
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hollow Sr/Rh-codoped TiO2 photocatalyst for efficient sunlight-driven organic compound degradation†
Chinh-Chien Nguyena,
Cao-Thang Dinhb and
Trong-On Do*a
aDepartment of Chemical Engineering, Laval University, Quebec, G1V 0A8, Canada. E-mail: trong-on.do@gch.ulaval.ca; Fax: +1-418-656-5993; Tel: +1-418-656-3774
bDepartment of Electrical and Computer Engineering, University of Toronto, Toronto, ON M5S 1A4, Canada
First published on 13th January 2017
Abstract
Sunlight-driven photocatalysis has emerged as a potential technology to address organic pollutant issues. Here, we report the first Rh-doped hollow-structured TiO2 photocatalyst, which is highly active in the photocatalytic decomposition of organic pollutants under solar light. We achieved this by introducing Sr2+ as a co-doping agent, which stabilized the hollow structure at high temperatures and enabled us to control the oxidation state of Rh. The designed photocatalyst exhibited strong visible light absorption (up to 600 nm), and a very high surface area (up to 140 m2 g−1). As a result, the Sr/Rh-doped TiO2 hollow photocatalysts demonstrated a photocatalytic efficiency (PE) of 0.242%, which was at least 8 times higher than that of commercial TiO2 (0.03%) and 25 times higher than that of bulk Sr/Rh–TiO2 (0.01%), in the photocatalytic decomposition of isopropanol under solar light irradiation.
Introduction
As a consequence of rapid global economic growth, environmental pollution by organic compounds has become one of the most critical issues for human society over the past four decades.1 Photocatalytic decomposition of organic species in air and water offers an efficient solution to solve this problem through the utilization of solar energy and high performance photocatalysts.2 Among the various photocatalysts developed to date, TiO2-based photocatalysts continue to receive considerable attention because of their abundance, nontoxicity, and stability under photochemical conditions, which makes them highly suitable for scale-up in environmental remediation.3Currently, most TiO2 photocatalysts are based on the anatase phase, which is generally the most active phase of TiO2.4 Compared to the anatase phase, the rutile phase is often overlooked due to its relatively low specific surface area originating from its high temperature synthesis, and its poor light absorption. Nevertheless, it has been demonstrated that the rutile phase is a promising candidate for the degradation of organic pollutants because of its strong reduction potential and high O2 adsorption capacity.5,6
One of the biggest limitations of TiO2 is its large bandgap (>3 eV), which makes it active only in the UV region, which accounts for less than 5% of the solar spectrum. There have been numerous attempts to overcome the poor light absorption of TiO2 under sunlight by methods including the sensitization of TiO2 with dyes or small bandgap semiconductors, doping, and coupling TiO2 with plasmonic nanostructures.7 Among them, doping TiO2 with Rh has emerged as a viable technique for the enhancement of visible light absorption.8–12 Both Rh3+ and Rh4+ introduce sub-bands into the forbidden band of TiO2. Rh3+ contributes a donor level to the valence band, thus reducing the band energy and shifting light absorption to the visible region, whereas Rh4+ introduces an electron acceptor level below the conduction band, which serves as a recombination site, reducing the activity of the materials, as shown in Fig. SI 1.†6,13 Therefore, controlling the oxidation state of Rh in Rh-doped TiO2 is critical to the photocatalytic enhancement of TiO2.
Another approach to improve the performance of TiO2 photocatalysts is through nanostructuring.14 For example, photocatalysts based on hollow structures have attracted considerable attention because they improve the photoactivity of the catalyst by enhancing the separation of photogenerated charge carriers, shortening the charge diffusion length, and increasing the accessibility of active sites for reactants.15 In addition, multiple reflections within the hollow cavity could improve the efficiency of light absorption, leading to the generation of additional electron–hole pairs.16
It is reasonable that combining the aforementioned approaches (i.e., Rh doping and a hollow structure) would result in an efficient TiO2-based photocatalyst with strong visible light absorption, good charge separation, and high surface active area. However, the design of such materials remains challenging because the introduction of Rh into the TiO2 lattice requires high temperatures (>1000 °C), at which the hollow structure collapses because high surface energy induces sintering effects. Therefore, using a stabilizer to maintain the surface area is critical and so far has not been explored in the development of high performance photocatalysts.
Here, we report the first Rh-doped hollow-structured TiO2 photocatalyst, which is highly active in the photocatalytic decomposition of organic pollutants under sunlight. We achieved this by introducing simultaneous rhodium and strontium. We found that Sr2+ is as a codoping agent which stabilized the hollow structure at high temperatures, and contribute to control the chemical state of Rh in doped material.16–18 The designed photocatalyst exhibited strong visible light absorption (up to 600 nm), and a very high surface area (up to 140 m2 g−1). As a result, the photoactivity of the photocatalyst was at least 8 times higher than those of commercial TiO2 and bulk Sr/Rh–TiO2 powders in the decomposition of isopropanol under solar radiation.
Results and discussion
Traditional methods for the synthesis of Rh-doped TiO2 photocatalysts mainly focus on improving the doping of Rh in the TiO2 lattice to enhance the visible light response of TiO2. However, high-temperature calcination requirements for the substitution of Rh for Ti4+ in the TiO2 matrix results in a photocatalyst with a very low surface area, which increases the charge recombination and decreases the number of active sites. Also, Rh3+, which serves a critical role in the photocatalytic activity of Rh-doped TiO2 under visible light irradiation, cannot be obtained by conventional methods. Therefore, Rh3+-doped TiO2 materials with high surface areas are highly desired. Compared to traditional bulk Rh-doped TiO2 photocatalysts, Sr/Rh-codoped TiO2 offer several advantages. First, they exhibit a high surface area, owing to the presence of nanoparticles on the hollow wall. Therefore, the reaction can proceed inside and outside of the hollow spheres. Second, the co-incorporation of Sr cations enabled us to control the oxidation state of Rh, which leads to a significant enhancement in the visible light absorption and charge separation. Third, the homogeneous distribution of dopants significantly lowers the doping temperature as compared to conventional synthetic routes.Hollow Sr/Rh–TiO2 was prepared via a multistep pathway (Scheme 1) comprised of the following steps: (i) the one-pot synthesis of carbon colloidal spheres@Sr,Rh (C@Sr,Rh); (ii) coating C@Sr,Rh with TiO2 by controlling the hydrolysis of tetraethyl titanium butoxide to obtain core–shell carbon sphere@Sr,Rh–TiO2 microspheres (C@Sr,Rh–TiO2); (iii) two-step calcination at 550 °C for 5 h to obtain anatase Sr/Rh–TiO2-500, followed by calcination at 900 °C for 10 h to form rutile Sr/Rh–TiO2-900. The calcination steps are critical for the substitution of Sr/Rh ions for Ti4+ in the TiO2 crystal lattice and for the formation of the hollow structure with high porosity.
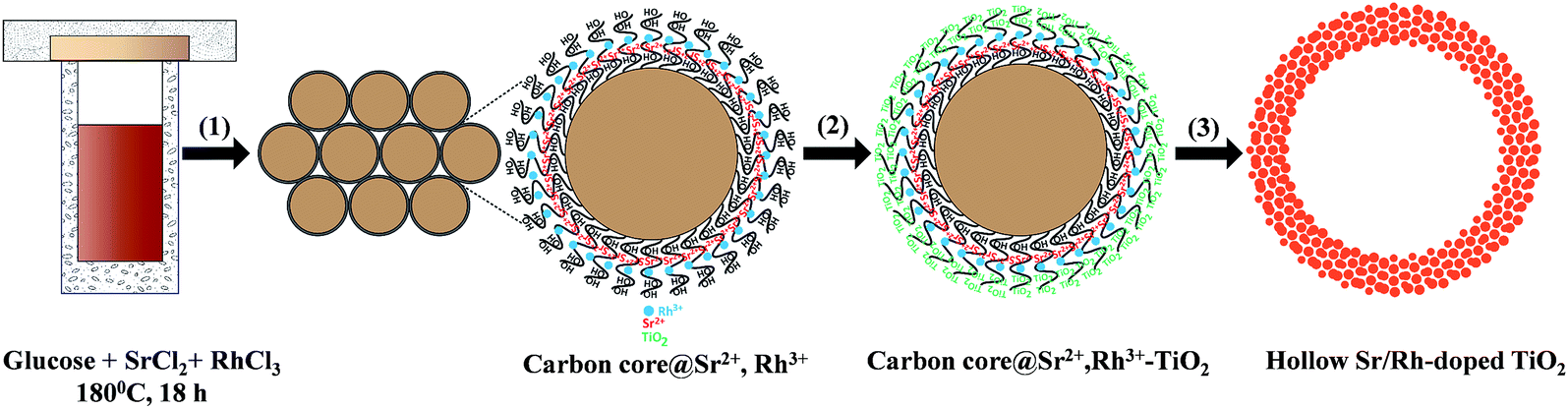 | ||
| Scheme 1 Schematic illustration of the synthesis of hollow Sr/Rh-doped TiO2 nanospheres: (1) one-pot synthesis of carbon colloidal spheres@Sr2+,Rh3+, (2) loading of TiO2 from the titanium alkoxide precursor, and (3) calcination at 550 °C for 5 h followed by calcination at 900 °C for 10 h. | ||
Fig. 1 shows the scanning electron microscopy (SEM) images of C@Sr,Rh–TiO2, hollow Sr/Rh–TiO2-550, and hollow Sr/Rh–TiO2-900. The size of the C@Sr,Rh–TiO2 microspheres was in the range of 500 nm to 2 μm prior to the two-step calcination (Fig. 1A). After calcination at 550 °C, the hollow structure was formed due to the combustion of the carbon colloidal spheres. Broken spheres were observed (Fig. 1B), confirming the formation of the hollow structure. The morphology and size of the hollow structures remained unchanged (500 nm to 2 μm) after calcination at 900 °C, as depicted in Fig. 1C. Fig. SI 2† shows a transmission electron microscopy (TEM) image of hollow Sr/Rh-doped TiO2 after calcination at 900 °C, indicating the formation of particles (100–200 nm in size) on the wall of the hollow structure. Interestingly, we found that the incorporation of Sr2+ increased the stability of the hollow structure at high temperatures. As shown in Fig. SI 3 and SI 4,† the samples prepared using the same procedure in the absence of Sr2+ (Rh–TiO2-900 and TiO2-900) exhibited completely destroyed structures. In contrast, hollow TiO2 structures were retained at high temperatures in the presence of Sr2+ with Rh3+ (Fig. 1C) or without Rh3+ (Fig. SI 5†). In general, the sintering-induced phase transformation of TiO2 from anatase to rutile during calcination at high temperatures is responsible for the change of morphology and the collapse of hollow structure.19,20 The addition of Sr2+ could lower the surface energy change during the TiO2 phase transformation at high calcination temperatures thus preventing the sintering effect, and as a consequence, maintaining the hollow structure in the resulting hollow Sr/Rh–TiO2-900.
 | ||
| Fig. 1 SEM images of (A) carbon colloidal spheres@Sr,Rh@TiO2 and hollow Sr/Rh–TiO2 after calcination at (B) 550 °C and (C) 900 °C. | ||
Elemental mapping by energy-dispersive X-ray microscopy (EDX) from SEM analysis of a single hollow Sr/Rh–TiO2-900 sphere clearly confirmed the homogeneous distribution of Ti, O, Rh, and Sr, as depicted in Fig. 2. The homogeneous distribution of these elements indicates efficient doping of Sr and Rh into the TiO2 lattice without a phase separation. In this approach, using carbon colloidal spheres with a high density of OH groups on the surface facilitated the adsorption of Sr2+ and Rh3+ cations, which resulted in a homogeneous distribution of doping elements in the final hollow sphere products. The chemical composition of the hollow Sr/Rh–TiO2-900 was further analyzed by inductively coupled plasma mass spectrometry (ICP-MS); Rh and Sr contents of 2.0 and 2.3%, respectively, confirmed the successful synthesis of the hollow Rh/Sr-doped TiO2 structure.
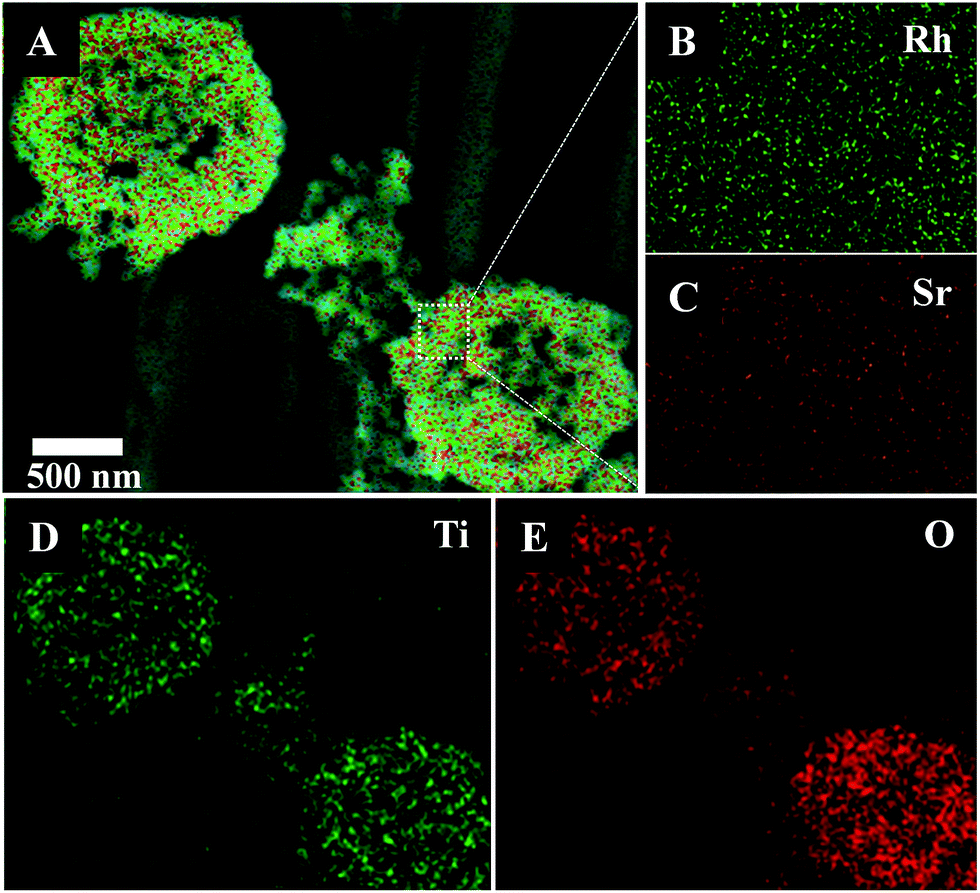 | ||
| Fig. 2 EDX elemental mapping of hollow Sr/Rh–TiO2-900: (A) SEM image; (B) Rh and (C) Sr at high magnification; (D) Ti; (E) O. | ||
The X-ray diffraction (XRD) pattern of hollow Sr/Rh–TiO2-900 (line a) and rutile TiO2 (line b) (Fig. 3A) indicate the presence of rutile TiO2 in the prepared photocatalysts, as neither SrTiO3 nor Rh were detected.21,22 The XRD peaks of hollow Sr/Rh–TiO2-900 were slightly shifted to lower angles as compared to non-doped rutile-TiO2, which could be due to the substitution of Rh3+ for Ti4+,11 as Rh3+ ions (0.805 Å) are larger than Ti4+ ions (0.745 Å).23 Because of the higher ionic radius of Sr2+ (1.12 Å) as compared to Ti4+ (0.61 Å), Sr2+ was unable to enter the TiO2 lattice. Hence, Sr2+ could be present on the TiO2 surface as Sr–O clusters.24,25
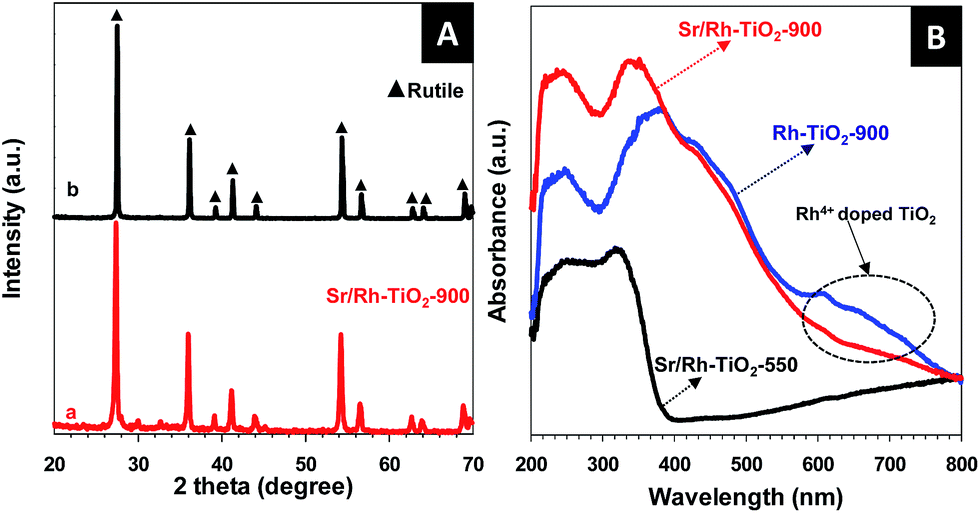 | ||
| Fig. 3 (A) XRD pattern of the hollow Sr/Rh–TiO2 sample after calcination at 900 °C and that of rutile TiO2. (B) UV-vis spectra of the various hollow samples: Sr/Rh–TiO2-550 (black line), Sr/Rh–TiO2-900 (red line), and Rh–TiO2-900 (blue line). | ||
Fig. 3B shows the ultraviolet-visible (UV-vis) spectra of hollow Sr/Rh–TiO2-550, hollow Sr/Rh–TiO2-900, and bulk Rh–TiO2-900. A remarkable enhancement in the light absorption along with a color alteration from white to yellow, as shown in Fig. SI 6,† was observed for hollow Sr/Rh–TiO2-900 and Rh–TiO2-900 compared to hollow Sr/Rh–TiO2-550, indicating the effect of calcination temperature on the light absorption properties of Rh-doped TiO2 materials. Higher calcination temperatures may favor the introduction of Rh to TiO2 crystallites forming Rh-doped TiO2 with a smaller bandgap. The UV-vis spectrum of Rh–TiO2-900 revealed a discontinuous level at ∼420 nm, which could be attributed to electron excitation from the donor levels formed by Rh3+ to the conduction band of rutile TiO2.11,26 In addition, a significant absorption band around 580–750 nm appeared in the UV-vis spectrum of this sample, indicating the presence of an acceptor level formed by Rh4+ in the TiO2 structure. Thus, both Rh3+ and Rh4+ were present in the bulk Rh–TiO2-900 sample. Compared to bulk Rh–TiO2-900, hollow Sr/Rh–TiO2-900 (in the presence of Sr) exhibited a simple absorption profile with an onset at ∼420 nm, and the disappearance of the absorption band at around 580–750 nm. This suggested that only Rh3+ was present in hollow Sr/Rh–TiO2-900.26–28 The presence of Rh3+ ions in the hollow Sr/Rh–TiO2-900 further confirm the important role of Sr2+ in this synthesis, as it not only prevented sintering induced by high temperature annealing, but also maintained the Rh3+ chemical state in the Rh-doped material. Fig. SI 7† shows the plot of (αhν)2 versus photon energy for the bandgap calculation of the samples. The band gap of hollow Sr/Rh–TiO2-900 was calculated to be 2.25 eV, which was significantly lower than that of the hollow sample calcined at 550 °C (3.37 eV). The increased band gap of hollow Sr/Rh–TiO2-550 as compared to anatase TiO2 alone (3.2 eV) could be ascribed to the presence of Sr2+ on the TiO2 surface, which reduced the particle size.29
To understand the surface composition and chemical states of the elements in the samples, X-ray photoelectron spectroscopy (XPS) was carried out for the hollow Sr,Rh-doped TiO2-900 and Rh-doped TiO2-900. The presence of Sr, Rh, Ti, and O in Sr/Rh–TiO2-900 was confirmed by XPS, as depicted in Fig. 4A. As shown in Fig. SI 8,† for the Sr 3d XP spectrum, the deconvoluted peaks centered at 133.1 and 134.87 eV could be attributed to Sr 3d5/2 and Sr 3d3/2 of Sr–O-modified TiO2 surface.30 Fig. 4B shows the deconvoluted peaks of Rh 3d XPS spectra in hollow Sr/Rh–TiO2-900 (a) and Rh–TiO2-900 (b). It should be noted that isolated observation of Rh3+ and Rh4+ is ambiguous by XPS analysis. However, it is essential to consider the shifting of their binding energies because it would help to evaluate the electron density of Rh atoms contributing to confirm the chemical states in each sample. For the Rh–TiO2-900, the Rh 3d XPS spectrum shows two peaks centered at 309.3 eV and 314.2 eV, which could be attributed to 3d5/2 and 3d3/2 of Rh3+ and Rh4+ chemical states, respectively.31,32 Interestingly, these peaks were shifted −0.3 eV and appeared at 390.0 and 313.9 eV for the hollow Sr/Rh–TiO2-900, which could be attributed to the higher electron density of Rh cations. In other words, the chemical states of Rh in the hollow Sr/Rh–TiO2-900 is lower than that in Rh–TiO2-900. This is associated with UV-vis analysis, which confirms mainly presence of Rh3+ chemical state in the hollow Sr/Rh–TiO2-900. Fig. 4C shows the Ti 2p XPS spectra of the hollow Sr/Rh–TiO2-900 (line a) and Rh–TiO2-900 (line b). The Rh–TiO2-900 exhibited two peaks at binding energies of 458.2 and 464.1 eV, which could be assigned to Ti 2p3/2 and Ti 2p1/2 of Ti4+ in the rutile phase.33 These peaks are −0.1 eV shifted compared to those of r-TiO2, suggesting the replacement of Ti4+ by both Rh3+ and Rh4+, which induced a higher electron density around Ti4+ atoms in the lattice, as depicted in Table SI-1.† In the case of hollow Sr/Rh–TiO2-900, the Ti 2p3/2 and Ti 2p1/2 peaks appeared at the binding energies of 457.8 and 463.7 eV, respectively, which are −0.4 eV shifted compared to those of Rh–TiO2-900. This indicates a significant increase in the electron density of Ti atoms in hollow Sr/Rh–TiO2-900 compared to that in Rh–TiO2-900 which further confirm the replacement of Ti4+ by Rh3+ in the hollow Sr/Rh–TiO2-900. These results, in combination with the data obtained from UV-vis, suggest that Rh is presented in the hollow Sr/Rh–TiO2-900 primarily in the form of Rh3+. Fig. 4D presents the O 1s XP spectra of the hollow Sr/Rh–TiO2-900 (line a) and Rh–TiO2-900 (line b). Two peaks with binding energies of 529.3 and 530.1 eV were observed in the deconvoluted peaks of O 1s in Rh–TiO2-900 (line b). The peak centered at 529.3 eV could be ascribed to the lattice oxygen in TiO2, while other one at 530.1 eV could be attributed to strongly bound Ti–OH. Using the O 1s of rutile-TiO2 alone as a reference (Table SI-1†), the O 1s peak (at 529.3 eV) of Rh–TiO2-900 exhibited a negative shift in comparison to that of pristine rutile-TiO2 (at 529.6 eV), which originated from the introduction of partially filled Rh4+ states within the band gap.13 This may cause the downward shifting of Fermi level towards the valence band, which may decrease energy bands along with the reduction potential of photogenerated electrons and induce a negative impact on the photoactivity.13,34,35 In contrast, a strong positive shift in O1s peak was observed (at 529.9 eV) for hollow Sr/Rh–TiO2-900 in comparison to rutile-TiO2 alone and Rh–TiO2-900, for which the O1s peaks were centered at 529.9 and 530.5 eV. The increased binding energy of O1s in hollow Sr/Rh–TiO2-900 could be assigned to the filling of Rh3+ states within the band gap, which caused the upward shifting of Fermi level towards the conduction band, which increase the energy bands along with the reduction potential of photogenerated electrons and has a positive impact on the photocatalytic activity.36,37 These differences could explain the disadvantages of Rh4+ and the critical role of Rh3+ in doped photocatalysts. Therefore, the introduction of Sr2+ to Rh-doped TiO2 favors the formation of Rh3+ in the doped TiO2. In the absence of Sr2+, Rh3+ oxidizes to Rh4+ to re-establish the charge balance, resulting in the formation of Rh4+-doped TiO2. In contrast, the presence of Sr2+ which could function as the critical factor limiting this oxidation process and leading to the formation of Rh3+-doped TiO2.
 | ||
| Fig. 4 XPS spectra of hollow Sr/Rh–TiO2-900 (a) in comparison to those of Rh–TiO2-900 (b) survey spectrum of hollow Sr/Rh–TiO2-900 (A); XPS spectra of Rh 3d (B), Ti 2p (C), and O 1s (D). | ||
The photocatalytic activity of a catalyst is strongly dependent on the number of active sites on its surface. Thus, surface and porosity properties are the important parameters governing the performance of a photocatalyst because high specific surface area and large pore volume facilitate mass transfer and effectively promote the kinetics of the photocatalytic reaction. Therefore, the photocatalytic activity is significantly enhanced.38,39 The specific surface areas of various samples were determined by N2 physical adsorption measurements at 77 K. As shown in Fig. SI 9A,† hollow Sr/Rh–TiO2-900 exhibits a high adsorption capacity in high relative pressure (P/P0: from 0.8 to 1), suggesting a large porous structure.40 The (Barrett–Joyner–Halenda) BJH pore size distribution obtained from the adsorption branch also reveals that the majority of pores are mainly from 2 to 10 nm, as depicted in Fig. SI 9B.† As shown in Table 1, the specific surface area of hollow Sr/Rh–TiO2-900 reached 140 m2 g−1, which was ∼50 times higher than that of Rh–TiO2-900 (3 m2 g−1). Also, the specific surface area of hollow Sr/Rh–TiO2-900 was significantly higher than those of r-TiO2 (10 m2 g−1) and commercial anatase TiO2 (P25) (50 m2 g−1), and ∼13 times that of bulk Sr/Rh–TiO2-900-B (Table 1). To the best of our knowledge, this hollow Sr/Rh–TiO2-900 photocatalyst has one of the highest surface areas among doped rutile TiO2 materials reported to date.22,41,42 The above analyses afforded two important findings about the roles of Sr2+: (i) the presence of Sr2+ functioned as a stabilizer to maintain the hollow structure leading to a high surface area photocatalyst; (ii) the chemical oxidation state of Rh3+ could be controlled in the doped TiO2 with the co-presence of Sr2+, which limited the oxidation of Rh3+ to Rh4+. We believe that these two unique properties have a close relation in the resulted photocatalyst.
| Photocatalyst | TiO2 phase | Morphology | % dopants (Rh3+, Sr2+) | Band gap energy (eV) | Specific surface area (m2 g−1) |
|---|---|---|---|---|---|
| Hollow Sr/Rh–TiO2-900 | Rutile | Hollow spheres | 2.0; 2.3 | 2.25 | 140 |
| Hollow Sr/Rh–TiO2-550 | Anatase | Hollow spheres | 2.0; 2.3 | 3.3 | 190 |
| Sr/Rh–TiO2-900-B | Rutile | Bulk crystals | 2.0; 2.3 | 2.25 | 11 |
| Rh–TiO2-900 | Rutile | Bulk crystals | 2.1 | 2.0 | 3 |
| r-TiO2 | Rutile | Bulk crystals | 0; 0 | 3.1 | 10 |
| TiO2 (P25) | Anatase-rutile | Nanoparticles | 0; 0 | 3.2 | 50 |
The photocatalytic activity of hollow Sr/Rh–TiO2-900 was evaluated in the photodecomposition of isopropanol in water. So far, the isopropanol photodegradation is used as a model reaction for photo-oxidation of organic compounds.43 Notably, Pt has been demonstrated to significantly improve the photocatalytic activity of TiO2-based photocatalysts.44 For free-Pt photocatalysts, only small amount of CO2 was evolved from the decomposition of isopropanol for the hollow Sr/Rh–TiO2-900 and no CO2 was detected for the rest of samples, as shown in Fig. SI 10.† This indicates the critical role of Pt particles as co-catalyst. Therefore, the evolution of CO2 from the decomposition of isopropanol under visible light (λ ≥ 420 nm) and solar light irradiation (see Experimental section) was monitored and compared with those observed using Sr/Rh–TiO2-B, Rh–TiO2-900 and r-TiO2 with the optimized amounts of Pt loading, which were found to be ∼1% Pt for the hollow Sr/Rh–TiO2-900, ∼0.3% for Sr/Rh–TiO2-B and r-TiO2 and ∼0.1% for Rh–TiO2-900, respectively. As shown in Fig. 5A, the photocatalytic activity of hollow Sr/Rh–TiO2-900 was superior to those of Sr/Rh–TiO2-B and Rh–TiO2-900, as indicated by the higher CO2 generation. The amount of CO2 produced over the hollow Sr/Rh–TiO2-900 under visible light was 27.5 μmol g−1 h−1, which was more than 23 times higher than that of Sr/Rh–TiO2-B (1.2 μmol g−1 h−1). Under similar reaction conditions, CO2 was not detected for the Rh–TiO2-900 and r-TiO2 samples, indicating their negligible photoactivities under visible light. As shown in Fig. 5B, hollow Sr/Rh–TiO2-900 also exhibited a higher photoactivity than bulk Sr/Rh–TiO2-B and r-TiO2 under simulated solar irradiation. The amount of CO2 evolved for hollow Sr/Rh–TiO2-900 was 146 μmol g−1 h−1, which was ∼25 times higher than that of Sr/Rh–TiO2-B (5.68 μmol g−1 h−1), whereas r-TiO2 and Rh–TiO2-900 showed very low photoactivities. Compared to hollow Sr/Rh–TiO2-550, hollow Sr/Rh–TiO2-900 exhibited ∼2.5 times higher CO2 generation (Fig. SI 11†), despite the higher surface area of hollow Sr/Rh–TiO2-550 (see Table 1). Moreover, the amount of CO2 produced with the hollow Sr/Rh–TiO2-900 catalyst was 6.6 times higher than that of commercial TiO2 (P25). Therefore, the superior activity of hollow Sr/Rh–TiO2-900 could be attributed to the synergistic effect of doping Rh3+ into the TiO2 structure in the presence of Sr2+, which causes strong solar light absorption, and a hollow structure with a high surface area. These features significantly enhanced the generation of electron–hole pairs and improved the charge separation during photocatalysis.
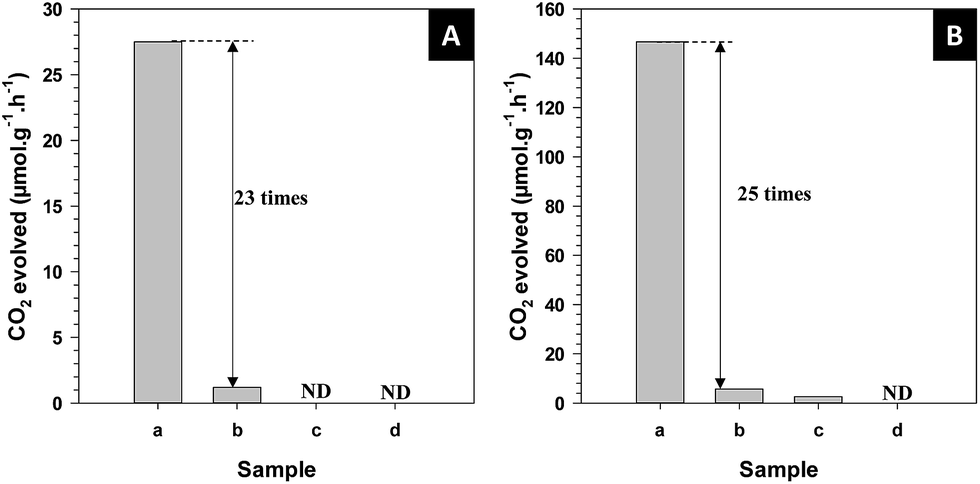 | ||
| Fig. 5 CO2 formation from the photodegradation of isopropanol of different types of photocatalysts with 1% Pt: (A) under visible light, (B) under solar simulator irradiation (AM 1.5 G, intensity 100 mW cm−2): (a) hollow Sr/Rh–TiO2-900; (b) bulk Sr/Rh–TiO2-B; (c) rutile TiO2; (d) Rh–TiO2-900. ND: not detected. | ||
The photocatalytic efficiency (PE) for the photocatalytic decomposition of isopropanol under solar irradiation was calculated using the following equation:
| PE = Nconsumed photons for CO2/Nincident photons | (1) |
The stability of the hollow Sr/Rh–TiO2-900 was also studied by performing multi-recycling experiments under the same conditions, as shown in Fig. SI 12.† No significant change in the production of CO2 was observed after 5 cycles. Also, the morphology was retained after the fifth cycle indicating the stability of the hollow Sr/Rh–TiO2-900, as shown in Fig. SI 13.†
It has been demonstrated that rutile-TiO2 contains a large amount of adsorbed oxygen on its surface.47–51 Therefore, the photodecomposition of organic pollutants should be mainly directed toward the electron usage route, which produces strong oxidants (O2˙−, H2O2) (ECB = −0.7 V vs. standard hydrogen electrode (SHE); O2 + e = O2˙−, −0.28 V vs. SHE; O2 + 2H2O + 2e− = H2O2 + 2OH−, +0.28 vs. SHE).47–51 A mechanism for CO2 generation with Sr/Rh-doped TiO2 is proposed in Fig. 6. Hollow Sr/Rh–TiO2-900 exhibited high photoactivity, which could be due to the synergistic contribution of efficient Rh3+ doping and high surface area. Indeed, efficient absorption of sunlight appeared to generate additional electron–hole pairs for the photodegradation of isopropanol into CO2. Moreover, the high surface area of the hollow structure increases the number of exposed active sites for the photoreactions.48 As a result, both as-photogenerated electron–hole pairs could contribute to produce strong oxidants inducing the efficient degradation of isopropanol.
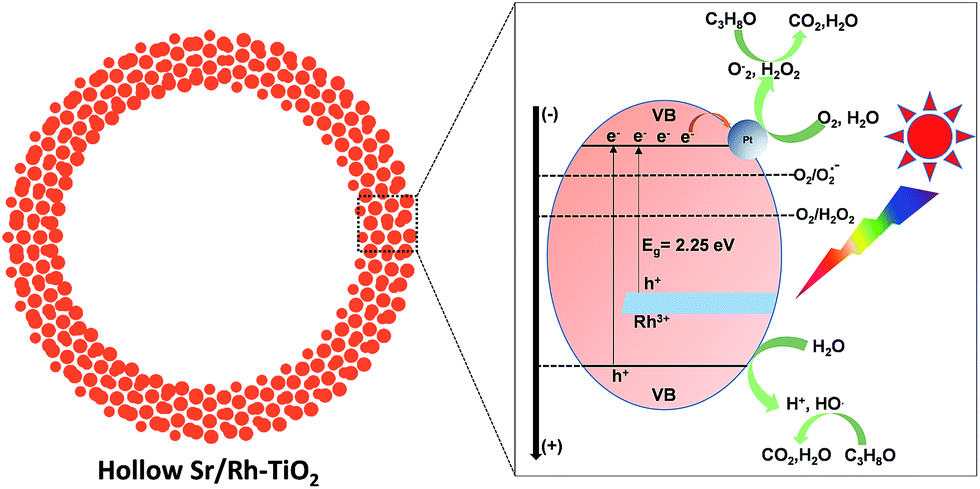 | ||
| Fig. 6 Schematic illustration of the photodecomposition of organic isopropanol using hollow Sr/Rh–TiO2-900 under solar light irradiation. | ||
Conclusions
We successfully prepared a novel type of hollow Sr/Rh-doped TiO2 using carbon colloidal spheres as the sacrificial template. For the first time, we demonstrated that the introduction of Sr2+ to Rh-doped TiO2 stabilizes the hollow structure and maintains the Rh3+ oxidation state of Rh in the doped material at high temperatures. The obtained photocatalyst exhibited high photoactivity in the degradation of isopropanol under visible light and simulated solar irradiation, owing to the synergistic contributions of strong solar light absorption and high surface area. Therefore, our hollow material has significant potential for applications in water purification under natural solar illumination. Furthermore, this synthetic strategy can be extended to other hollow systems for a wide range of applications in catalysis and energy conversion. Work into gaining further insight into the mechanistic aspects of this process is in progress.Experimental section
Chemicals
D-(+)-Glucose, hexachloroplatinic acid hexahydrate, strontium chloride, rhodium(III)nitrate, and tetraethyl titanium butoxide were purchased from Sigma-Aldrich. All reagents were used as received without further purification.Material syntheses
Characterization
Transmission electron microscopy (TEM) images of the samples were obtained using a JOEL JEM 1230 operated at 120 kV. Scanning electron microscopy (SEM) images were obtained using a JEOL 6360 operated at 15 kV. Powder X-ray diffraction (XRD) patterns of the samples were obtained using a Bruker SMART APEXII X-ray diffractometer equipped with a Cu Kα radiation source (λ = 1.5418 Å). X-ray photoelectron spectroscopy (XPS) measurements were carried out in the ion-pumped chamber (evacuated to 10−9 Torr) of a photoelectron spectrometer (Kratos Axis-Ultra) equipped with a focused X-ray source (Al Kα, hν = 1486.6 eV). UV-vis spectra were recorded on a Cary 300 Bio UV-visible spectrophotometer. N2 adsorption–desorption isotherms of the samples were obtained at 77 K using a Quantachrome Autosorb-1 MP analyzer. Prior to the measurements, the samples were outgassed under vacuum for 6 h at 150 °C.Photocatalytic tests
The photocatalytic decomposition of isopropanol was carried out in a top-irradiated reactor at ambient temperature and pressure. Prior to the photocatalytic tests, Pt was photodeposited on the catalyst. In a typical photocatalytic experiment, 20 mg of the photocatalyst, the optimum amount of photocatalyst for this photocatalytic reactor system, was dispersed in a 200 ppm solution of isopropanol in water. The reaction cell was then filled with fresh synthetic air (Prax air), stirred for 1 h to get steady-state regime prior to be illuminated with a solar simulator 150 W Xe lamp (AM 1.5 G, 100 mW cm−2) for 3 h. To evaluate the photocatalytic activity of the samples under visible light, a UV cut-off filter (λ ≥ 420 nm) was used to produce visible light from the solar simulator. The amount of CO2 gas generated during the reaction was analyzed using a gas chromatograph (Agilent 7820A) equipped with a thermal conductivity detector (TCD) and HP-PLOT U column, using helium as the carrier gas.Photocatalytic efficiency (PE) calculations
Photocatalytic efficiency can be measured by the ratio of the number of product formed to the number of incident photons on the system and in a wavelength range λ1–λ2.18,45,46 Under full solar irradiation, the number of incident photons calculated by integrating the intensity at each wavelength was 2.75 × 1017 photon per s per cm2. A 4.5 cm2 area of the sample was irradiated for 3 h. Therefore, the number of incident photons was calculated as follows:| Nincident photons = (2.75 × 1017) × 3 × 4.5 × 3600 = 1.31 × 1022 photons | (2) |
For CO2 generation, assuming that the conversion of isopropanol (C3H8O) to CO2 takes place on hollow Sr/Rh–TiO2-900 according to the following equation, (six photons are required to produce one CO2 molecule):
| C3H8O + 9O2 + 18H+ + 18e− → 3CO2 + 13H2O (2eCB− + O2 + 2H+ → H2O2; C3H8O + 9H2O2 → 3CO2 + 13H2O); 2H2O + 4H+ → O2 + 4H+ |
The amount of CO2 generated under simulated solar light was determined to be 8.8 μmol. Therefore, the number of photons consumed to produce CO2 under solar light illumination over 3 h was calculated as follows:
| Nconsumed photons for CO2 = 6 × (8.8 × 10−6) × (6.023 × 1023) = 3.18 × 1019 | (3) |
| PE = Nconsumed photons for CO2/Nincident photons = (3.18 × 1019/1.31 × 1022) × 100 = 0.242% | (4) |
The same procedure was used to determine the PE of commercial TiO2 (P25) and Sr/Rh–TiO2-900-B.
Acknowledgements
This work was supported by the Natural Science and Engineering Research Council of Canada (NSERC) through the Collaborative Research and Development with EXP Inc. (CRD) and Discovery Grants.Notes and references
- L. G. Devi and R. Kavitha, Appl. Catal., B, 2013, 140, 559–587 CrossRef.
- X. Li, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603–2636 RSC.
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 169–189 CrossRef CAS.
- G. Odling and N. Robertson, ChemSusChem, 2015, 8, 1838–1840 CrossRef CAS PubMed.
- S. Yurdakal, G. Palmisano, V. Loddo, V. Augugliaro and L. Palmisano, J. Am. Chem. Soc., 2008, 130, 1568–1569 CrossRef CAS PubMed.
- Q. Sun and Y. Xu, J. Phys. Chem. C, 2010, 114, 18911–18918 CAS.
- M. Nolan, A. Iwaszuk, A. K. Lucid, J. J. Carey and M. Fronzi, Adv. Mater., 2016, 28, 5425–5446 CrossRef CAS PubMed.
- L. R. Grabstanowicz, S. Gao, T. Li, R. M. Rickard, T. Rajh, D.-J. Liu and T. Xu, Inorg. Chem., 2013, 52, 3884–3890 CrossRef CAS PubMed.
- J. Tao, M. Yang, J. W. Chai, J. S. Pan, Y. P. Feng and S. J. Wang, J. Phys. Chem. C, 2014, 118, 994–1000 CAS.
- Y. Matsumoto, T. Shimuzu and E. Sato, Electrochim. Acta, 1982, 27, 419–424 CrossRef CAS.
- R. Niishiro, R. Konta, H. Kato, W.-J. Chun, K. Asakura and A. Kudo, J. Phys. Chem. C, 2007, 111, 17420–17426 CAS.
- B. Liu, H. M. Chen, C. Liu, S. C. Andrews, C. Hahn and P. Yang, J. Am. Chem. Soc., 2013, 135, 9995–9998 CrossRef CAS PubMed.
- E. Glover, S. Ellington, G. Sankar and R. Palgrave, J. Mater. Chem. A, 2016, 4, 6946–6954 CAS.
- X. Chen and A. Selloni, Chem. Rev., 2014, 114, 9281–9282 CrossRef CAS PubMed.
- C. C. Nguyen, N. N. Vu and T.-O. Do, J. Mater. Chem. A, 2015, 3, 18345–18359 CAS.
- C.-T. Dinh, H. Yen, F. Kleitz and T.-O. Do, Angew. Chem., Int. Ed., 2014, 53, 6618–6623 CrossRef CAS PubMed.
- M.-H. Pham, C.-T. Dinh, G.-T. Vuong, N.-D. Ta and T.-O. Do, Phys. Chem. Chem. Phys., 2014, 16, 5937–5941 RSC.
- C.-C. Nguyen, N.-N. Vu and T.-O. Do, J. Mater. Chem. A, 2016, 4413–4419 CAS.
- J.-G. Yu, H.-G. Yu, B. Cheng, X.-J. Zhao, J. C. Yu and W.-K. Ho, J. Phys. Chem. B, 2003, 107, 13871–13879 CrossRef CAS.
- Q. Mao, Y. Ren, K. H. Luo and S. Li, J. Phys. Chem. C, 2015, 119, 28631–28639 CAS.
- Y. Cao, X. Li, Z. Bian, A. Fuhr, D. Zhang and J. Zhu, Appl. Catal., B, 2016, 180, 551–558 CrossRef CAS.
- L. Li, J. Yan, T. Wang, Z.-J. Zhao, J. Zhang, J. Gong and N. Guan, Nat. Commun., 2015, 6, 5881, DOI:10.1038/ncomms6881.
- R. T. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- L. Escriche-Tur, J. Jover, M. Font-Bardia, G. Aullón and M. Corbella, Inorg. Chem., 2015, 54, 11596–11605 CrossRef CAS PubMed.
- Q. Wu, Q. Zheng and R. van de Krol, J. Phys. Chem. C, 2012, 116, 7219–7226 CAS.
- S. Kawasaki, K. Akagi, K. Nakatsuji, S. Yamamoto, I. Matsuda, Y. Harada, J. Yoshinobu, F. Komori, R. Takahashi and M. Lippmaa, J. Phys. Chem. C, 2012, 116, 24445–24448 CAS.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992–8995 CrossRef CAS.
- Q. Wang, T. Hisatomi, S. S. K. Ma, Y. Li and K. Domen, Chem. Mater., 2014, 26, 4144–4150 CrossRef CAS.
- L. Kumaresan, M. Mahalakshmi, M. Palanichamy and V. Murugesan, Ind. Eng. Chem. Res., 2010, 49, 1480–1485 CrossRef CAS.
- S. Riedel, J. Neidhardt, S. Jansen, L. Wilde, J. Sundqvist, E. Erben, S. Teichert and A. Michaelis, J. Appl. Phys., 2011, 109, 094101 CrossRef.
- C. Li, J. Liu, W. Gao, Y. Zhao and M. Wei, Catal. Lett., 2013, 143, 1247–1254 CrossRef CAS.
- S. Parres-Esclapez, I. Such-Basañez, M. J. Illán-Gómez, C. Salinas-Martínez de Lecea and A. Bueno-López, J. Catal., 2010, 276, 390–401 CrossRef CAS.
- W. Q. Fang, X. L. Wang, H. Zhang, Y. Jia, Z. Huo, Z. Li, H. Zhao, H. G. Yang and X. Yao, J. Mater. Chem. A, 2014, 2, 3513–3520 CAS.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- D. Lang, F. Cheng and Q. Xiang, Catal. Sci. Technol., 2016, 6, 6207–6216 CAS.
- L. Li, P. A. Salvador and G. S. Rohrer, Nanoscale, 2014, 6, 24–42 RSC.
- J. Zhang, F. Ren, M. Deng and Y. Wang, Phys. Chem. Chem. Phys., 2015, 17, 10218–10226 RSC.
- Q. Liang, Z. Li, Z. H. Huang, F. Kang and Q. H. Yang, Adv. Funct. Mater., 2015, 6885–6892 CrossRef CAS.
- T. Weller, J. Sann and R. Marschall, Adv. Energy Mater., 2016, 6, 1600208 CrossRef.
- J. Zhang, J. Yu, Y. Zhang, Q. Li and J. R. Gong, Nano Lett., 2011, 11, 4774–4779 CrossRef CAS PubMed.
- H. Hou, M. Shang, L. Wang, W. Li, B. Tang and W. Yang, Sci. Rep., 2015, 5, 15228 CrossRef CAS PubMed.
- R. Kaplan, B. Erjavec, G. Dražić, J. Grdadolnik and A. Pintar, Appl. Catal., B, 2016, 181, 465–474 CrossRef CAS.
- Y. Li, S. Ouyang, H. Xu, X. Wang, Y. Bi, Y. Zhang and J. Ye, J. Am. Chem. Soc., 2016, 138, 13289–13297 CrossRef CAS PubMed.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- M. Liu, X. Qiu, M. Miyauchi and K. Hashimoto, J. Am. Chem. Soc., 2013, 135, 10064–10072 CrossRef CAS PubMed.
- S. E. Braslavsky, A. M. Braun, A. E. Cassano, A. V. Emeline, M. I. Litter, L. Palmisano, V. N. Parmon and N. Serpone, Pure Appl. Chem., 2011, 83, 931–1014 CrossRef CAS.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave and I. P. Parkin, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- M. Buchalska, M. Kobielusz, A. Matuszek, M. Pacia, S. Wojtyła and W. Macyk, ACS Catal., 2015, 5, 7424–7431 CrossRef CAS.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
- Y. Kakuma, A. Y. Nosaka and Y. Nosaka, Phys. Chem. Chem. Phys., 2015, 17, 18691–18698 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25987a |
| This journal is © The Royal Society of Chemistry 2017 |