradiation, the closed gas circulation system and reaction cell were well evacuated. The amounts of evolved gases were analyzed by an on-line gas chromatograph (GC1690, Hangzhou Kexiao Scientific Instruments Co. Ltd) equipped with a thermal conductivity detector (TCD) and a KX-112 packed column (Lanzhou Institute of Chemical Physics).
Results and discussion
Characterization of photocatalysts
The XRD patterns of Ca2NiW1−xMoxO6 (x = 0, 0.01, 0.02, 0.03, 0.04, 0.05) are shown in Fig. 1. For the undoped sample, the positions and relative intensities of all diffraction peaks matched well with the standard data of Ca2NiWO6 (JCPDS No. 47-0026), indicating that a pure perovskite phase was obtained. | ||
| Fig. 1 XRD patterns of Ca2NiW1−xMoxO6 (x = 0, 0.01, 0.02, 0.03, 0.04, and 0.05). | ||
After doping with Mo6+ ions, the XRD patterns did not show significant differences from that of the pure Ca2NiWO6, even at a relatively high Mo doping level (x = 0.05). The results indicated that the doping of Mo6+ ions did not cause a significant change in the host structure or introduce any impurity phase. The Mo-doped Ca2NiWO6 still maintained the perovskite structure. It is suggested that the doped Mo6+ ions can be substituted into the W6+ sites in the structure, due to their similar ionic radii. Further evidence came from the XAFS measurements, which can provide detailed information on the local coordination structure of center atoms.
Fig. 2 shows the Fourier transforms of Mo K-edge EXAFS of the Ca2NiW1−xMoxO6 (x = 0.01, 0.03 and 0.05) and standard compounds MoO2 and MoO3. For comparison, the theoretical radial distribution function (RDF) of W atoms of the Ca2NiWO6 crystals was also calculated and presented in the figure. The fitting results are listed in Table 1. From the figure, it can be seen that the RDFs of the Mo-doped Ca2NiWO6 were completely different from that of the MoO3 and MoO2. In particular, no significant Mo–Mo coordination peaks assigned to MoO3 and MoO2 were found in the doped samples. Furthermore, the RDF curves of doped samples were very similar to those of pure Ca2NiWO6. The Mo–O and Mo–Ca coordination peaks at about 1.41 Å and 2.55 Å exhibited similar characteristics to those of W–O and W–Ca coordination peaks in Ca2NiWO6. These results confirmed that no isolated molybdenum oxides were formed at the present Mo doping levels. The Mo ions entered the crystal lattice of Ca2NiWO6, substituting for the hexa-coordinated W6+ sites.
 | ||
| Fig. 2 RDFs of Ca2NiW1−xMoxO6 (x = 0.01, 0.03, and 0.05), Ca2NiWO6, MoO2, and MoO3 samples. | ||
| Sample | Shell | Ra (Å) | Nb | σc (Å2) |
|---|---|---|---|---|
| a Coordination distance.b Coordination number.c Debye–Waller factor. | ||||
| Ca2NiWO6 | W–O | 1.411 | 6.0 | — |
| W–Ca(I) | 2.546 | 6.0 | — | |
| W–Ca(II) | 3.221 | 2.0 | — | |
| W–Ni | 3.344 | 4.0 | — | |
| x = 0.01 | Mo–O | 1.411 | 5.21 | 0.001 |
| Mo–Ca(I) | 2.577 | 6.00 | 0.011 | |
| x = 0.03 | Mo–O | 1.411 | 5.13 | 0.001 |
| Mo–Ca(I) | 2.608 | 6.00 | 0.012 | |
| x = 0.05 | Mo–O | 1.442 | 4.87 | 0.001 |
| Mo–Ca(I) | 2.608 | 6.00 | 0.010 | |
According to the fitting structural parameters, the schematic diagrams of the crystal lattice of the samples were drawn by Materials Studio as shown in Fig. 3. For the pure Ca2NiWO6 (Fig. 3a), each W atom was coordinated with six O atoms (the first shell) and six Ca atoms (the second shell). The Mo doping induced internal stresses and exerted a certain effect on the structure of Ca2NiWO6, leading to the lattice distortion, as illustrated in Fig. 3b. The coordination distances of Mo–O and Mo–Ca in Ca2NiW1−xMoxO6 were both larger than that of W–O and W–Ca in pure Ca2NiWO6. Furthermore, the coordination number of Mo–O atoms also decreased, leading to the formation of oxygen vacancies.
 | ||
| Fig. 3 Schematic of the crystal structure of (a) pure Ca2NiWO6 and (b) Mo-doped Ca2NiWO6. | ||
The UV-Vis DRS of Ca2NiW1−xMoxO6 (x = 0, 0.01, 0.02, 0.03, 0.04, 0.05) are shown in Fig. 4a. The pure Ca2NiWO6 had three distinct absorption bands in the range of 443–535 nm. The two steep absorption bands at 443 and 492 nm were assigned to the charge transition from the VB to the W 5d level and Ni 3d level, respectively.32 The absorption band located at 535 nm may be due to the charge transition from the VB to the impurity or defect level. For the doped samples, the steps between different absorption edges were gradually elevated with an increase in Mo concentration, and finally combined into a gentle absorption edge. Furthermore, the absorption edge was shifted to longer wavelengths with increasing Mo content. Accordingly, the color of the samples changed from light green to yellow (the inset in Fig. 4a). The band gaps, Eg, of the samples were calculated using the Tauc equation.36,37
| αhν = C1(hν − Eg)2 | (1) |
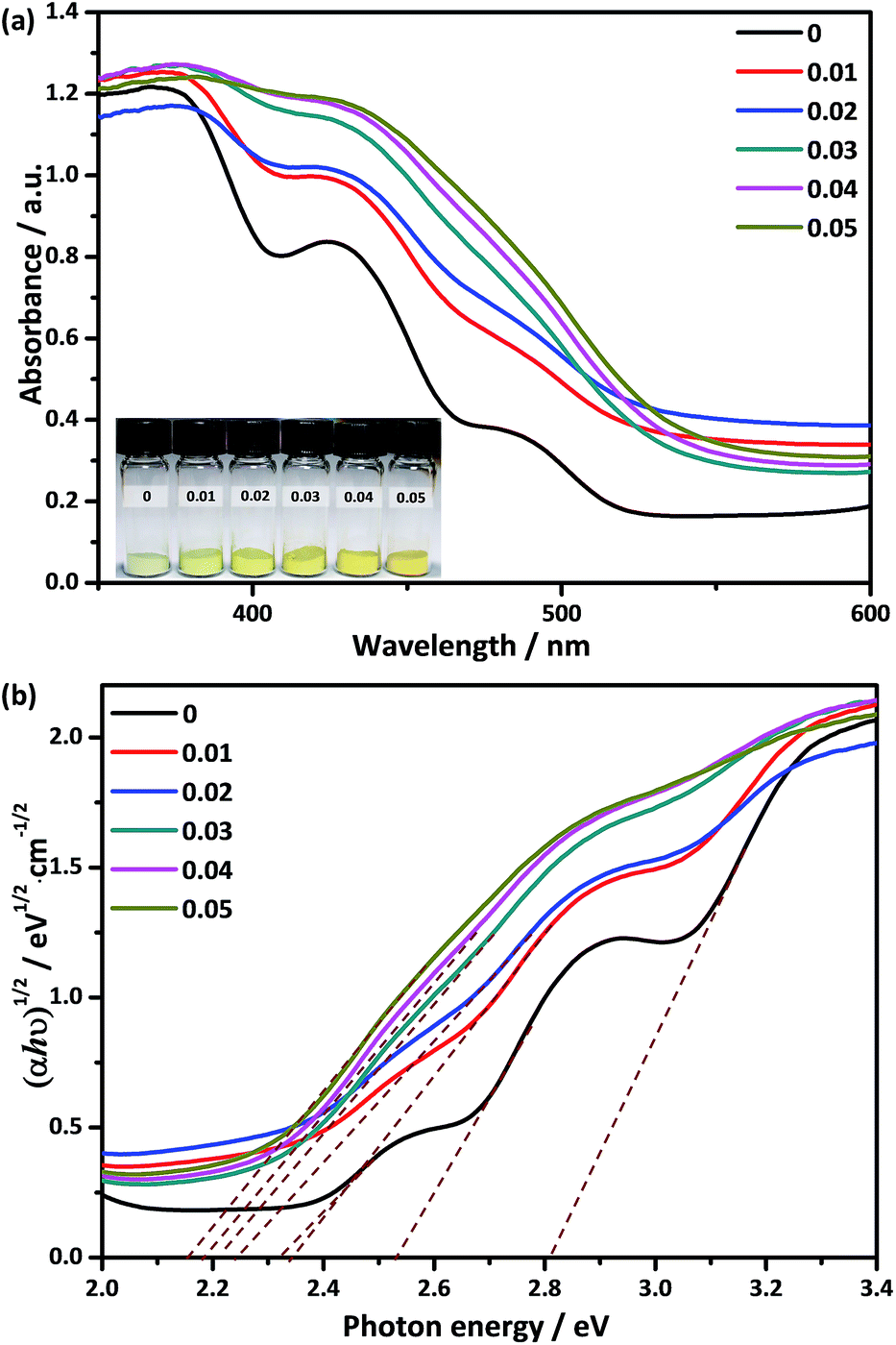 | ||
| Fig. 4 (a) UV-visible DRS of Ca2NiW1−xMoxO6 (x = 0, 0.01, 0.02, 0.03, 0.04, 0.05), the inset shows the color change after Mo doping; (b) the corresponding Kubelka–Munk transformed diffuse reflectance spectra. | ||
To deeply understand the effect of Mo doping on the electronic structure of Ca2NiWO6, the band structure and the density of states (DOS) of the pure and doped samples were calculated and the results are shown in Fig. 5. For the pure Ca2NiWO6, shown in Fig. 5a, the partial density of states of the individual atoms clearly indicated that the Ni 3d level was split into two main segments corresponding to the Ni t2g and Ni eg levels, respectively. The W 5d level was located at a higher energy position than that of the Ni 3d level. From the total density of states curves, the CB contained two discrete sub-bands. The high energy one consisted of the W 5d orbital, and another was mainly derived from the Ni eg level. The VB consisted of the hybridization of O 2p and Ni t2g levels. The calculation results indicated that there were two intrinsic band gaps in the pure perovskite Ca2NiWO6, consistent with the DRS experiment (Fig. 4). Similar results were also reported by Zou et al.32 The Mo doping had a significant impact on the band structure of Ca2NiWO6. As shown in Fig. 5b, the doped sample exhibited a higher DOS and more complex band structure with respect to pure Ca2NiWO6, confirming that the Mo doping formed impurity energy levels in the band gap of Ca2NiWO6. The CB of the Ca2NiW1−xMoxO6 mainly arose from the hybridization of Mo 3d, W 5d and Ni eg orbitals. The electronic structure of CB changed from discrete to continuous with the Mo doping, consistent with the DRS result. It should be noted that the top of the VB remained unchanged after the Mo6+ doping, whilst the bottom of the CB was shifted to more positive potentials, compared to that of the pure Ca2NiWO6. The result indicated that the Mo dopant narrowed the band gap, thus extended the absorption in the visible light region, as revealed by UV-Vis DRS results.
 | ||
| Fig. 5 Band structure and density of states of (a) pure Ca2NiWO6 and (b) Mo-doped Ca2NiWO6. | ||
Photocatalytic activity
The photocatalytic O2 evolution from water under visible irradiation was applied to evaluate the photocatalytic performance of the as-prepared photocatalysts and the results are shown in Fig. 6a. The pure Ca2NiWO6 had a rather low O2 evolution rate of 1.38 μmol h−1 g−1. The Mo doping significantly enhanced the oxygen production. With an increase in the Mo doping, the oxygen evolution rate increased and reached the highest level at the Mo doping content of 3 at% (Ca2NiW0.97Mo0.03O6). On this catalyst, the O2 evolution rate was 2.65 μmol h−1 g−1, approximately 2 times higher than that of pure Ca2NiWO6. Fig. 6b shows the cyclic tests of oxygen production of the optimized Ca2NiW0.97Mo0.03O6 photocatalyst. No great change in the photocatalytic activity was observed after three cyclic tests, which suggested that the doped sample had good stability. | ||
| Fig. 6 (a) OER activity of Ca2NiW1−xMoxO6 (x = 0, 0.01, 0.02, 0.03, 0.04, and 0.05) under visible irradiation; (b) cyclic tests of oxygen production of the optimized Ca2NiW0.97Mo0.03O6 photocatalyst. | ||
The XRD and XAFS results indicated that the Mo6+ ions entered the crystal lattice of Ca2NiWO6, substituting for the hexa-coordinated W6+ sites and promoting the formation of the oxygen vacancies. It is well known that the oxygen vacancies play an important role in the photocatalytic reaction. The surface oxygen vacancies can directly provide the formation sites of the active species for the photocatalytic reaction and thus increase the photocatalytic activity.38 Furthermore, from the DRS and DOS results, the Mo doping introduced impurity energy levels into the band structure of Ca2NiWO6, leading to a lower band gap energy and higher absorbance in a wider visible light region, compared to that of pure Ca2NiWO6. These characteristics contributed to the generation of more photogenerated carries and enhanced the photocatalytic activity. Based on the characterization results, the band structures of the as-prepared photocatalysts are proposed and shown in Fig. 7. The pure Ca2NiWO6 had two discrete CBs, consistent with a previous report by Zou et al.32 The high energy band consisted of the W 5d orbital and the low energy band was derived from the Ni eg level. The VB was composed of the hybridization of O 2p and Ni t2g levels. Accordingly, there existed two intrinsic band gaps (Fig. 7a). For the Mo-doped Ca2NiWO6, shown in Fig. 7b, its CB was formed by the hybridization of the Mo 3d orbital with the W 5d and Ni 3d orbitals. With respect to the pure Ca2NiWO6, the CB of the Mo doped sample was continuous, due to the formation of impurity energy levels. The bottom of the CB of the doped sample was more positive, while the top level of the VB remained unchanged. The results indicated that the Mo doping broadened the absorption band in the visible light region, but had no influence on the oxidation potential; therefore it was conducive to the oxygen evolution reaction. The photocatalytic activity of the Mo-doped Ca2NiWO6 decreased when the Mo doping content exceeded 3 at%. The reason may be that the high dopant level increased the extent of defects in the oxide structure, which acted as electron–hole recombination centers, with a consequent decrease in the photocatalytic reaction rate.39 Further investigation of the defects and electronic structure are in progress. This work provides some new insight into the dopant effects on the crystal, band structure and performance of photocatalysts.
 | ||
| Fig. 7 Schematic of the band structure of (a) pure Ca2NiWO6 and (b) Mo-doped Ca2NiWO6. | ||
Conclusions
The Ca2NiW1−xMoxO6 (x = 0, 0.01, 0.02, 0.03, 0.04, 0.05) photocatalysts with a double perovskite structure were successfully synthesized by a solid-state reaction method. The Mo-doped samples exhibited much higher photocatalytic activity for the oxygen evolution reaction under visible light irradiation than that of the pure Ca2NiWO6. The characterization results indicated that the Mo6+ ions can be substituted into the hexa-coordinated W6+ ions in the Ca2NiWO6 lattice, leading to a certain degree of lattice distortion and the formation of oxygen vacancies. The Mo doping introduced impurity energy levels in the band structure of Ca2NiWO6, forming a continuous CB. The bottom of the CB was more positive, while the top level of the VB remained unchanged as compared to the pure Ca2NiWO6. As a result, the Mo-doped Ca2NiWO6 had a lower energy band gap and higher absorbance in the wider visible light region, compared to that of pure Ca2NiWO6. These characteristics contributed to the generation of more photogenerated carriers and enhanced the photocatalytic activity of the oxygen evolution reaction.Acknowledgements
This work was supported by National Nature Science Foundation of China (U1632273, 11205159) and Chinese Academy of Sciences (CX3430000001).Notes and references
- Z. G. Zou, J. H. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625–627 CrossRef CAS PubMed.
- X. Zong, G. Wu, H. Yan, G. Ma, J. Shi, F. Wen, L. Wang and C. Li, J. Phys. Chem. C, 2010, 114, 1963–1968 CAS.
- H. Husin, H. M. Chen, W. N. Su, C. J. Pan, W. T. Chuang, H. S. Sheu and B. J. Hwang, Appl. Catal., B, 2011, 102, 343–351 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 118, 7970–7973 CrossRef.
- J. Ran, J. Zhang, J. Yu, M. Jaroniecc and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- W. N. Sua, D. W. Ayele, V. Ochie, C. J. Pan and B. J. Hwang, Appl. Catal., B, 2014, 150–151, 363–369 CrossRef.
- X. Y. Yu, Y. Feng, B. Guan, X. W. Lou and U. Paik, Energy Environ. Sci., 2016, 9, 1246–1250 CAS.
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- Y. Li, Z. Liu, L. Liu and W. Gao, J. Am. Chem. Soc., 2010, 132, 13008–13015 CrossRef CAS PubMed.
- S. Chen, S. Shen, G. Liu, Y. Qi, F. Zhang and C. Li, Angew. Chem., Int. Ed., 2015, 54, 3047–3051 CrossRef CAS PubMed.
- H. Kato and A. Kudo, J. Phys. Chem. B, 2002, 106, 5029–5034 CrossRef CAS.
- D. Wang, R. Li, J. Zhu, J. Shi, J. Han, X. Zong and C. Li, J. Phys. Chem. C, 2012, 116, 5082–5089 CAS.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940–4947 CAS.
- P. Kush, K. Deori, A. Kumar and S. Deka, J. Mater. Chem. A, 2015, 3, 8098–8106 CAS.
- W. Eerenstein, N. D. Mathur and J. F. Scott, Nature, 2006, 442, 759–765 CrossRef CAS PubMed.
- A. Kudo, H. Kato and S. Nakagawa, J. Phys. Chem. B, 2000, 104, 571–575 CrossRef CAS.
- R. Hu, C. Li, X. Wang, Y. Sun, H. Jia, H. Su and Y. Zhang, Catal. Commun., 2012, 29, 35–39 CrossRef CAS.
- T. Hatakeyama, S. Takeda, F. Ishikawa, A. Ohmura, A. Nakayama and Y. Yamada, J. Ceram. Soc. Jpn., 2010, 118, 91–95 CrossRef CAS.
- Y. Okamoto, S. Ida, J. Hyodo, H. Hagiwara and T. Ishihara, J. Am. Chem. Soc., 2011, 133, 18034–18037 CrossRef CAS PubMed.
- J. W. Liu, G. Chen, Z. H. Li and Z. G. Zhang, J. Solid State Chem., 2006, 179, 3704–3708 CrossRef CAS.
- Y. Yang, Q. Chen, Z. Yin and J. Li, J. Alloys Compd., 2009, 488, 364–369 CrossRef CAS.
- H. G. Kim, P. H. Borse, J. S. Jang, E. D. Jeong and J. S. Lee, Mater. Lett., 2008, 62, 1427–1430 CrossRef CAS.
- P. H. Borse, K. T. Lim, J. H. Yoon, J. S. Bae, M. G. Ha, E. H. Chung, E. D. Jeong and H. G. Kim, J. Korean Phys. Soc., 2014, 64, 295–300 CrossRef CAS.
- F. K. Meng, Z. L. Hong, J. Arndt, M. Li, M. J. Zhi, F. Yang and N. Q. Wu, Nano Res., 2012, 5, 213–221 CrossRef CAS.
- X. Cheng, E. Fabbri, M. Nachtegaal, I. E. Castelli, M. EI Kazzi, R. Haumont, N. Marzari and T. J. Schmidt, Chem. Mater., 2015, 27, 7662–7672 CrossRef CAS.
- A. Grimaud, K. J. May, C. E. Carlton, Y. L. Lee, M. Risch, W. T. Hong, J. G. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- D. Li, J. Zheng and Z. Zou, J. Phys. Chem. Solids, 2006, 67, 801–806 CrossRef CAS.
- B. Ravel and M. Newville, Phys. Scr., T, 2005, 115, 1007–1010 CrossRef.
- K. Yang, W. Q. Huangn, L. Xu, K. W. Luo, Y. C. Yang and G. F. Huang, Mater. Sci. Semicond. Process., 2016, 41, 200–208 CrossRef CAS.
- N. N. Wang, H. Y. Lu, Z. T. Lv, W. Wang, J. Q. He, L. Geng and S. Chen, J. Low Temp. Phys., 2015, 181, 242–252 CrossRef CAS.
- W. Zhou, G. Du, P. Hu, G. Li, D. Wang, H. Liu and J. Wang, J. Mater. Chem., 2011, 21, 7937–7945 RSC.
- V. M. Huxter, T. Mirkovic, P. S. Nair and G. D. Scholes, Adv. Mater., 2008, 20, 2439–2443 CrossRef CAS.
- M. Kong, Y. Li, X. Chen, T. Tian, P. Fang, F. Zheng and X. Zhao, J. Am. Chem. Soc., 2011, 133, 16414–16417 CrossRef CAS PubMed.
- S. H. Shen, L. Zhao, Z. H. Zhou and L. J. Guo, J. Phys. Chem. C, 2008, 112, 16148–16155 CAS.
| This journal is © The Royal Society of Chemistry 2017 |