
Total synthesis of natural products via iridium catalysis
Changchun
Yuan
*a and
Bo
Liu
 *b
*b
aSchool of Chemical Engineering and Technology, North University of China, Taiyuan 030051, PR China. E-mail: ycc543700483@nuc.edu.cn
bKey Laboratory of Green Chemistry & Technology of Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, PR China. E-mail: chembliu@scu.edu.cn
First published on 18th September 2017
Abstract
Catalysis with transition metals is a powerful synthetic tool for achieving a high degree of molecular complexity from relatively simple building blocks. Among these transition metals employed, iridium has attracted significant attention owing to its multifold roles in catalysis of various synthetically significant methodologies, and thus iridium catalysts are widely used in natural product synthesis. This review aims to comprehensively summarize recent accomplishments in total synthesis of natural products using iridium as the catalyst.
 Changchun Yuan | Changchun Yuan was born in Shandong Province, China in 1987. He received his B.Sc. degree in 2010 from Qufu Normal University. Then he pursued his M.Sc. degree and Ph.D. degree in the laboratory of Prof. Bo Liu at the College of Chemistry, Sichuan University. After receiving his PhD, he joined the School of Chemical Engineering and Technology, North University of China. |
 Bo Liu | Bo Liu was born in Chongqing, China, in 1976. He obtained his B.Sc. in Environmental Chemistry from Southwest Normal University (currently Southwest University) in 1998. He then began his research career under the supervision of Prof. Xiaoming Feng at the Chengdu Institute of Organic Chemistry (CIOC, CAS). After he obtained his M.Sc. in 2001, he joined Prof. Wei-Shan Zhou's group at the Shanghai Institute of Organic Chemistry (SIOC, CAS) to work on the total synthesis of steroids and polyketides. He obtained his Ph.D. in 2004. After postdoctoral research from 2004 to 2007, supervised by Prof. Jef K. De Brabander at the University of Texas Southwestern Medical Center at Dallas, he joined the College of Chemistry at Sichuan University as a faculty member. In 2014, he received a Thieme Chemistry Journal Award. His current research interests are focused on the total synthesis of natural products, synthesis-oriented methodology, and medicine synthesis. |
1 Introduction
Great achievements have been made in the research field of metal catalysis,1 since the early work on heterogeneous platinum group metals in the nineteenth century.2 At present, transition metal catalysts with peculiar properties, have become the most powerful tools in modern organic synthesis, which successfully revolutionize synthetic strategies increasing molecular complexity in a straightforward fashion.The early history of iridium catalysis has been detailed previously, featuring a very tortuous process.2 At the beginning, iridium was not considered as important as Pd, Pt, Ru, Rh etc., because it showed no special catalytic advantages.2 However, the situation of Ir as an unessential heterogeneous catalyst gradually changed, after it was employed as a homogeneous catalyst, which contributed to the understanding of the mechanism of oxidative addition, a fundamental reaction step in organometallic chemistry.1,3 Today, iridium catalysis plays a dominant role in organic synthesis, and a wide range of iridium-catalyzed reactions have been developed, including asymmetric hydrogenation with chiral ligands,4 hydrogen transfer reactions,5 functionalization of C–H bonds,6 asymmetric allylic substitution,7 cycloaddition reactions,8 enantioselective polycyclization reaction,9 photoredox catalysis reaction,10 and so on.11 Moreover, compared to other metal catalysts such as Pd, Pt, Ru, and Rh, the Ir complex does show special properties resulting in excellent enantioselectivity, short reaction time and a high conversion rate (Fig. 1).
 | ||
| Fig. 1 Advantage of an iridium catalyst. | ||
Transition metal catalysis, involving iridium catalysis, contributes well to the state of the art of natural product synthesis.12 The aim of this review is to highlight application of iridium catalysis in total synthesis of complex natural products on the basis of a collection of recent literature studies. In this review, we have summarized some elegant work in natural product total synthesis, which were classified into five categories according to the above-mentioned key iridium-catalyzed step used.
2 Asymmetric hydrogenation reaction
Asymmetric hydrogenation is one of the most important catalytic reactions for synthesis of optically active compounds, which can provide various synthetic intermediates leading to biologically active compounds and naturally occurring substances. In recent decades, asymmetric hydrogenation with noble metal catalysts has been proven effective for all types of olefins. In particular, iridium-catalyzed asymmetric hydrogenation draws a lot of attention owing to its low catalyst loading, excellent catalytic activity and impressive ability of enantioselective introduction.4 Thus iridium catalysts are widely employed in total synthesis of natural products.2.1 Zhou's synthesis of crispine A, (S)-equol, (S)-(+)-homoisoflavone, (R)-aristelegone A, (R)-curcumene and (R)-xanthorrhizol
Zhou's group has been working on the study of iridium-catalyzed asymmetric hydrogenation, and has obtained great achievements.4e In 2009, Zhou and his coworkers developed a methodology on asymmetric hydrogenation of cyclic enamines 1 by using monodentate spiro-phosphoramidite (Ra,S,S)-3 as the ligand to afford excellent enantioselectivities (Scheme 1A).13 It prompted them to utilize this asymmetric hydrogenation in total synthesis of the isoquinoline alkaloid, crispine A,14 which was isolated from Carduus crispus, and showed significant cytotoxic bioactivities.15 The tricyclic enamine 7 could be conveniently synthesized from 2-(3,4-dimethoxyphenyl)ethanamine (4) in three steps by following the literature method.14b The asymmetric hydrogenation of enamine 7 was performed under the optimized conditions using the catalyst (Sa,R,R)-3 to produce crispine A in 97% yield with 90% ee (Scheme 1B).13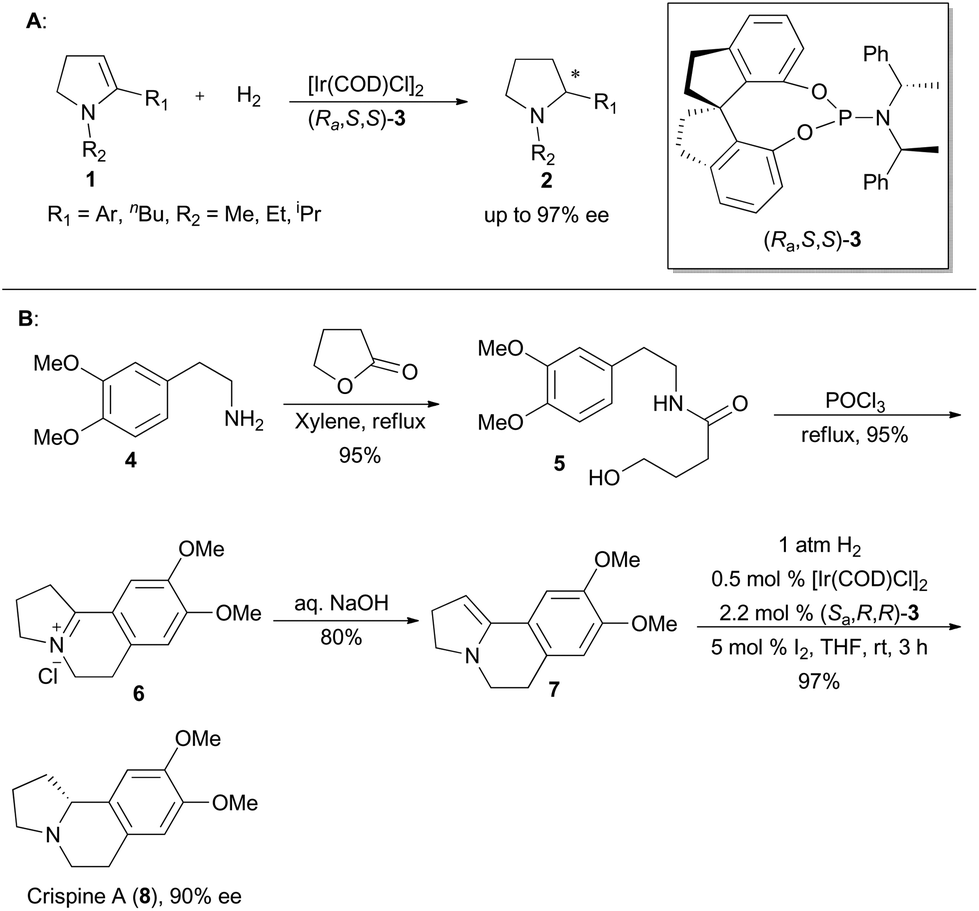 | ||
| Scheme 1 Zhou's synthesis of crispine A. | ||
In addition, Zhou's group also realized asymmetric hydrogenation of unsaturated carboxylic acid with iridium complexes involving chiral spiro-phosphine-oxazoline ligands or chiral spirophosphine-benzylamine ligands.4e The substrate scope can be extended to α,β-disubstituted acrylic acids, trisubstituted acrylic acids, α-substituted acrylic acids, and heterocyclic α,β-unsaturated acids.
(S)-Equol (15, Scheme 2A) is an important soy isoflavonoid metabolite, which has attracted increasing attention due to its phytoestrogenic activity and potential use in menopausal hormone replacement therapy.16 In Zhou's synthesis of this natural product, (E)-3-(2,4-dimethoxyphenyl)-2-(4-methoxyphenyl)acrylic acid 11 was firstly prepared from commercially available 2,4-dimethoxyphenylaldehyde (9) and 2-(4-methoxyphenyl)acetic acid (10) (Scheme 2A). With asymmetric hydrogenation of 11 by means of the Ir-(Ra)-12 catalyst as the key step, the enantioselective synthesis of (S)-equol was accomplished in six steps with 48.4% overall yield from commercially available starting materials (Scheme 2A).17
 | ||
| Scheme 2 Zhou's synthesis of (S)-equol and (S)-(+)-homoisoflavone. | ||
Natural homoisoflavonoids possess remarkable and abundant biological activities, and thus isolation of such kinds of new compounds from nature arouses great attention from phytochemists.18 In 2006, the (S)-(+)-homoisoflavone (Scheme 2B) was isolated from Chlorophytum inornatum, which bears a stereogenic center at the C3 position and displays significant antibacterial activity.19 Oudeyer and Levacher et al. accomplished asymmetric total synthesis of (S)-(+)-homoisoflavone in 12 steps with 7% yield and 81% ee by using an organo-catalyzed enantioselective protonation reaction as the key step.20 Alternatively, Zhou and coworkers conducted its total synthesis to demonstrate the utility of asymmetric hydrogenation with the catalyst (Sa)-12. Firstly, α-aryloxymethylcinnamic acid (16) was prepared from commercially available 4-methoxybenzaldehyde in high yield through a series of transformations, including Perkin condensation, esterification, bromination with NBS, Williamson etherification and basic hydrolysis. Asymmetric hydrogenation of 16 was performed using the catalyst (Sa)-12 to afford compound 17 in nearly quantitative yield with extremely high enantioselectivity (99.5% ee), which was converted into the final natural product in high efficacy. Thus, Zhou's group accomplished asymmetric total synthesis of (S)-(+)-homoisoflavone in 7 steps with 59% overall yield and 97% ee (Scheme 2B).21 These two examples in Scheme 2 on successful asymmetric total synthesis by Zhou's group indicated its high application potential of this iridium-catalyzed asymmetric hydrogenation.
Moreover, concise total syntheses of natural products (R)-aristelegone A, (R)-curcumene and (R)-xanthorrhizol were accomplished as well by Zhou's group through asymmetric hydrogenation of unfunctionalized alkene (Scheme 3).22 With compound 19a as the starting material catalyzed by the iridium complex (Sa,S)-12′ as the chiral catalyst, product 20a was obtained in 97% yield with 94% ee. Finally, (R)-aristelegone A, a natural metabolite isolated from the root and stem of Aristolochia elegans in 2002,23 was synthetically achieved through a Brønsted acid-catalyzed Friedel–Crafts acylation and a subsequent demethylation (Scheme 3).
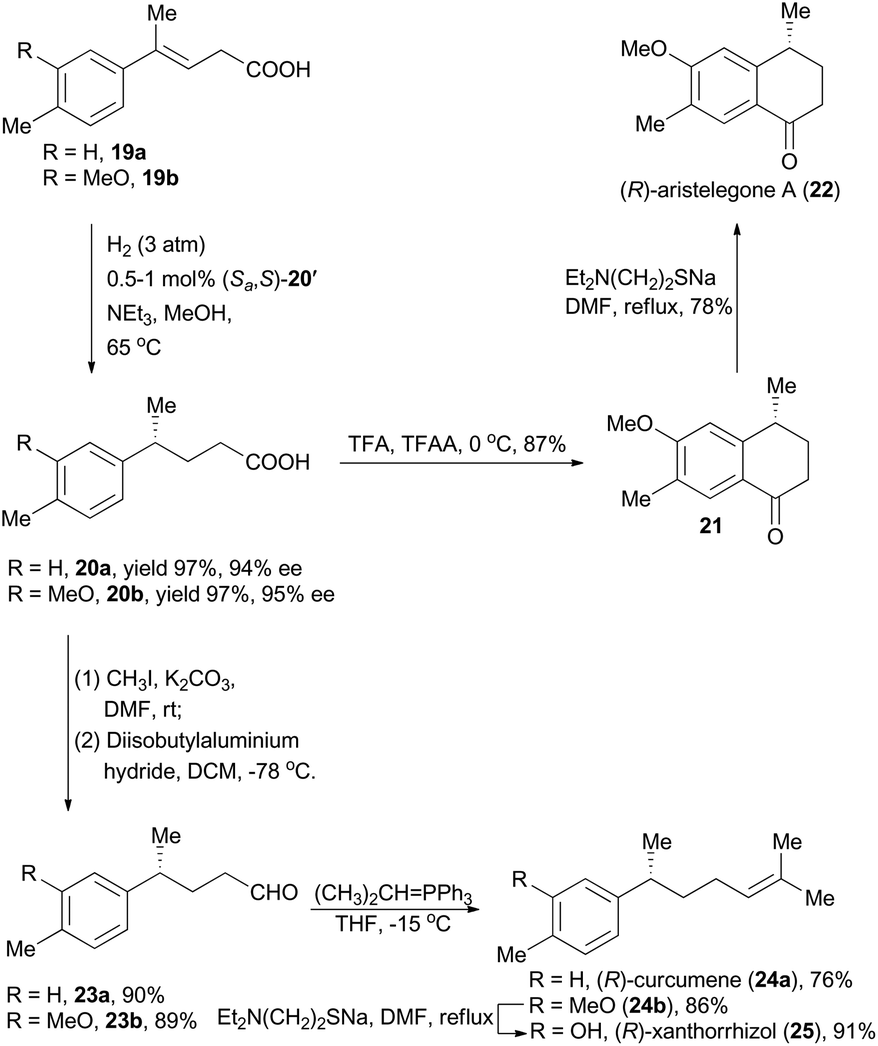 | ||
| Scheme 3 Zhou's synthesis of (R)-aristelegone-A, (R)-curcumene and (R)-xanthorrhizol. | ||
(R)-Curcumene (24a) represents a typical constitute of a large number of essential oils, and was first detected in the essential oil from the rhizomes of Curcuma aromatica Salisb.24 (R)-Xanthorrhizol (25), structurally similar to (R)-curcumene, was isolated from the rhizomes of Curcuma xanthorrhiza Roxb.25 Zhou and coworkers accomplished asymmetric total synthesis of these two natural products by following a conceptually similar strategy from the hydrogenation products 20a and 20b respectively. Accordingly, the chiral acids 20a and 20b were transformed to the corresponding aldehydes 23a and 23b by means of esterification and a subsequent reduction. Wittig reactions were then performed to introduce the alkene moiety, delivering (R)-curcumene in 76% yield and compound 24b in 86% yield. Finally, (R)-xanthorrhizol was obtained by demethylation of 24b.
Besides the above-mentioned research work, Zhou and his coworkers have elegantly synthesized a number of other chiral natural products and pharmaceuticals by employing iridium-catalyzed asymmetric hydrogenation as the key step.4e,26
2.2 Pfaltz's synthesis of demethyl calamenene and (+)-mutisianthol
In 2007, Pfaltz and coworkers reported a new class of chiral iridium complexes as catalysts for enantioselective hydrogenation of olefins.4d The synthetic applicability of this methodology was showcased by his research group in total synthesis of demethyl calamenene (Scheme 4),27 a potent cytotoxic agent against human adenocarcinoma A 549.28 Commercially available 6-methoxytetralone (26) was converted into dihydronaphthalene 27 in 43% yield after a ZnCl2-catalyzed nucleophilic addition of isopropyl Grignard reagent and an acid-catalyzed dehydration of the resultant tertiary alcohol. Asymmetric hydrogenation of compound 27 in the catalysis of Ir-28 afforded compound 29 in 98% yield with 93% ee. After subsequent Vilsmeier formylation and hydrogenolysis, asymmetric total synthesis of demethyl calamenene (31) was finally accomplished in only 4 steps. The target molecule was obtained in 21% overall yield with 98% ee, which is superior to other reported routes with regard to synthetic steps, overall yield and purification of the intermediates.29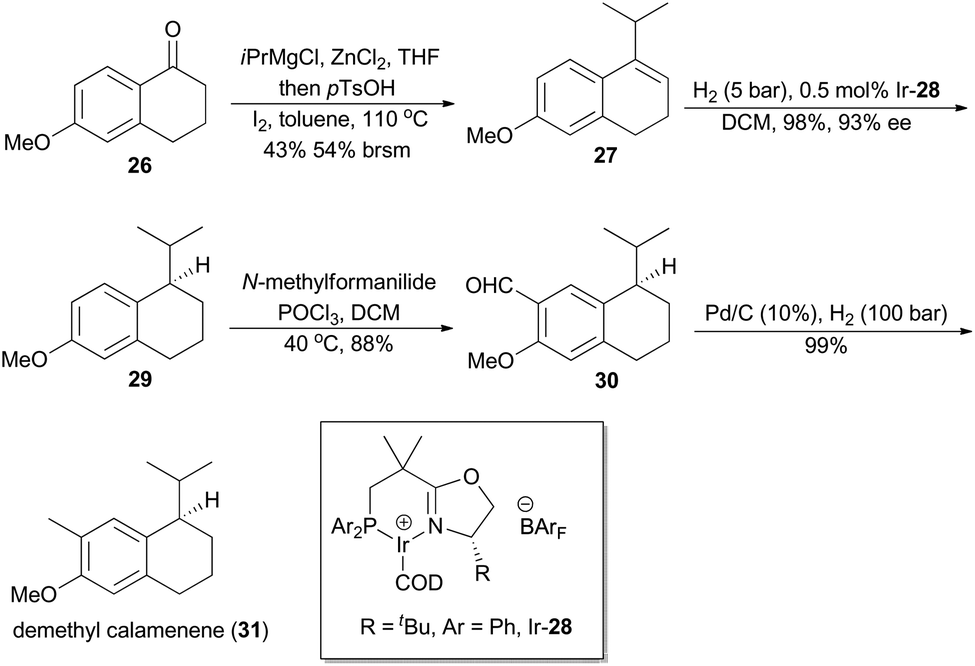 | ||
| Scheme 4 Pfaltz's synthesis of demethyl calamenene. | ||
The phenolic sesquiterpene mutisianthol (40, Scheme 5), isolated from the roots of Mutisia homoeantha as the dextrorotatory enantiomer by Bohlmann and coworkers in 1979, possesses a cis-1,3-disubstituted cyclopentyl unit.30 In order to assign its absolute configuration via total synthesis, Pfaltz and coworkers conducted asymmetric hydrogenation with chiral iridium catalyst Ir-34 as the key step (Scheme 5).31 So the alkene 33, available in four steps from commercially available 2-methyl-anisole (32) in 56% overall yield, was transformed into chiral compound 35 in 97% yield with 90% ee. It was then converted into (+)-mutisianthol (40) in six steps involving benzylic oxidation, reduction, dehydration, ring contraction, Wittig reaction and deprotection. Through total synthesis, the absolute configuration of (+)-mutisianthol was determined to be (1S,3R).
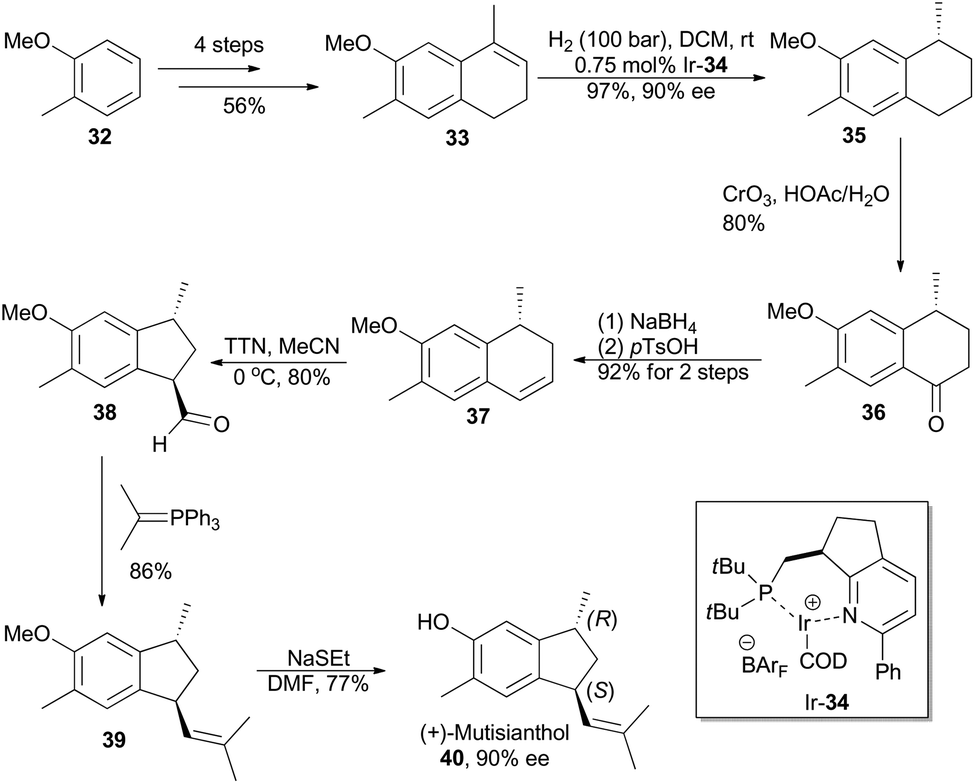 | ||
| Scheme 5 Pfaltz's synthesis of (+)-mutisianthol. | ||
2.3 Schneider's synthesis of (+)-vittatalactone and (+)-norvittatalactone
(+)-Vittatalactone and (+)-norvittatalactone, isolated by Francke and coworkers in 2005 from male striped cucumber beetles, Acalymma vittatum,32 possess an all-syn-configured 1,3,5,7-tetramethyl-substituted alkyl chain and a trans-configured β-lactone (Scheme 6). Key elements in Schneider's total synthesis of these two products include a thermal oxy-Cope rearrangement, an iridium-catalyzed asymmetric hydrogenation, and an auxiliary-controlled enolate methylation, all of which proceed with excellent stereocontrol.33 Thus, aldol reaction between 41 and 42 and subsequent protection provided chiral 1,5-diene 43, which underwent a thermal Cope rearrangement to afford compound 44. Subsequent asymmetric hydrogenation with chiral iridium complex Ir-45 furnished 46 in 98% yield with 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 dr. After diastereoselective methylation with KHMDS and MeI, compound 46 was converted into 47. Alternatively, the conjugate double bond in compound 44 was firstly chemoselectively hydrogenated with a Stryker-type copper hydride species developed by Lipshutz and coworkers34 in excellent yield. The enol benzoate 48 was then α-methylated as described above and furnished the enol benzoate 49 in 84% yield as a single diastereomer, which was then subjected to the hydrogenation conditions to afford compound 47.
:3 dr. After diastereoselective methylation with KHMDS and MeI, compound 46 was converted into 47. Alternatively, the conjugate double bond in compound 44 was firstly chemoselectively hydrogenated with a Stryker-type copper hydride species developed by Lipshutz and coworkers34 in excellent yield. The enol benzoate 48 was then α-methylated as described above and furnished the enol benzoate 49 in 84% yield as a single diastereomer, which was then subjected to the hydrogenation conditions to afford compound 47.
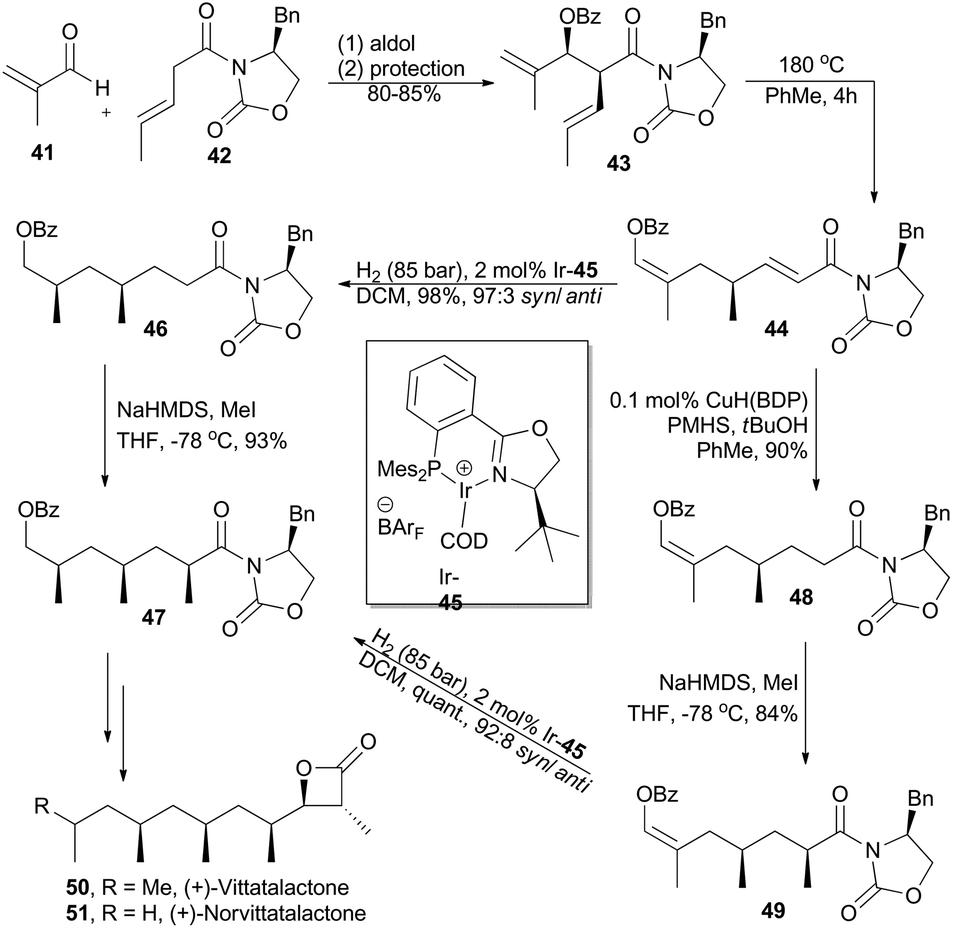 | ||
| Scheme 6 Schneider's synthesis of (+)-vittatalactone and (+)-norvittatalactone. | ||
2.4 Burgess's synthesis of (−)-spongidepsin
(−)-Spongidepsin (52), a novel macrolide with 9-hydroxy-2,4,7-trimethyltetradeca-14-ynoic acid and N-methylphenylalanine residues connected in a 13-membered macrocycle, was isolated in 2001 from a Vanuatu marine sponge, Spongia sp., by Riccio and coworkers (Scheme 7). This natural product showed cytotoxic activity against J774.A1, WEHI-164 and HEK-293 cancer cell lines with IC50 values in the sub-mM range.35 A routine total synthesis toward such a polyketide-derived macrolide might involve asymmetric aldol36 and alkylation reactions37 often with chiral auxiliaries. Instead, Burgess and coworkers applied chiral iridium complex Ir-56 as the catalyst to achieve two pivotal chirons (54 and 55) in their total synthesis of (−)-spongidepsin.38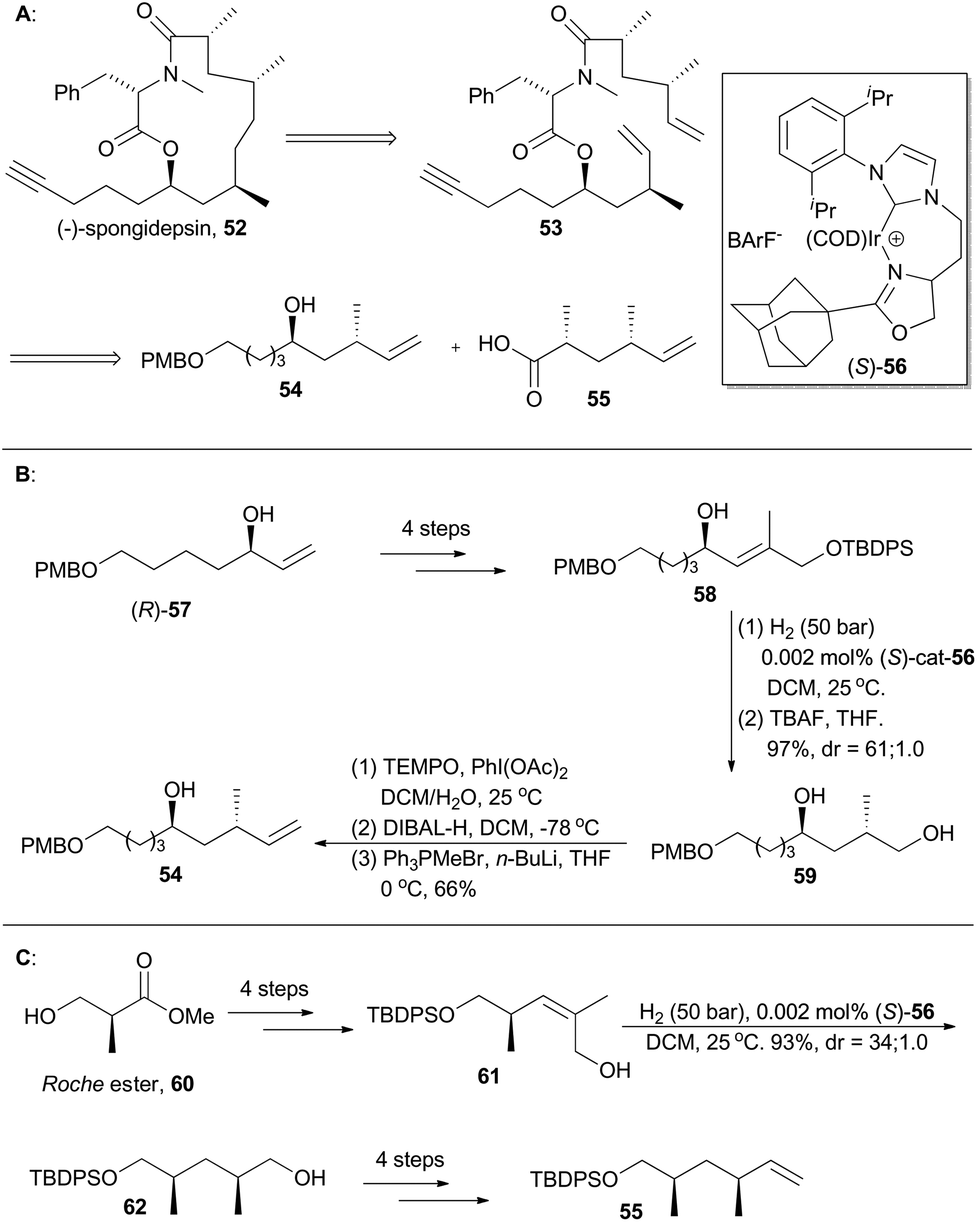 | ||
| Scheme 7 Burgess's synthesis of (−)-spongidepsin. | ||
Accordingly, an allylic alcohol (R)-57, prepared via Sharpless kinetic resolution, was converted into compound 58 after routine 4-step transformations. It was then enantioselectively hydrogenated and deprotected to afford compound 59 with high diastereoselectivity. Chiron 54 was then generated from 59via a series of treatments involving oxidation, reduction and Wittig olefination. The other 1,3-dimethyl chiron, 55, can be similarly prepared via asymmetric iridium-catalyzed hydrogenation of compound 61, which is available from the Roche ester 60 in four steps (Scheme 7C).
2.5 Gademann's synthesis of (+)-torrubiellone C
Torrubiellones C, a pyridone and tetramic acid alkaloid, was isolated from the spider pathogenic fungus Torrubiella sp. in 2010.39 From a synthetic viewpoint of Gademann,40 this natural alkaloid could be constructed via a modified Horner–Wadsworth–Emmons reaction between a known functionalized pyridine-type phosphonate 64 and an unsaturated aldehyde 65 (Scheme 8),41 while 65 can be generated by Stille coupling of the fragment 66 and the stannane 67. Accordingly, compound 68 was subjected to asymmetric hydrogenation with the iridium catalyst prepared from ligand 69 to afford compound 70 in nearly quantitative yield with 90% ee. The subsequent three-step routine transformations, involving reduction of ester, oxidation to aldehyde and Takai reaction, gave a hydroxyl-protected alkenyl iodide 71. Gademann's asymmetric total synthesis of (+)-torrubiellone C employing compound 71 also resulted in assignment of the absolute configuration of this natural product.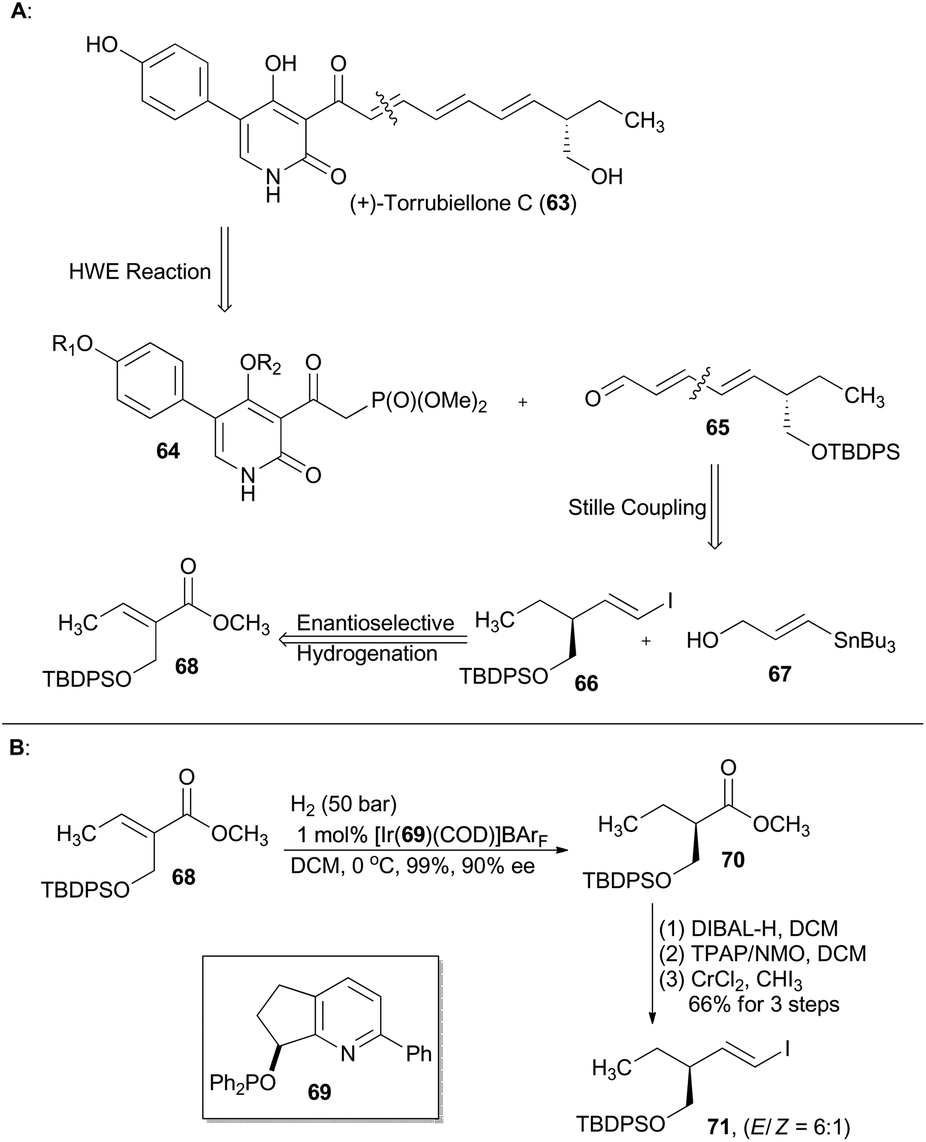 | ||
| Scheme 8 Gademann's synthesis of (+)-torrubiellone C. | ||
From the above-mentioned and other reported examples,42 it has been nicely illustrated that asymmetric hydrogenation with a chiral iridium catalyst can make great contribution to total synthesis of natural products and pharmaceuticals.
3 Hydrogen transfer reaction
Oxidation or reduction of organic compounds is one of the most fundamental technologies for organic synthesis. Many oxidation reactions associated with the hydrogen transfer process have been well developed in the last few decades,43 among which iridium-catalyzed hydrogen transfer reactions provide an alternative to direct hydrogenation of a range of substrates, such as ketones, aldehydes, alkenes and imines.5a,443.1 Zhou's synthesis of sclerotigenin, pegamine, deoxyvasicinone, mackinazolinone and rutaecarpine
Quinazolinones are a type of prevalent natural product which exhibit a variety of important biological and pharmacological activities.45 In 2011, Zhou and coworkers described a one-pot oxidative cyclization of primary alcohols with o-aminobenzamides to synthesize 3-unsubstituted quinazolinones via an Ir-catalyzed hydrogen transfer reaction.46 The wide substrate scope of this methodology ensures construction of the core structures of sclerotigenin, pegamine, deoxyvasicinone, mackinazolinone, and rutaecarpine (Scheme 9).47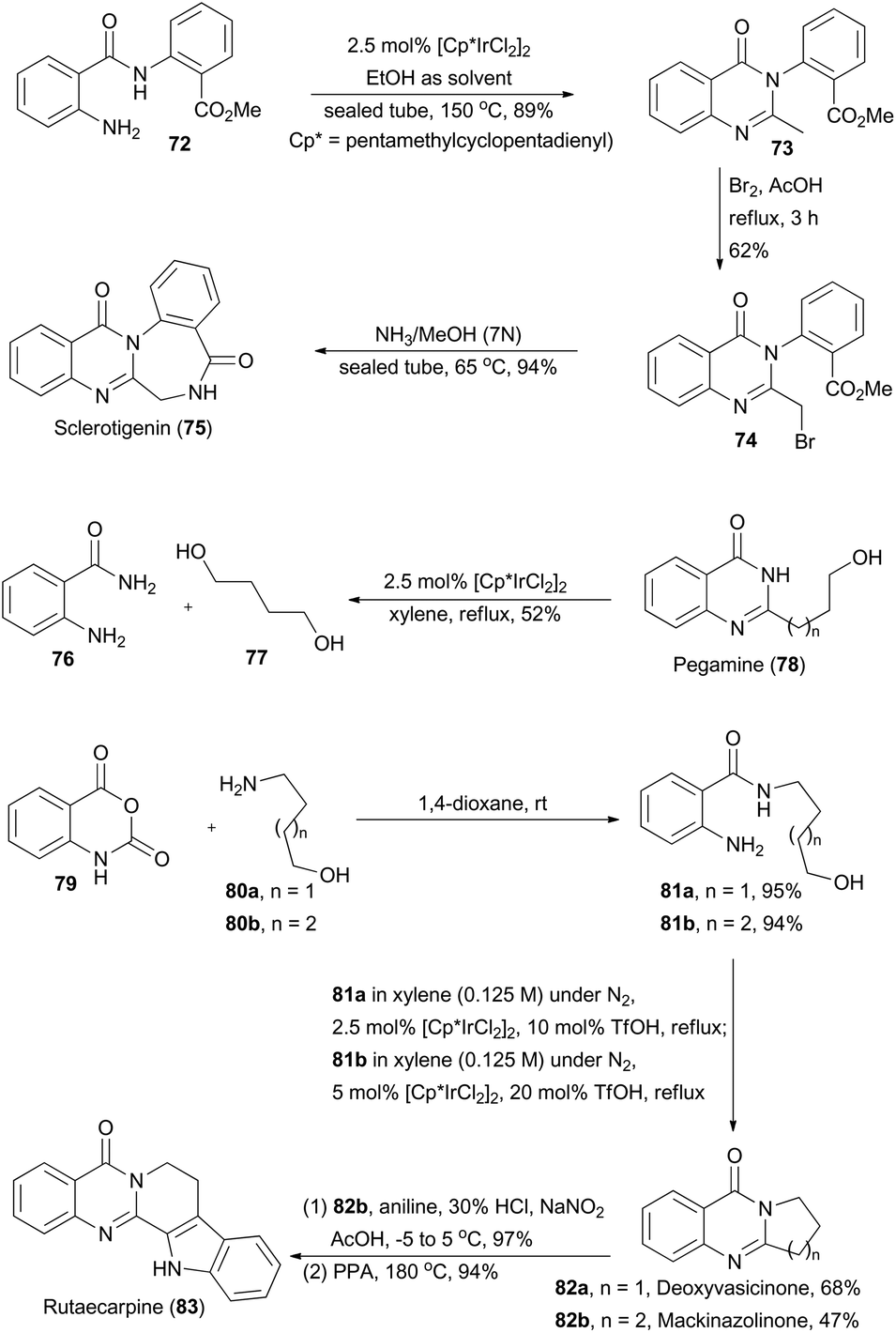 | ||
| Scheme 9 Zhou's synthesis of sclerotigenin, pegamine, deoxyvasicinone, mackinazolinone and rutaecarpine. | ||
In the presence of ethanol, N-substituted aminobenzamide 72 was transformed via catalysis of [Cp*IrCl2]2 (Cp* = pentamethyl-cyclopentadienyl) to quinazolinone 73 in 89% yield. After sequential bromination and ammonolysis, sclerotigenin 75, an alkaloid isolated from sclerotia of Penicillium sclerotigenum (NRRL 3461),48 was generated. Pegamine (78), a natural product isolated from Peganum harmala,49 was prepared from 2-aminobenzamide 76 and an unprotected diol 77 in the presence of the iridium catalyst. Furthermore, Zhou's group aimed at total synthesis of deoxyvasicinone (82a) and mackinazolinone (82b),50 which are natural quinazolinones fused with pyrrolidine and piperidine respectively and show antimicrobial, anti-inflammatory and anti-depressant bioactivities.51 Thus, aminolysis of compound 79 with 80a and 80b afforded 81a and 81b in 95% and 94% yield respectively. Then 81a and 81b underwent the iridium-catalyzed hydrogen transfer process to generate deoxyvasicinone (82a) and mackinazolinone (82b). Additionally, by following a known literature procedure,52 mackinazolinone (82b) was transformed to rutaecarpine through oxidative hydrazone formation and Fisher indole synthesis. Similarly, deoxyvasicinone (82a) could also be utilized for synthesis of rutaecarpine.53
3.2 Krische's synthesis of rifamycin S, 6-deoxyerythronolide B, (+)-zincophorin methyl ester, oridamycin A, triptoquinones B & C, and isoiresin
Although alkene hydroformylation has already been a landmark industrial application for homogeneous catalysis, exploration on new C–C bond-forming reactions under metal-catalyzed hydrogenations and transfer hydrogenation conditions54 is still stimulating continuous motivation in chemists. In 2011, the Krische group55 reported their novel iridium-catalyzed methodology to expeditiously construct polyketide motifs via double diastereo- and enantio-selective crotylation of 1,3-diols (Scheme 10). Under the optimized conditions with the iridium catalyst (R)-86, prepared from [Ir(COD)Cl]2, allyl acetate, 4-cyano-3-nitrobenzoic acid, and (R)-SEGPHOS, 2-methyl-1,3-propanediol (85) went through transfer hydrogenation and double crotylation to deliver stereopolyads 87 characterized with pseudo-C2-symmetry, with exceptional diastereoselective and enantioselective control (Scheme 10). Desymmetrization of 87 is readily achieved via iodoetherification to form pyran 88.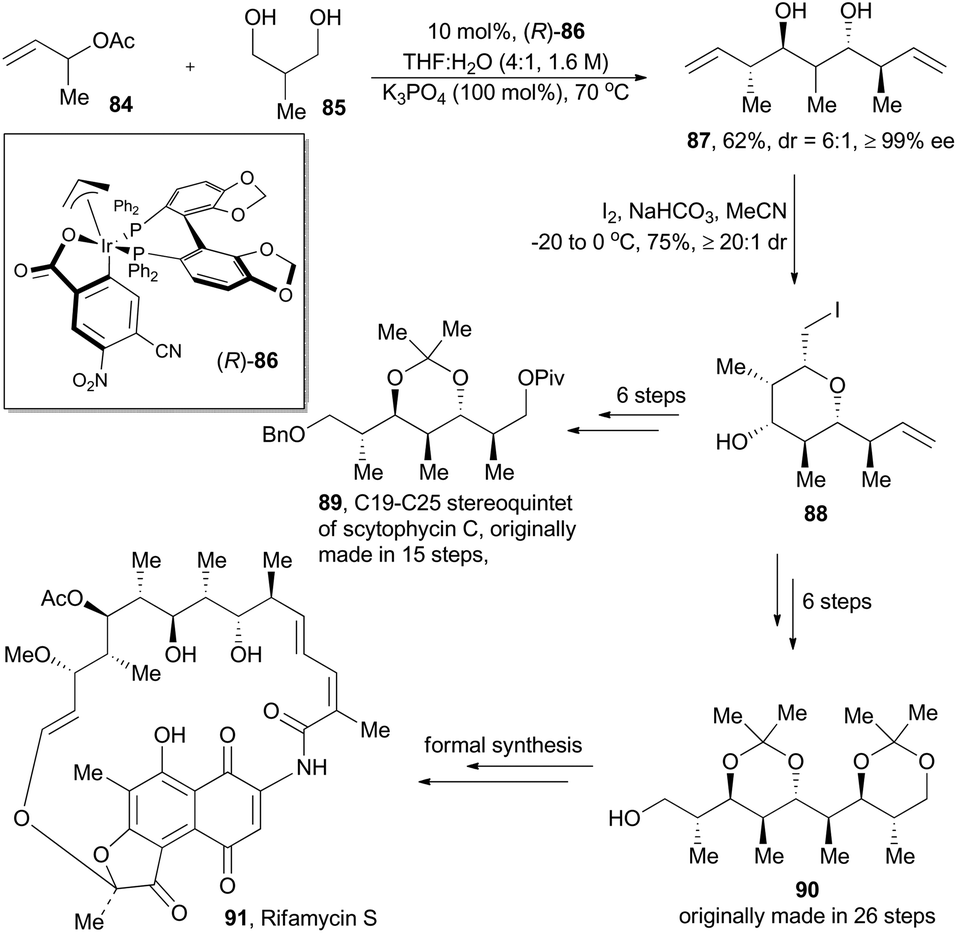 | ||
| Scheme 10 Krische's formal synthesis of rifamycin S. | ||
Generation of 87 in such a straightforward fashion enables a much more concise synthesis than those previously established ones. Scytophycin C is a natural product isolated from the terrestrial blue-green algae Scytonema pseudohofmanni and exhibits both antifungal and cytotoxic properties.56 The C19–C25 segment of scytophycin C, originally prepared in 15 steps,57 can be easily constructed in only 8 steps. Meanwhile, formal synthesis of rifamycin S (91), a famous antibiotic isolated from the fermentation medium of Norcardia mediterranei,58 can be accomplished via construction of its C19–C27 segment (90) in 8 steps (originally prepared in 26 steps58b).
Furthermore, to benchmark the utility of the above methodology in double diastereo- and enantio-selective crotylation of 2-methyl-1,3-propanediol (85) to form polypropionate motifs, this asymmetric iridium catalysis was applied in total synthesis of 6-deoxyerythronolide B,59 (+)-zincophorin,60a cryptocaryol A,60b (+)-roxaticin,60c and cyanolide A60d by Krische and coworkers. This methodology has also been employed by De Brabander and coworkers in total synthesis of psymberin and irciniastatin A,60e by Fürstner and coworkers in total synthesis of mandelalide A,60f,g as well as by Menche and coworkers in total synthesis of leupyrrins A1 and B1,60h demonstrating powerful application prospects.
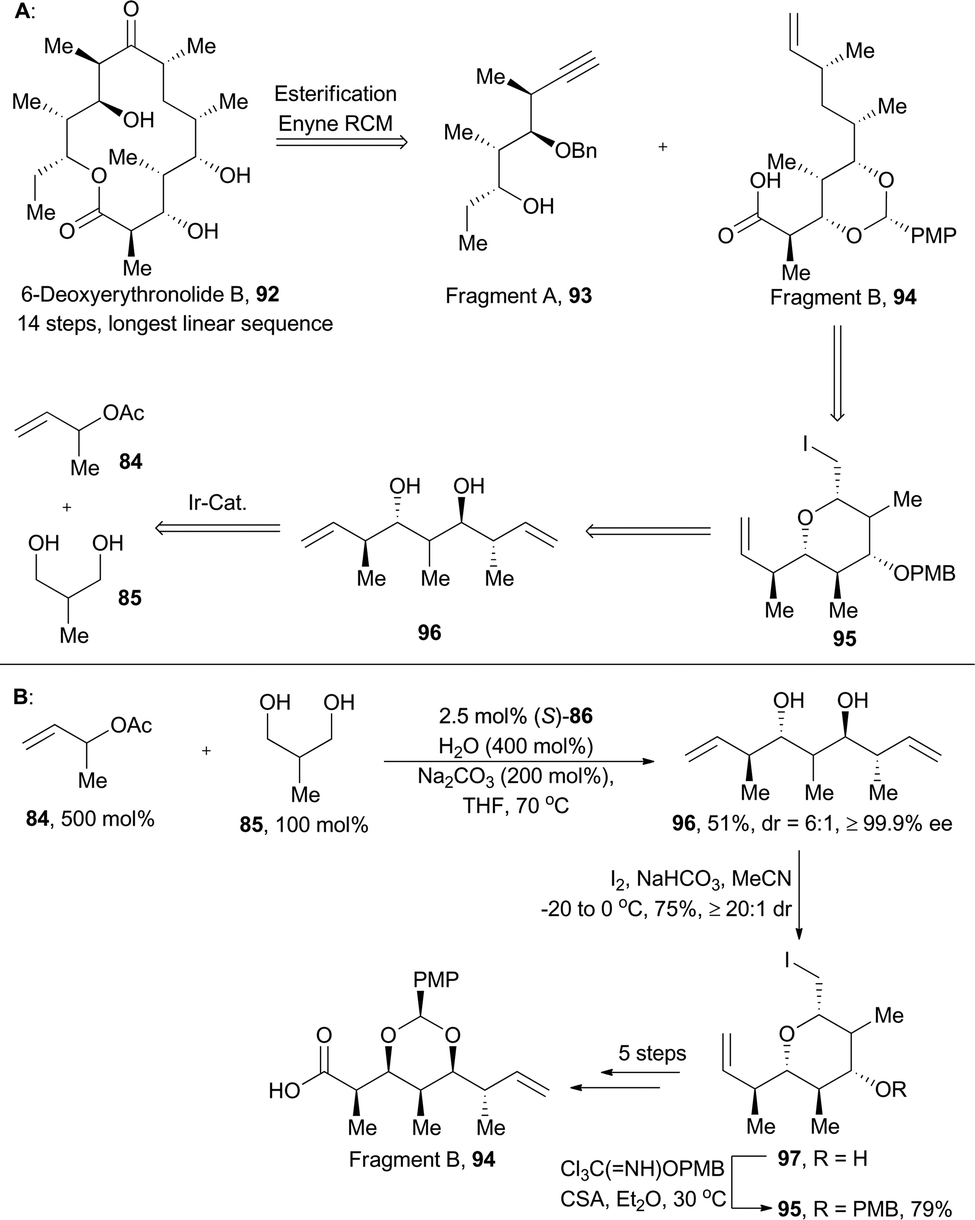
6-Deoxyerythronolide B (92), with ten stereogenic centers embedded in a 14-membered macrolactone, is produced by blocked mutants of Streptomyces erythreus.61 Its challenges in chemical synthesis and impressive medicinal potential greatly attracted synthetic chemists’ attention.61a–e In Krische's retrosynthetic analysis (Scheme 11),59 6-deoxyerythronolide B was devised to be installed through a convergent assembly of fragments A (93) and B (94) with the macrolide constructed by esterification and enyne RCM; the fragment B (94), embedded with five contiguous stereogenic centers, might be prepared from pyran 95. Accordingly, double crotylation of 85 catalyzed by (S)-86 furnished the pseudo-C2-symmetric diol 96, an enantiomeric antipode of 87. Then desymmetrization of 96via iodoetherification gave product 97, an enantiomeric antipode of 88. Then the fragment B was achieved after subsequent five synthetic steps.
 | ||
| Scheme 11 Krische's synthesis of 6-deoxyerythronolide B. | ||
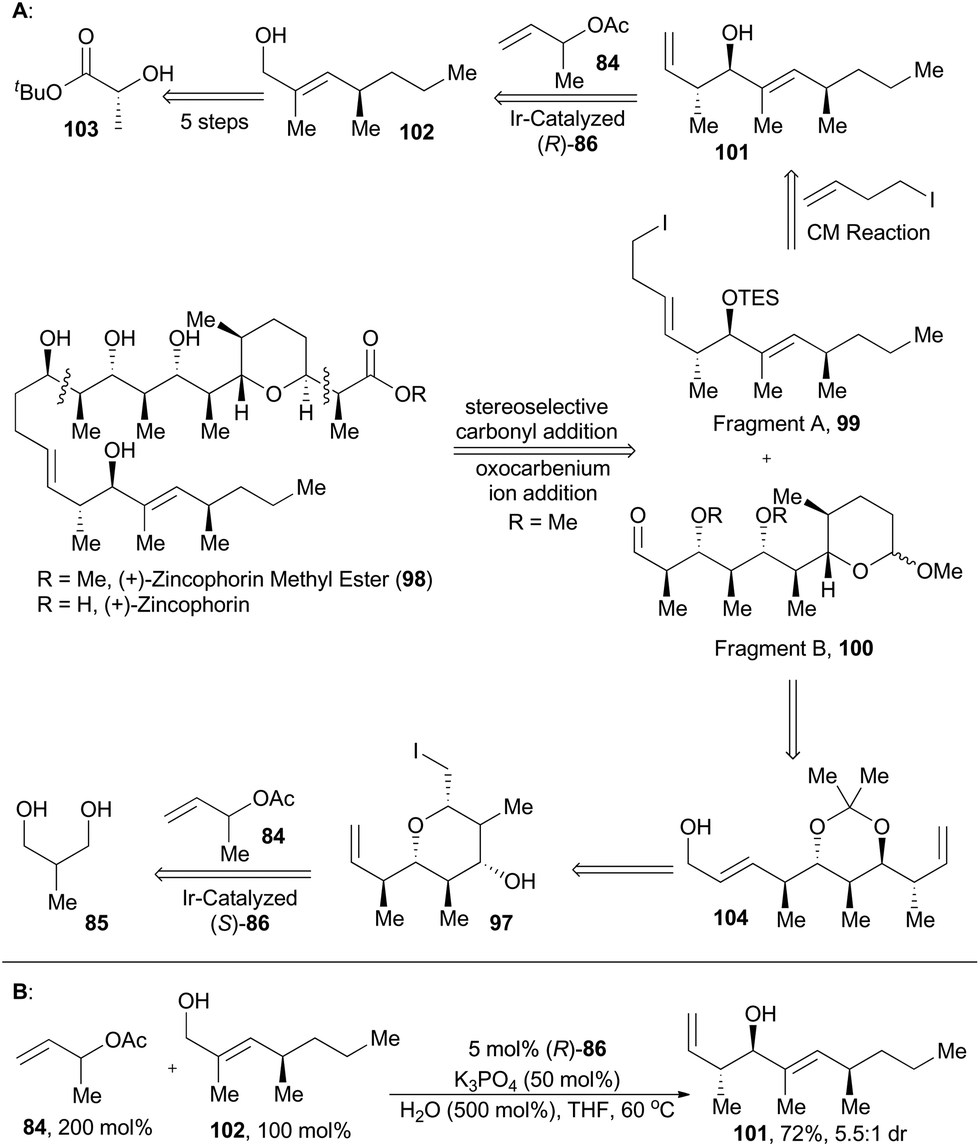
Zincophorin is an iconic polyketide ionophore antibiotic and was isolated from strains of Streptomyces griseus in 1984. It exhibited promising in vitro bioactivities against Gram-positive bacteria and thus was named for its exceptionally high affinity to divalent metals, especially zinc(II).62 Interestingly, its methyl ester also possesses antiviral activity, albeit with reduced host cell toxicity compared to the free acid.63 So it is not surprising that (+)-zincophorin and its methyl ester have been hot target molecules since their isolation.64 Retrosynthetically, a convergent assembly of (+)-zincophorin methyl ester from fragments A (99) and B (100) was conceived through stereoselective carbonyl addition followed by oxocarbenium ion addition.60a Synthetically, key steps for construction of 99 include Breit's method for the stereospecific substitution of the triflate of compound 103 with Grignard reagent, stereoselective Wittig olefination and direct redox-triggered anti-crotylation of the allylic alcohol 102 catalyzed by the iridium complex (R)-86 to afford 101. The fragment B (100) was accessible from compound 104via routine transformations, which was achievable from compound 97, the same intermediate as in total synthesis of 6-deoxyerythronolide B (92) (Scheme 12).
 | ||
| Scheme 12 Krische's synthesis of (+)-zincophorin methyl ester. | ||
In 2014, Krische and coworkers reported a methodology of direct regio-, diastereo-, and enantio-selective C–C coupling between primary alcohols and isoprene oxide, through Ir-catalyzed oxidation of primary alcohol followed by reductive C–O bond cleavage of isoprene oxide (Scheme 13A).65 This reaction generated a functionalized motif embracing an all-carbon quaternary stereocenter which is found to be embedded in molecular structures of over 2000 terpenoid natural products. In particular, diols 105 and isoprene oxide 106 were subjected to catalysis of Ir-catalyst (R)-107 or (S)-107 to afford the corresponding products 108 and 109 in excellent stereo-control. Based on this methodology, compound 113, a common intermediate in the total synthesis of oridamycin A,66 triptoquinones B and C,67 and isoiresin68 can be constructed in only four steps from compound 110 (Scheme 13B).69 It is worth noting that exposure of allylsilane 112 to ZnCl2 provided 113 as a single diastereomer in 92% yield. When ent-112 was treated with a stronger Lewis acid such as BF3·OEt2, elimination would directly take place to generate iso-113 in 63% yield. To further illustrate its synthetic applicability, Krische and coworkers connected 113 with 115 or 118 respectively with Suzuki cross-coupling conditions and then Friedel–Crafts cyclization as the key transformations, to accomplish total synthesis of oridamycin A and triptoquinones B & C. Additionally, intermolecular Diels–Alder cycloaddition between iso-113 and 120 furnished the core structure of isoiresin and thus contributed well to its final total synthesis (Scheme 14).
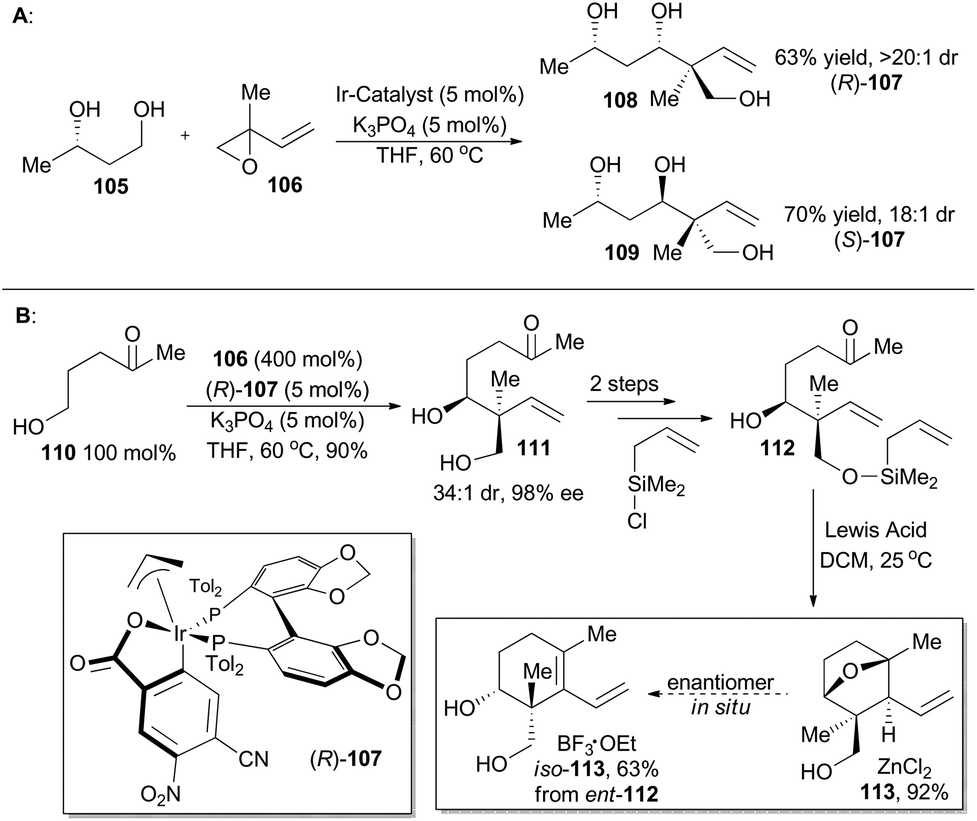 | ||
| Scheme 13 Krische's synthesis of key intermediates 113 and iso-113. | ||
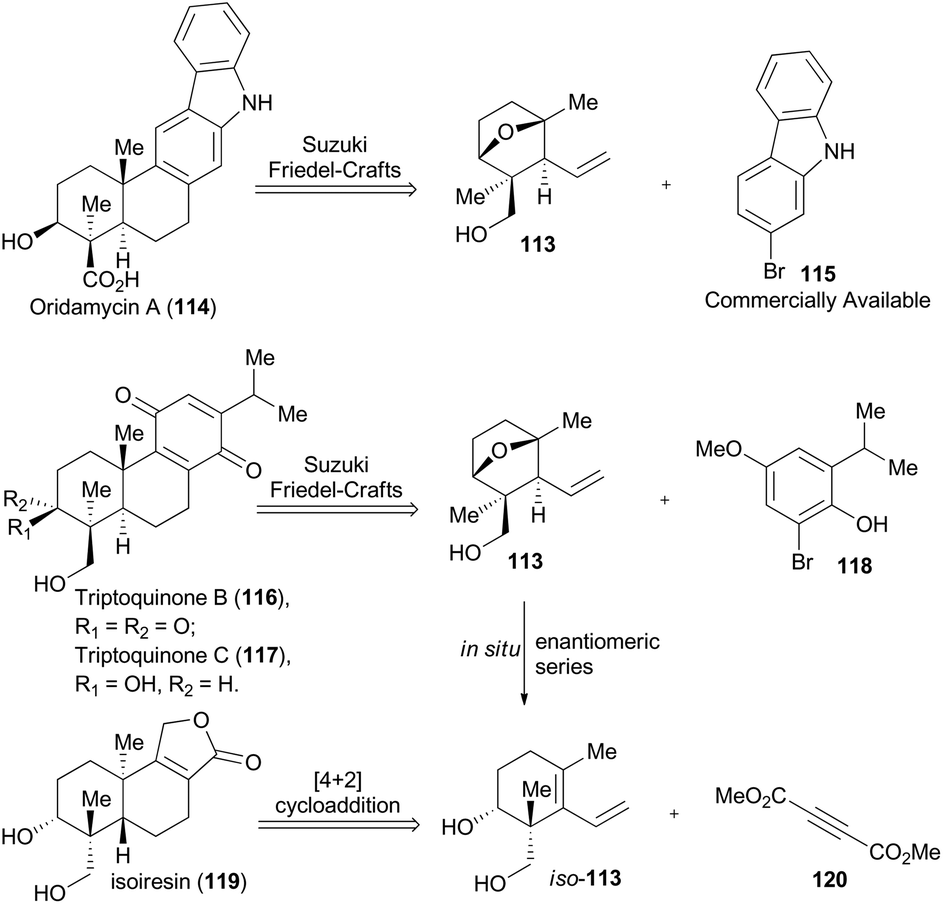 | ||
| Scheme 14 Krische's retrosynthetic analysis of oridamycin A, triptoquinones B and C, and isoiresin. | ||
4 Functionalization of the C–H bond
Functionalization of the C–H bond represents an important organic transformation to form useful synthetic intermediates and medicinally significant compounds. Transition metal complexes have shown great application prospects in selective functionalization of the C–H bond, which pleasingly prompts efficacy of step-economy synthesis of complex organic molecules.70 Among them, iridium catalysts have also proven highly effective for various C–H functionalization involving borylation of C–H bonds,6a,b,71 C–C bond coupling6c,72 and C–Si bond coupling.73 To date, direct C–H bond activation catalyzed by iridium has been used in pivotal transformations of total synthesis of natural products.4.1 Sarpong's synthesis of (+)-complanadine A
Complanadine A (128), a lycodine-type dimeric alkaloid, was isolated from the club moss Lycopodium complanatum by Morita et al. in 2000.74 Featuring its unique and complex molecular structure, complanadine A also shows enhancement in mRNA expression for NGF and the production of NGF in human glial cells.75 In 2010, Sarpong and coworkers completed asymmetric total synthesis of (+)-complanadine A with an iridium-catalyzed C–H functionalization of pyridine as the key step (Scheme 15).76a First, a combination of the known compounds 121 and 122 provided N-desmethyl α-obscurine 123 after an acid-promoted cascade cyclization.77 Subsequent selective protection of 123 with Boc afforded 124, which was converted into 125 after oxidative aromatization with lead tetraacetate and treatment with triflic anhydride. The Pd-catalyzed removal of the triflate group from 125 afforded compound 126. Relying on the effective Ir(I)-catalyzed C–H functionalization methodology, developed by the Hartwig group,76b compound 126 was transformed to the boronic ester 127 with excellent site-selectivity. Finally, palladium-catalyzed Suzuki cross-coupling between 127 and 125 and acid-mediated global deprotection accomplished total synthesis of complanadine A (128).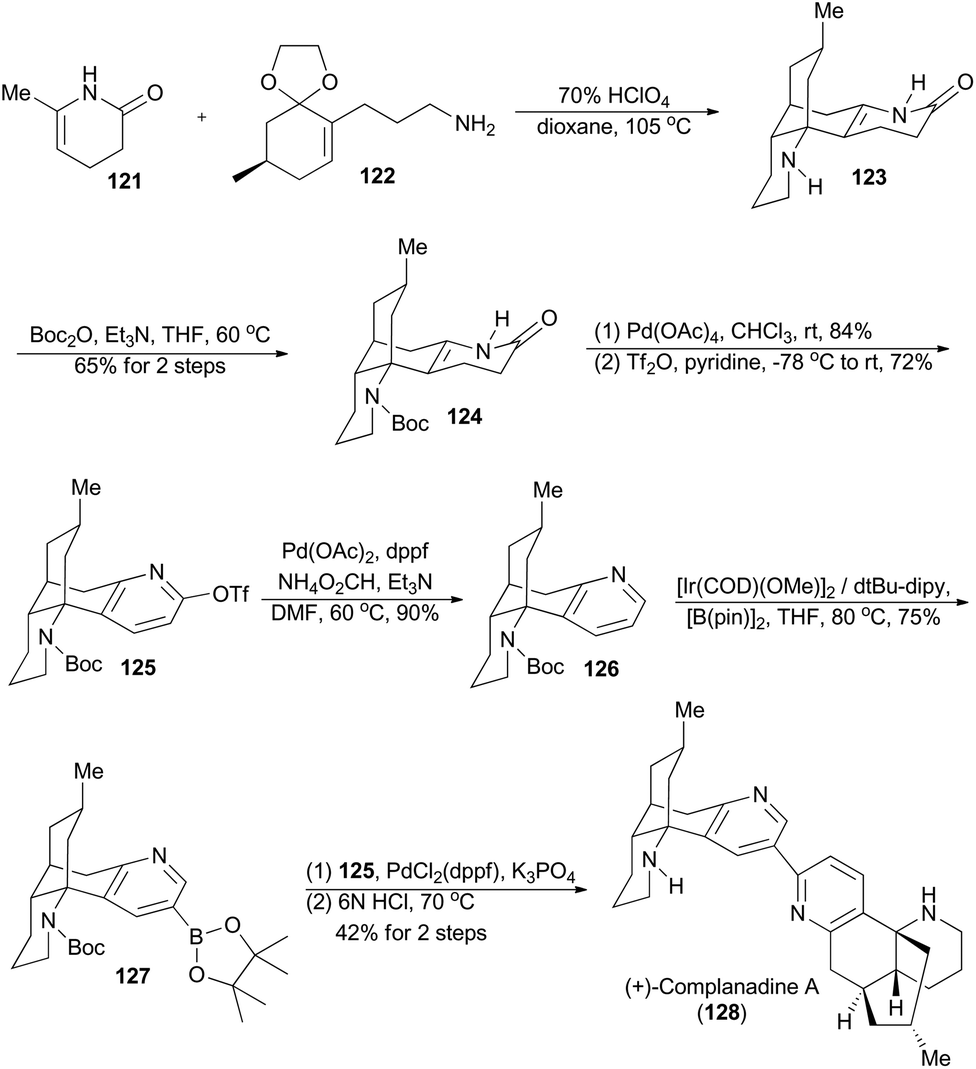 | ||
| Scheme 15 Sarpong's total synthesis of (+)-complanadine A. | ||
4.2 Hartwig's synthesis of (−)-taiwaniaquinone H and (−)-taiwaniaquinol B
In 2007, Hartwig and coworkers reported a methodology that different 3,5-disubstituted aryl halides could be prepared from simple arenes through sterically controlled iridium-catalyzed borylation and subsequent halogenation.71c Accordingly, compound 130 was accessed from 129 in 75% yield over two steps (Scheme 16A), setting the ground for total synthesis of (−)-taiwaniaquinone H (135) and (−)-taiwaniaquinol B (137).71b Structurally, both natural products contain a [6,5,6]-abeo-abietane skeleton and a benzylic quaternary stereogenic center.78 Then, 130 and 131 were subjected to asymmetric palladium-catalyzed α-arylation of ketone to afford 132 in 80% yield with 94% ee. The epoxide 133 was easily achieved in four steps from 132. Treatment of 133 with BF3·OEt2 or a Bi(III) salt triggered a semi-pinacol rearrangement/Friedel–Crafts cascade to provide 134 smoothly. By following a known deprotection–oxidation procedure,79134 was converted into (−)-taiwaniaquinone H (135) in 51% yield (Scheme 16A). Alternatively, hydroboration–oxidation of 134, followed by oxidation with IBX, provided the ketone 136 in 80% yield over two steps. Then according to the procedure reported by Fillion and Trauner,79,80 selective demethylation of 136 with BCl3, followed by oxidation–reduction treatment of the resulting intermediate, afforded (−)-taiwaniaquinone B (137) in 52% yield over two steps (Scheme 16B).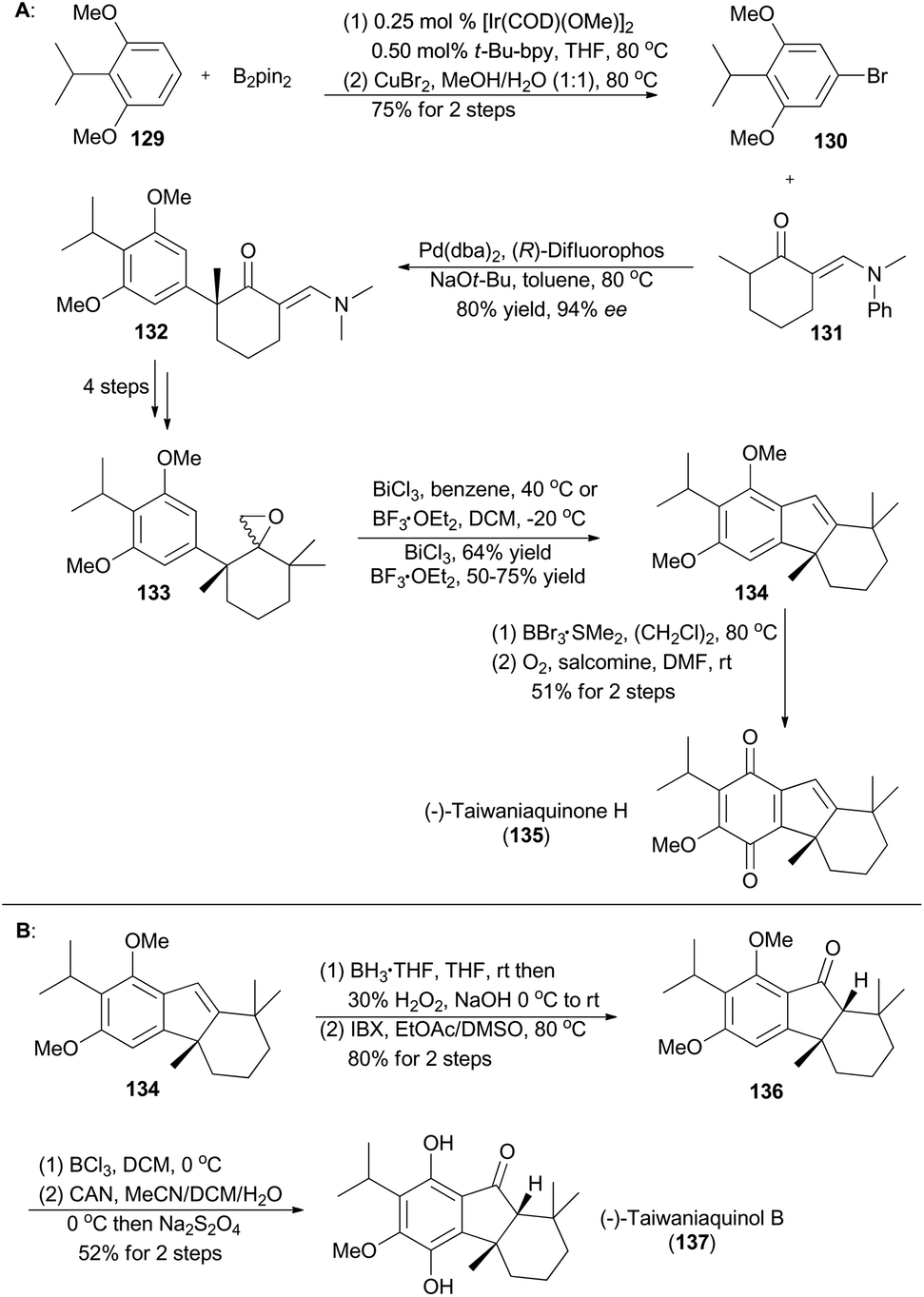 | ||
| Scheme 16 Hartwig's total synthesis of (−)-taiwaniaquinone H and (−)-taiwaniaquinol B. | ||
4.3 Baran's synthesis of verruculogen and fumitremorgin A
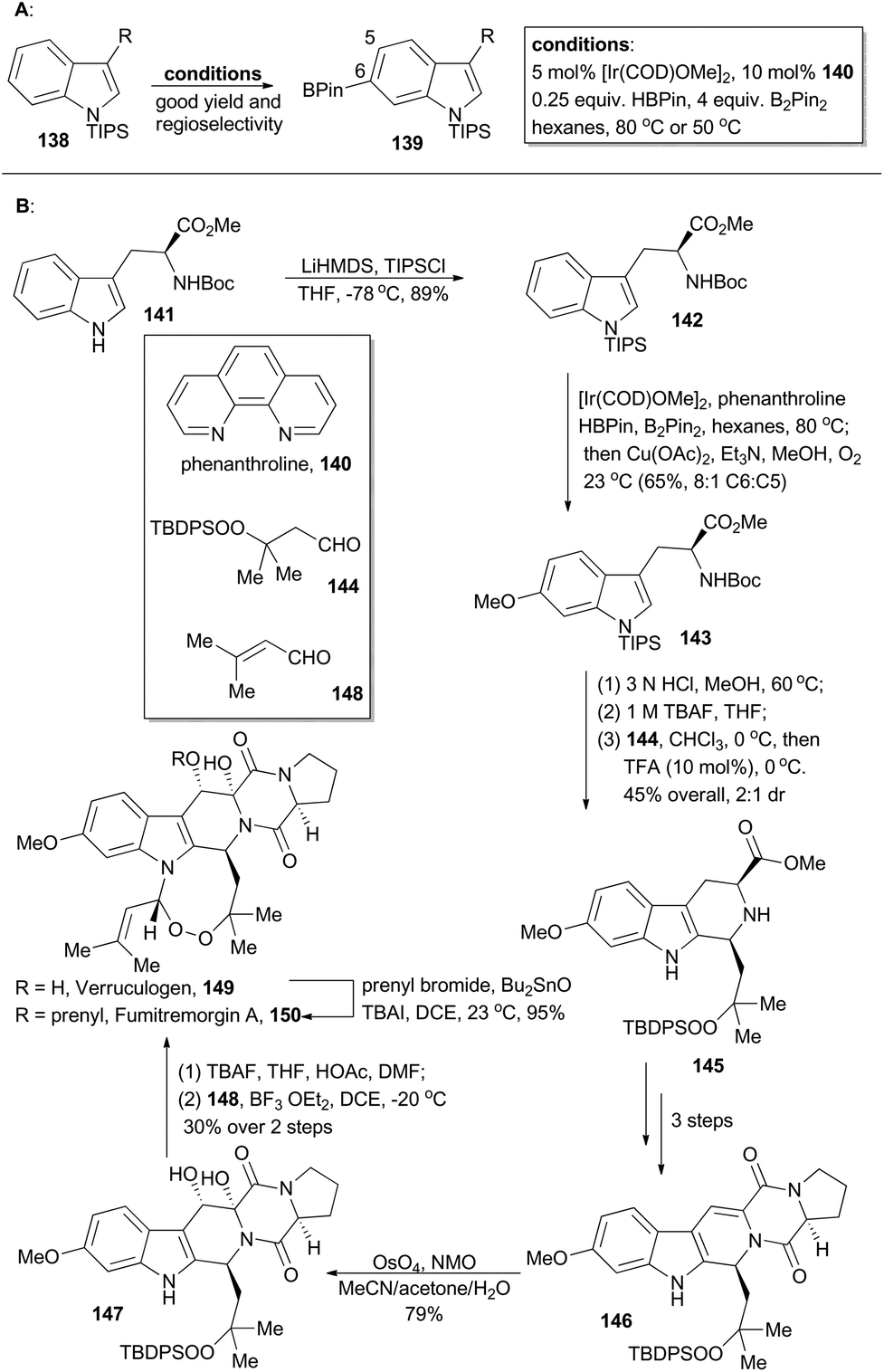
Verruculogen81 (149) and fumitremorgin A82 (150) are two bioactive alkaloids embracing a complex polycyclic framework, of which an eight-membered endoperoxide is the most astounding feature. These two polycyclic tryptophan-based natural products were first identified due to their tumor-inducing activity in mice.81,82 Moreover, the fumitremorgins display potent inhibitory activity against multi-drug resistant (MDR) cancer cell lines83 and show anti-HIV bioactivity as well.84 After their isolation about 40 years ago, the first syntheses of verruculogen and fumitremorgin A were completed by the Baran group,85 enabled by an iridium-catalyzed C–H borylation. Compound 142 could be derived from a chiral and commercially available compound, Boc-L-Trp-OMe (141). It was subjected to iridium-catalyzed C–H borylation in the presence of phenanthroline (140) (Scheme 17A). Notably, the presence of a large blocking group TIPS on the indole nitrogen is necessary to reach site-selective control at the C6 position of indole. Accordingly, 142 was selectively borylated at the C6 position for the first time and then immediately underwent Chan–Lam coupling,86 to give 143 in 65% yield with 8:1 C6:C5 selectivity. Sequential treatment of 143 with HCl and TBAF removed the silyl and Boc protective groups respectively, and the following Pictet–Spengler reaction with the peroxy-aldehyde 144 afforded the tricyclic peroxide 145 in 45% overall yield with 2:1 dr. After three routine steps, the pentacyclic peroxide 146 could be obtained, which went through diastereoselective dihydroxylation with OsO4 to yield 147. After desilylation with TBAF and acid-catalyzed formation of endoperoxide with 148, verruculogen (149) was achieved in 30% yield over two steps. Furthermore, selective prenylation of 149 afforded fumitremorgin A (150) in 95% yield.
 | ||
| Scheme 17 Baran's total synthesis of verruculogen and fumitremorgin A. | ||
4.4 Jia's synthesis of (−)-goniomitine
(−)-Goniomitine (161), isolated from the root bark of Gonioma malagasy by Husson and coworkers in 1987, is a new type of the well-known aspidosperma family of indole natural products.87 Owing to its unique structure as well as potential biological activities, goniomitine became a popular synthetic target.88 In 2015, Jia and coworkers reported their total synthesis of (−)-goniomitine (161), featuring a chiral pool approach to introduce a quaternary chiral center, an Ir-catalyzed C–H borylation at the C-2 position of an indole and a Suzuki reaction to couple two key intermediates (155 and Scheme 18).89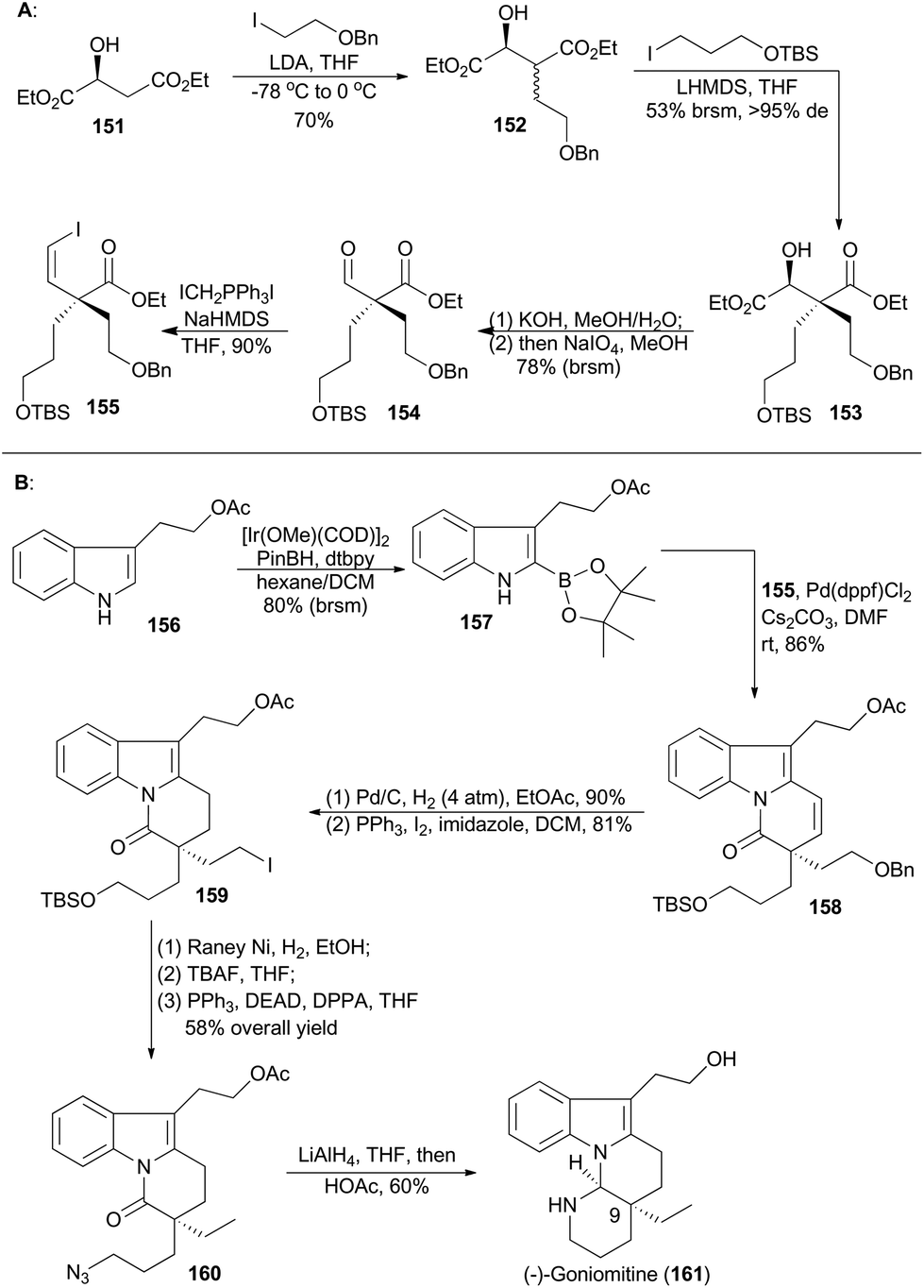 | ||
| Scheme 18 Jia's total synthesis of (−)-goniomitine. | ||
Double α-alkylation of diethyl L-malate (151) with different iodides gave compound 153 in a substrate-controlling fashion with excellent diastereoselectivity. Selective saponification of the diester 153,90 and then oxidative cleavage of the resulting α-hydroxy acid provided the desired aldehyde 154.91 Finally, the Z-vinyl iodide 155 was accomplished in 90% yield via the Stork–Wittig reaction of 154.92 At the same time, borylation of the indole 156 with PinBH, [Ir(OMe)(COD)]2 and 4,4′-di-tert-butyl-2,2′-bipyridine (dtbpy) afforded the corresponding 2-indoleboronate 157 in 80% yield.93 Coupling of the boronate 157 and the vinyl iodide 155 furnished a tricyclic compound 158 in 86% yield. This coupling featured a Suzuki coupling/lactamization sequence. Hydrogenolysis of alkene and benzylic ether and iodination of the resulting alcohol produced 159, which was transformed to the azide 160 after three routine steps in 58% overall yield. Ultimately, reduction of azide and lactam with LiAlH4, followed by acidic treatment, furnished the natural product (−)-goniomitine (161) in 60% yield.
4.5 Sperry's synthesis of 5-methoxyindole-4,7-quinone, 6-methoxyindole-4,7-quinone and scalaridine A
The indolequinone pharmacophores are widely found in bioactive natural alkaloids, such as anticancer agent mitomycin C,94 superficial bladder cancer therapeutic agent apaziquone,95 the topoisomerase II inhibitor BE-10988,96 as well as the antimicrobial indolequinones 165 and 169.97 Sperry realized a synthetic strategy toward natural indolequinones on the basis of a late-stage indole functionalization approach,98a thereby avoiding commonly employed fragment-based approaches to indolequinones.99 The Hartwig group developed a novel methodology for borylation of N-hydrosilylindoles exclusively at the C7 position,100 without a substituent at C2 to block a competitive reaction at this site.101 Inspired by this iridium catalysis, Sperry and coworkers successfully applied this methodology in total synthesis of the natural products, 165 and 169 (Scheme 19).98a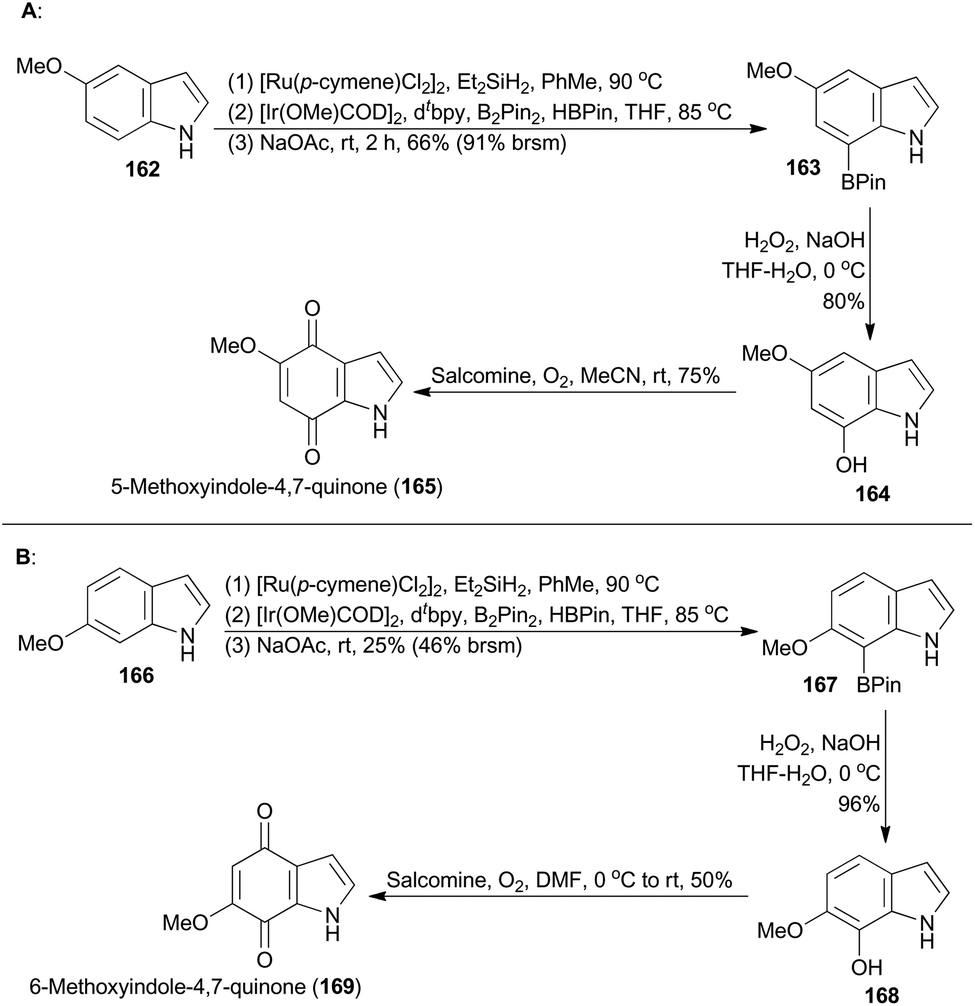 | ||
| Scheme 19 Sperry's total synthesis of 5-methoxyindole-4,7-quinone and 6-methoxyindole-4,7-quinone. | ||
Commercially available 5-methoxyindole (162) was subjected to ruthenium-catalyzed hydrosilylation, iridium-catalyzed borylation and desilylation sequentially, providing 5-methoxy-7-borylindole (163) (Scheme 19A). Oxidative treatment of 163 gratifyingly afforded the indolequinone natural product 165. Then Sperry and coworkers completed total synthesis of the indolequinone natural product 169 by following the same synthetic route (Scheme 19B), although the C–H borylation of 166 to 167 proceeded in low yield.
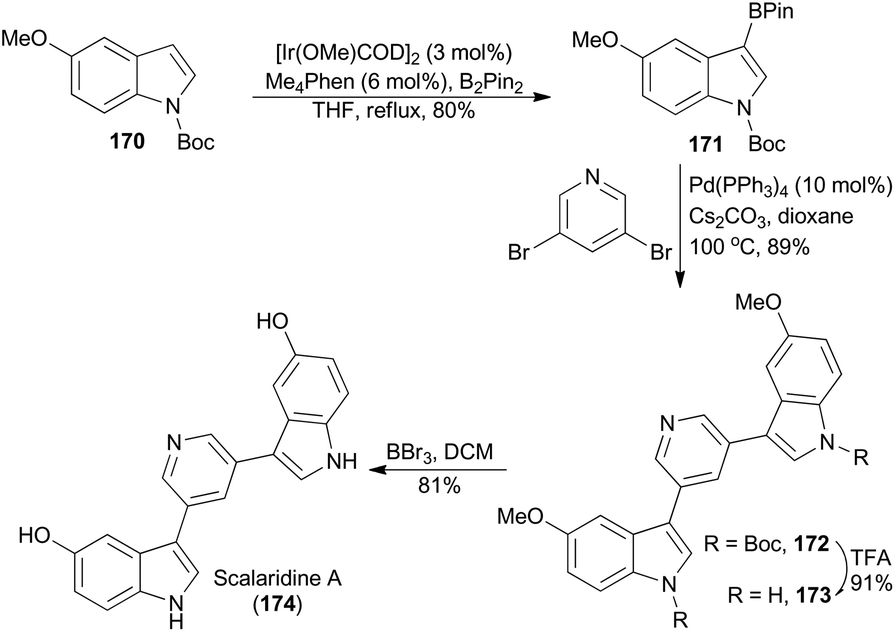
In 2014, another insightful study on borylation of heteroarene was reported by Hartwig and coworkers.102 They found that several heteroarenes containing multiple heteroatoms were amenable to site-selective C–H borylation catalyzed by a combined iridium-Me4Phen system. Scalaridine A is a symmetrical bis-indole with pyridine linker isolated from a Dokdo marine sponge Scalarispongia sp.103 Application of this catalytic system by Sperry and coworkers resulted in completion of total synthesis of scalaridine A (Scheme 20).98b Besides the directed C–H borylation of N-Boc-5-methoxyindole (170) leading to 171,104 the synthesis featured the double Suzuki–Miyaura cross-coupling to install the entire heteroaromatic framework to afford 172. Deprotection of both Boc groups in the bisindole 172 gave 173, which underwent global demethylation using boron tribromide to afford scalaridine A (174).
 | ||
| Scheme 20 Sperry's total synthesis of scalaridine A. | ||
4.6 Brimble's synthesis of (±)-thysanone
In 2012, Brimble and coworkers reported their methodology on site-selective C–H borylation of various polyoxygenated naphthalenes in the presence of catalytic [Ir(COD)OMe]2 and di-tert-butylbipyridine.105 Based on this chemistry, they completed racemic total synthesis of thysanone (182).106 (−)-Thysanone was isolated from a fungus named Thysanophora penicilloides, and was demonstrated an effective inhibitor of HRV 3C protease (IC50 = 13 μg mL−1).107Brimble's route began with an oxa-Pictet–Spengler reaction of compound 175 with dimethoxymethane, which produced the naphthopyran 176 in 89% yield (Scheme 21).108 Applying the key iridium-catalyzed borylation,105,109176 was connected with freshly prepared pinacolborane to afford the boronate 177 as a crude mixture, which was oxidatively transformed to the phenol 178. Silyl protection of the phenol and oxidative demethylation with freshly prepared silver(II) oxide gave the pyranonaphthoquinone 180, which underwent three transformations to accomplish total synthesis of (±)-thysanone (182).
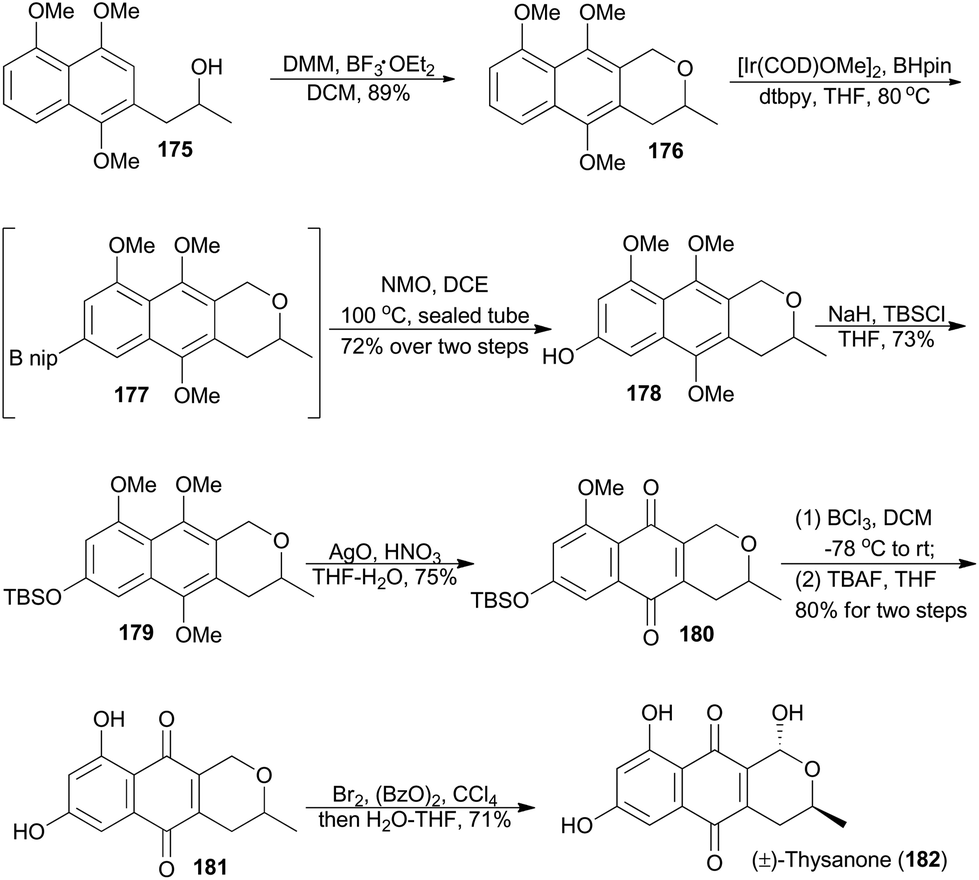 | ||
| Scheme 21 Brimble's total synthesis of (±)-thysanone. | ||
4.7 Shibata's synthesis of cis-clavicipitic acid
A 4-substituted tryptophan skeleton was usually used as a key intermediate for the total synthesis of clavicipitic acid110 and other alkaloids,111 but introduction of a substituent at the C-4 position on indole is generally difficult.111d,112 In 2015, Shibata and coworkers achieved 4-substituted tryptophan derivatives via Ir-catalyzed C–H bond activation of β-keto aniline.113 Based on this methodology, they synthesized cis-clavicipitic acid from a commercially available and cheap compound (183).113 It was transformed to 184 in three routine steps (Scheme 22). Then after activation of the acid with thionyl chloride, treatment with trimethylsilyldiazomethane and bromination with HBr, α-keto bromide 185 was accessed. Subsequent amination with compound 186 afforded the β-keto aniline 187. With the carbonyl group on the phenyl ring as a directing group, cyclodehydration of β-keto aniline 187via C–H bond activation was realized in the presence of an iridium catalyst, prepared from [Ir(COD)2]BArF and rac-BINAP at 135 °C in PhCl. The corresponding 4-substituted tryptophan 188 was obtained in 79% yield. Deprotection of 188 in HBr/HOAc gave the free amine 189, which was subjected to the intramolecular reductive amination conditions to afford the tricyclic azepinoindole product 190. The final basic hydrolysis of 190 achieved cis-clavicipitic acid, an ergot alkaloid isolated from Claviceps strain SD-58 and Claviceps fusiformis.114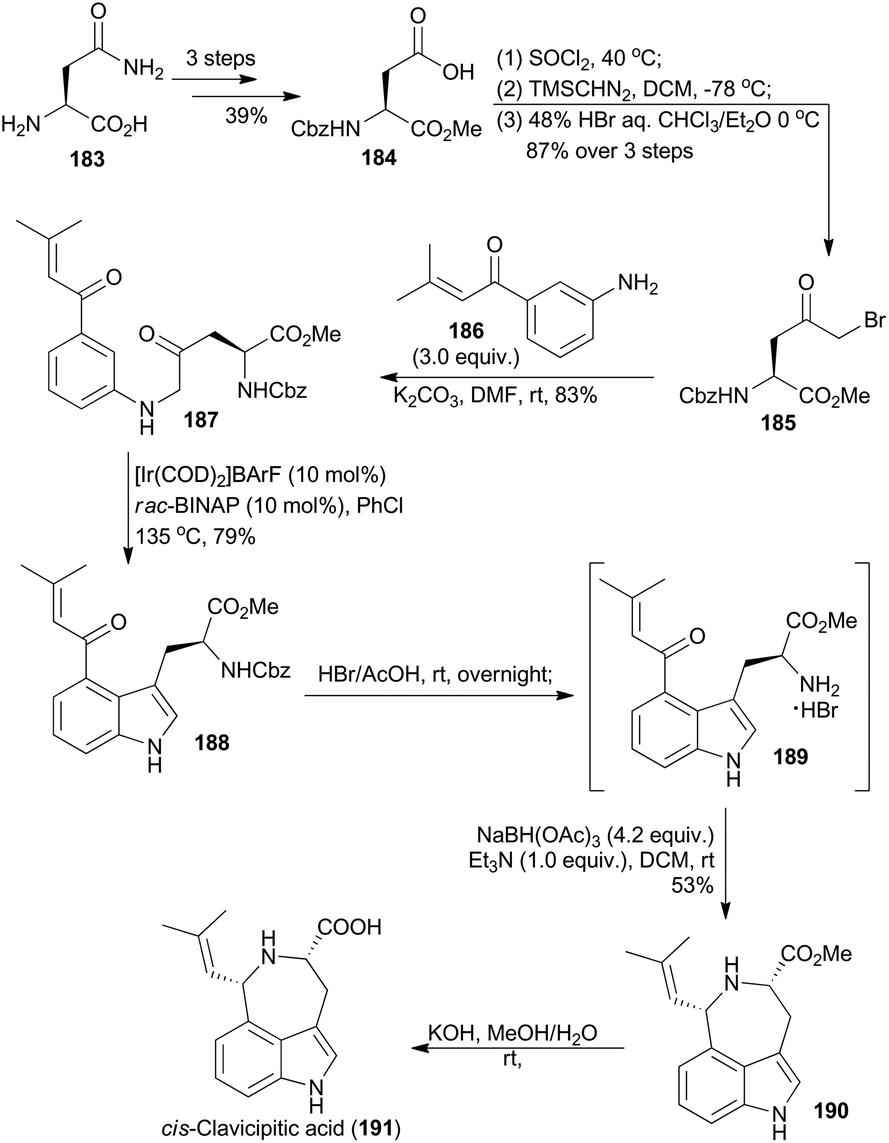 | ||
| Scheme 22 Shibata's total synthesis of cis-clavicipitic acid. | ||
Thus the iridium-catalyzed C–H bond activation strategy has been widely applied in total synthesis of natural products. Besides the above elegant research work, there are other excellent examples of total synthesis of natural products as well, with iridium as the catalyst for the C–H bond activation catalysis.115
5 Asymmetric allylic substitution
Transition metal-catalyzed asymmetric allylic substitution reactions are among the most powerful transformations currently available for the enantioselective formation of C–C bonds, C–N bonds, C–S bonds and C–O bonds.116 In this regard, various transition metals such as Ni, Fe, Mo, Pd, Ir, Ru, Au and Cu have proven efficient.117 In particular, iridium-catalyzed allylic substitution reactions have recently caught much attention due to the wide substrate scope, low catalyst loading and impressive stereochemical control.7,117 Recently, with iridium-catalyzed asymmetric allylic substitution as a key step, some elegant total syntheses of natural products have been reported. Herein we provide an overview of such application of iridium catalysis in natural product synthesis.5.1 Carreira's synthesis of (+)-aszonalenin and (−)-brevicompanine B
In 2014, Carreira and coworkers developed a catalytic system combining [Ir(COD)Cl]2 and a simple phosphoramidite ligand (194), to prompt the C3 reverse prenylation of 3-substituted indoles (192) with 193 (Scheme 23A).118 The reaction simultaneously installed two vicinal quaternary centers and furnished the adduct with high regio- and chemo-selectivities. Accordingly, this methodology has been utilized in total synthesis of (+)-aszonalenin (199) and (−)-brevicompanine B (198).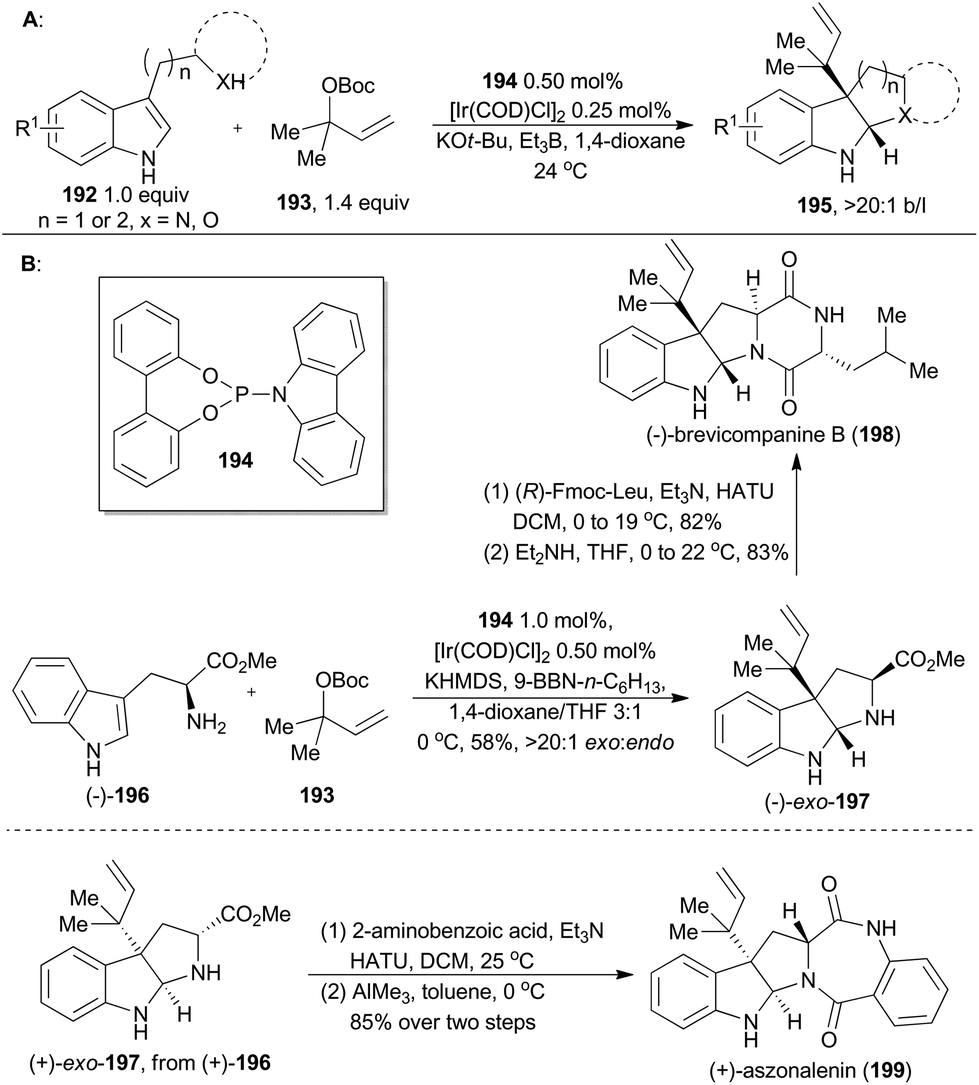 | ||
| Scheme 23 Carreira's total synthesis of (+)-aszonalenin and (−)-brevicompanine B. | ||
Brevicompanine B (198), isolated from Penicillium brevicompactum, is a plant growth regulator.119 Aszonalenin (199), isolated from Aspergillus zonatus,120 displays remarkable activity as an inhibitor for the human neurokinin-1 receptor.121 First, (−)-exo-197 was accomplished by Carreira's group according to their iridium-catalyzed methodology, but in an improved procedure (Scheme 23B). Coupling of (−)-exo-197 with (R)-Fmoc-Leu and subsequent deprotection/cyclization sequence readily gave (−)-brevicompanine B. This total synthesis was completed in only three steps from a commercially available starting material and was in higher overall yield than other reported syntheses.122 In a similar synthetic route, (+)-aszonalenin (199) can be obtained by coupling (+)-exo-197 to 2-aminobenzoic acid, followed by cyclization.
5.2 You's formal synthesis of (+)-gephyrotoxin and synthesis of (−)-lycopladine
The catalytic asymmetric dearomatization (CADA) reaction has attracted enormous attention because of its potential impact on synthetic organic chemistry.123 In particular, the transition-metal-catalyzed allylic dearomatization reaction124 has emerged as an innovative and powerful CADA reaction. In recent years, the You group has achieved Ir-catalyzed asymmetric allylic dearomatization of indoles, pyridines, pyrazines, quinolines, and isoquinolines in an intramolecular fashion (Scheme 24A).125 By employing a chiral Ir-catalyst formed in situ from [Ir(COD)Cl]2 with ligands 203 and 204, a series of dearomatized products was easily prepared in high yields (up to 99%) with excellent enantioselectivities (up to 99% ee). Through Ito's intermediate (212),126 You and coworkers achieved total synthesis of gephyrotoxin, which was isolated from the skin extract of the Columbian frog, Dendrobates histrionicus,127 and exhibited mild muscarinic activity and neurological activities.128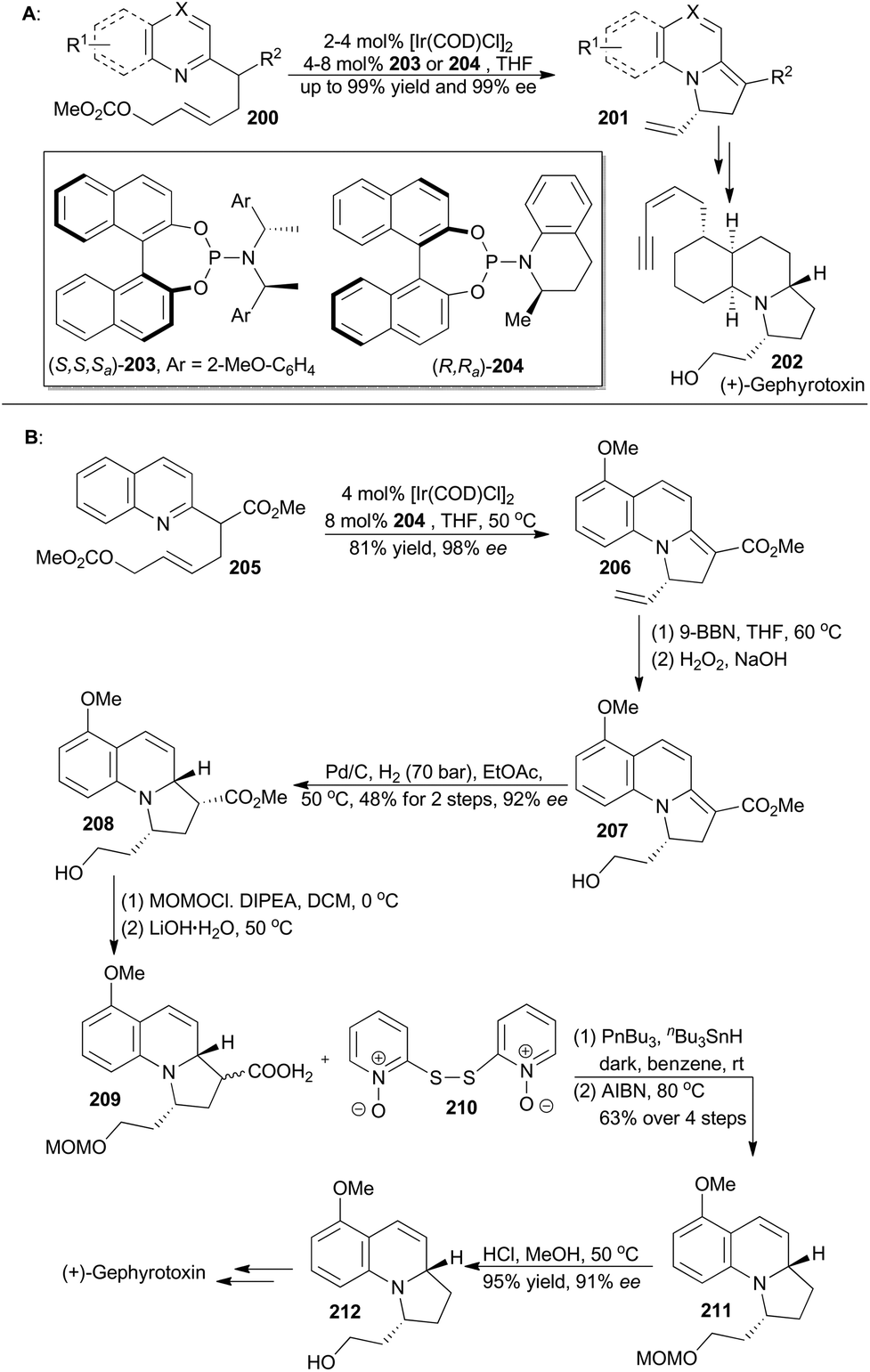 | ||
| Scheme 24 You's formal synthesis of (+)-gephyrotoxin. | ||
First, catalytic dearomatization of compound 205 by the iridium catalyst provided 2,3-dihydroindolizine 206 in 81% yield with 98% ee.125b Then elaboration of the terminal alkene to a primary alcohol and partial hydrogenation afforded compound 208 as a single diastereomer with 92% ee. Protection and subsequent hydrolysis with LiOH·H2O afforded 209, which was decarboxylated under modified Barton decarboxylation conditions to afford compound 211.129 Removal of the MOM ether produced Ito's intermediate (212), which could be converted to gephyrotoxin.
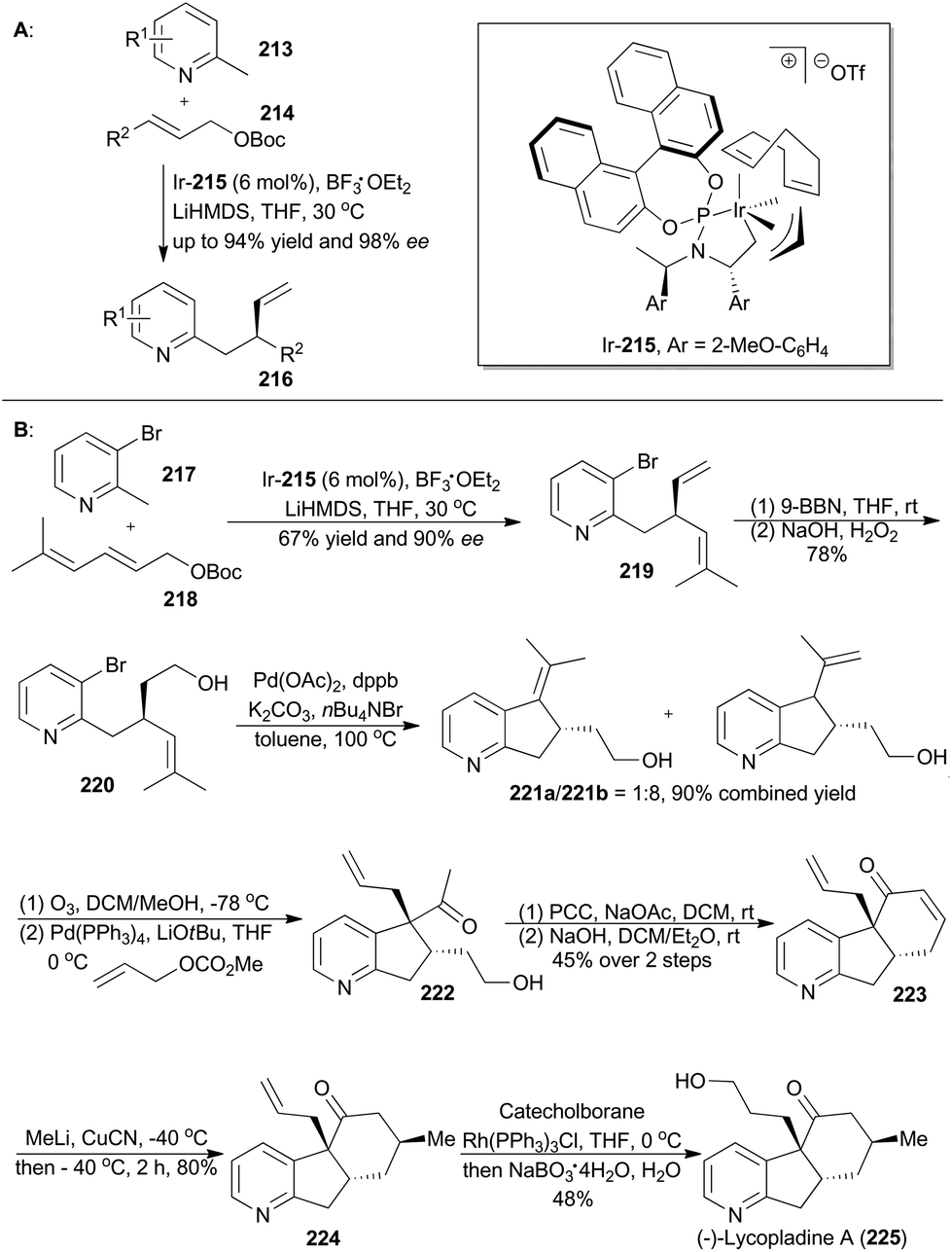
The derivatives of pyridine have been widely applied in many fields.130 In order to obtain diverse functionalization of pyridine, You's group developed an enantioselective iridium-catalyzed allylic substitution with a set of nucleophiles generated in situ from 2-methylpyridines (213) (Scheme 25A).131 Thus a series of enantioenriched 2-substituted pyridines (216) was easily synthesized in high yields and enantioselectivities. In addition, utility of this method promoted synthesis of (−)-lycopladine A (225) and a key intermediate of a reported Na+/H+ exchanger inhibitor.132 Lycopladine A, isolated from the club moss Lycopodium complanatum, is a C16N type of alkaloid featuring a pyridyl-fused hydrindanone core and three stereocenters.133 It exhibits weak cytotoxicity against murine lymphoma L1210 cells (IC50 = 7 μg mL−1).
 | ||
| Scheme 25 You's synthesis of (−)-lycopladine A. | ||
Asymmetric total synthesis of (−)-lycopladine A was triggered by asymmetric allylic alkylation of 217 with 218, enabled by the chiral catalyst Ir-215, in 67% yield with 90% ee to afford compound 219 (Scheme 25B). This 2-substituted pyridine 219 was then subjected to hydroboration and oxidation of the terminal alkene to give 220, which underwent a palladium-catalyzed Heck reaction and thus afforded compound 221b. Cleavage of the double bond and subsequent palladium-catalyzed allylic alkylation provided compound 222. Its primary alcohol was oxidized with pyridinium chlorochromate (PCC) to an aldehyde, which went through an intramolecular condensation to produce the tricyclic intermediate 223. Michael addition with in situ generated [Me2Cu(CN)Li2] yielded 224 with perfect stereo-control. Finally, its terminal alkene was transformed into an alcohol to complete total synthesis of lycopladine A (225).
5.3 Yang's synthesis of (−)-aurantioclavine, (−)-alstoscholarisine A, (−)-aspidophylline A and (−)-communesin F
Iridium-catalyzed asymmetric amination of allylic organic compounds with nucleophilic N sources is a powerful method for the construction of nitrogen-containing building blocks.117q In 2015, Yang and coworkers reported a synthetic method to generating indoleazepines such as 230via an Ir-catalyzed asymmetric amination of secondary allylic alcohols in an intramolecular fashion, using a combination of [Ir(COD)Cl]2, (S)-229 and Sc(OTf)3 as an efficient catalytic system (Scheme 26A).134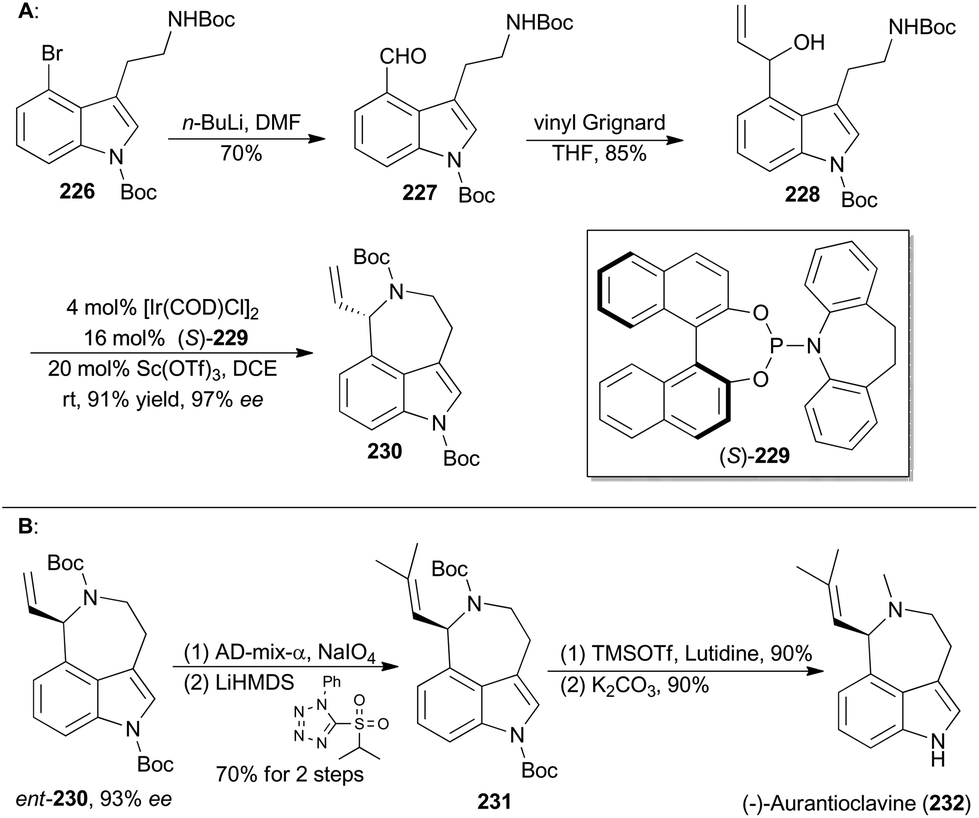 | ||
| Scheme 26 Yang's synthesis of (−)-aurantioclavine. | ||
(−)-Aurantioclavine (232) was isolated from Penicillium aurantiovirens in 1981.135 Structurally, aurantioclavine embraces an indoleazepine motif with a chiral center, whose configuration was assigned to R by Stoltz and coworkers through total synthesis.136 Yang's synthesis of (−)-aurantioclavine began with a known 4-bromo-tryptamine derivative (226),137 from which compound 228 was prepared after two steps (Scheme 26A). Then ent-230 was achieved through the above-mentioned Ir-catalyzed asymmetric allylic amination intramolecularly. Oxidative cleavage of the terminal olefin of ent-230 and the following Julia–Kocienski olefination afforded compound 231. Finally, global deprotection completed total synthesis of (−)-aurantioclavine (232). Notably, there is another successful total synthesis of (−)-aurantioclavine featuring a Pd-catalyzed asymmetric allylic amination by the Takemoto group in 2014.138
Alstoscholarisines A–E, isolated from Alstonia scholaris by Luo and coworkers in 2014, are five monoterpenoid indole alkaloids possessing a 6/5/6/6/6 polycyclic framework decorated with five continuous chiral centers.139 All of them displayed significant bioactivity in promoting adult neuronal stem cell (NSC) proliferation, and alstoscholarisine A was the most active one at a concentration of 0.1 μg mL−1 in a dosage-dependent manner.
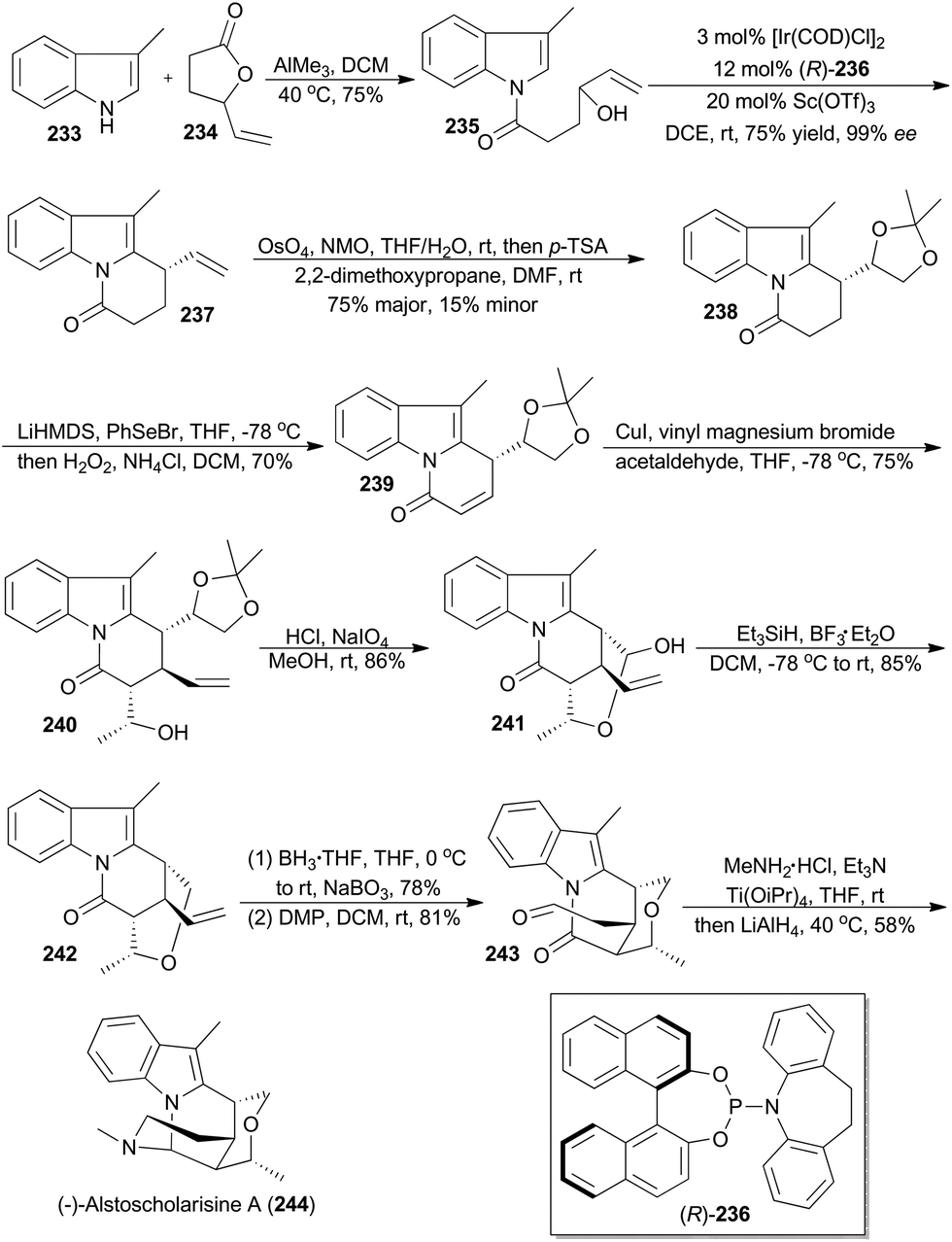
In 2016, Yang and coworkers reported a concise and enantioselective total synthesis of (−)-alstoscholarisine A (244), featuring an asymmetric intramolecular Friedel–Crafts alkylation of indole 235 with a secondary allylic alcohol,140 which was achieved by taking advantage of Carreira's iridium catalysis.9a,141 As depicted in Scheme 27, coupling of 3-methylindole (233) with the known 4-vinylbutyrolactone (234), promoted by trimethylaluminum, produced 235 in 75% yield. Exposure of 235 to [Ir(COD)Cl]2 and (R)-236, in the presence of Lewis acid Sc(OTf)3 as a promoter, afforded the tricyclic product 237 in 75% yield with 99% ee. This key transformation constructed the first stereogenic center, upon which the remaining three contiguous chiral centers would be readily set in a substrate-controlling manner. Dihydroxylation and subsequent protection provided the acetonide 238. After transforming 238 to the conjugated ketone 239, a Michael addition–aldol reaction cascade was realized to afford compound 240 with three contiguous chiral centers properly installed. After deprotection of the acetonide in 240 and oxidative cleavage of the resultant diol, the hemiacetal 241 was obtained. Reduction of the hemiacetal by employing triethylsilane-BF3·Et2O gave 242. Hydroboration–oxidation and Dess–Martin oxidation of the resulting primary alcohol afforded the aldehyde 243. Finally 243 was converted into (−)-alstoscholarisine A (244) after a one-pot reductive amination/cyclization sequentially.
 | ||
| Scheme 27 Yang's synthesis of (−)-alstoscholarisine A. | ||
(−)-Aspidophylline A, one of the notable akuammiline alkaloids, was isolated from Malayan Kopsia singapurensis by Kam and coworkers in 2007.142 Structurally, this pentacyclic alkaloid embraces a densely substituted cyclohexyl ring bearing five contiguous stereogenic centers and a bridged [3,3,1] bicycle. Biologically, it was found to reverse drug-resistance in drug-resistant KB cells.
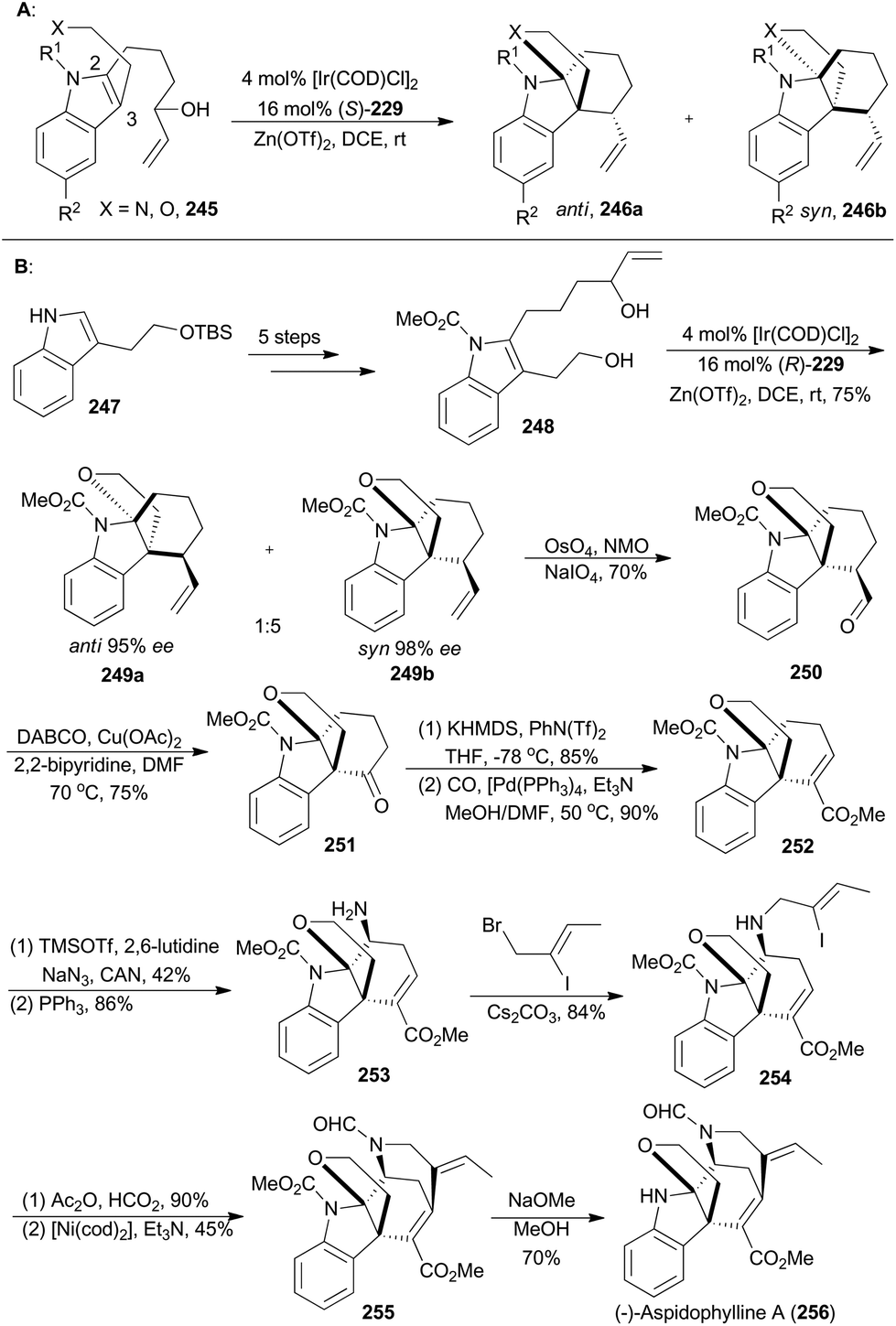
On the basis of enantioselective allylic substitution processes with branched allylic alcohols, the Yang group completed asymmetric total synthesis of (−)-aspidophylline A as well in 2016 (256, Scheme 28B). This synthesis featured a highly enantioselective allylic alkylation/iminium cyclization cascade with the indole 245 in the presence of [Ir(COD)Cl]2, (S)-229 and Zn(OTf)3.143 Beginning with the known compound 247, the 2,3-disubstituted indole 248 could be easily prepared in five steps, which was subjected to iridium-catalyzed cyclization with (R)-229 as a ligand to afford an inseparable diastereomeric mixture combining 249a and 249b. These two isomers were in a 1:5 ratio and both with excellent ee values. After oxidative cleavage of the terminal alkenes of the inseparable mixture, the aldehyde 250 could be isolated in 70% yield. Then 250 was subjected to copper catalysis, developed by the Van Rheenen group,144 to produce compound 251. Then enolization and palladium-catalyzed methoxycarbonylation afforded the α,β-unsaturated ester 252. By following a two-step strategy, an amino group was installed to produce the amine 253, which was alkylated with (Z)-1-bromo-2-iodobut-2-ene to afford 254. N-Formylation of 254, followed by a nickel-catalyzed Heck-type cyclization,145 yielded the pentacyclic product 255. Finally, removal of the carbamate delivered (−)-lycopladine A (256).
 | ||
| Scheme 28 Yang's synthesis of (−)-aspidophylline A. | ||
Communesin F is a member of communesins (A–H),146 a family of complex polycyclic indole alkaloids isolated from marine and terrestrial Penicillium fungi. These communesins showed significant cytotoxicity and insecticidal activity. Complexity of these polycyclic indole alkaloids was structurally indicated by their heptacyclic core arrayed with two aminal groups and at least five stereogenic centers. Among these natural products, communesin F was selected as a hot synthetic target and has been elegantly synthesized by several research groups.147
In 2017, Yang and coworkers reported another asymmetric total synthesis of (−)-communesin F (269) via the tetracyclic aminal 259, accessible from an iridium-catalyzed asymmetric cascade cyclization of indole in an intermolecular fashion (Scheme 29).148N-Methylation of 259 with tBuOK and MeI, followed by deprotection of the TBS ether, provided the desired tetracyclic primary alcohol 260. The phthalimide 261 was then obtained in 80% yield via a Mitsunobu reaction. Sequential oxidative cleavage, Pinnick oxidation and esterification with CH2N2 provided compound 262. Removal of the phthalimide group from 262 facilitated a spontaneous lactamization, and the subsequent protection smoothly afforded the N-Boc lactam 263. After several steps, compound 263 was transformed to 266, a key intermediate to construct a heptacyclic core. After the Heck reaction with 2-methyl-3-buten-2-ol and intramolecular nitrogen trapping of an allylic mesylate, compound 267 was generated. Finally, formation of a highly reactive iminium 268, intramolecular trapping by acetamide nitrogen and N-Boc deprotection afforded (−)-communesin F (269).
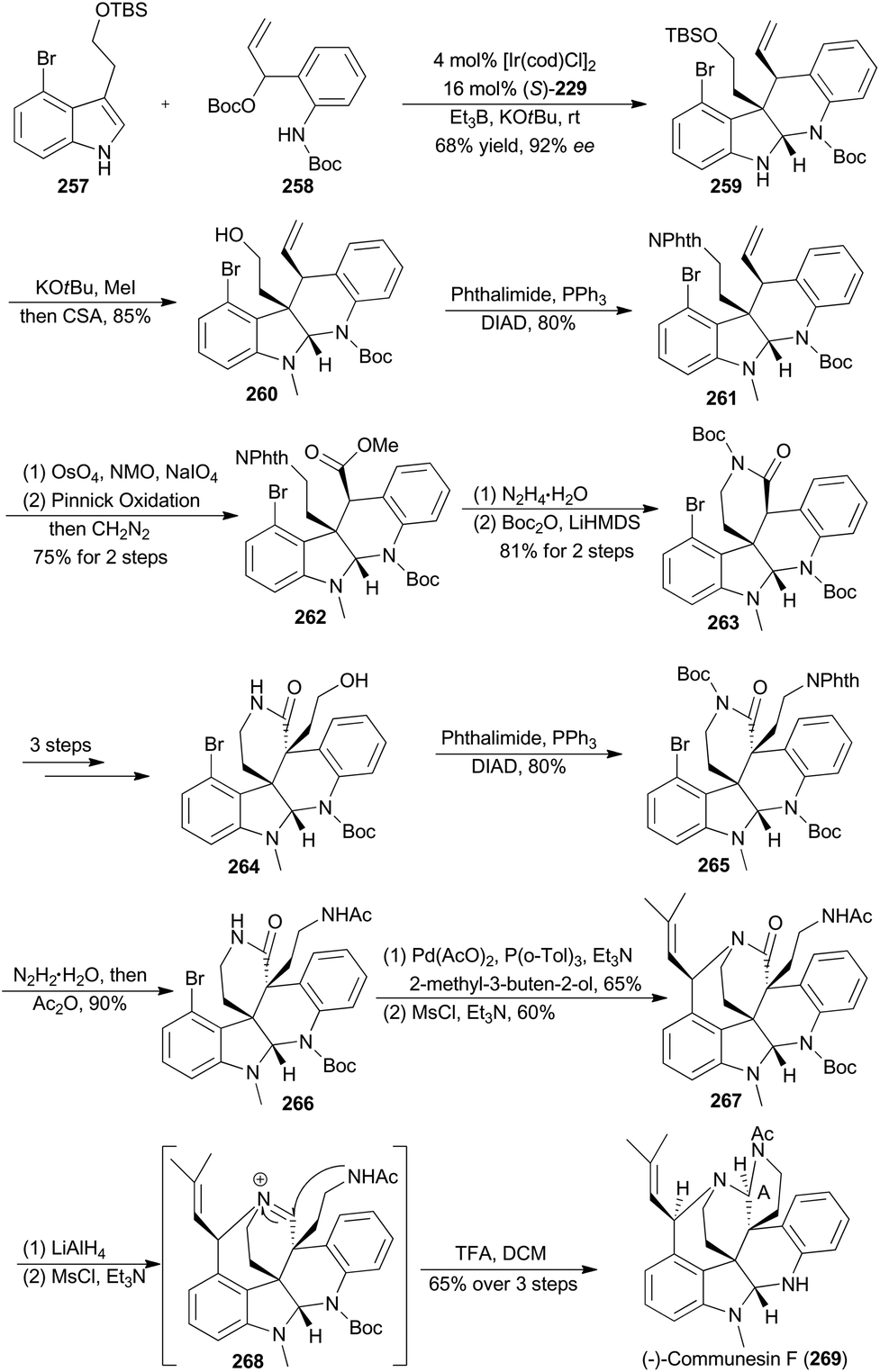 | ||
| Scheme 29 Yang's synthesis of (−)-communesin F. | ||
5.4 Hartwig's synthesis of fesoterodine
In 2015, Hartwig and coworkers reported an iridium-catalyzed enantioselective allylic substitution reaction with enol silanes of vinylogous esters and amides.149 Thus a variety of allylated esters 274 or amides 275 can be synthesized with high enantioselectivities and excellent branched-to-linear selectivities (Scheme 30A). Synthetic utility of this methodology was demonstrated in total synthesis of fesoterodine, an antimuscarinic drug (Scheme 30B).150 Accordingly, compound 276, obtained via this iridium catalysis methodology, was first transformed to a diene and then underwent [4 + 2] cycloaddition with ethyl propiolate as the dienophile to afford compound 277. Reduction of ethyl ester of 277 with DIBAL, followed by protection of the resulting alcohol with TBS, provided compound 278. After hydroboration–oxidation and Dess–Martin oxidation, the aldehyde 279 was generated, which was converted into the amine 280 through reductive amination and global deprotection of the silyl groups. By following a known procedure,150a final selective acylation with fumaric acid led to formation of fesoterodine (281).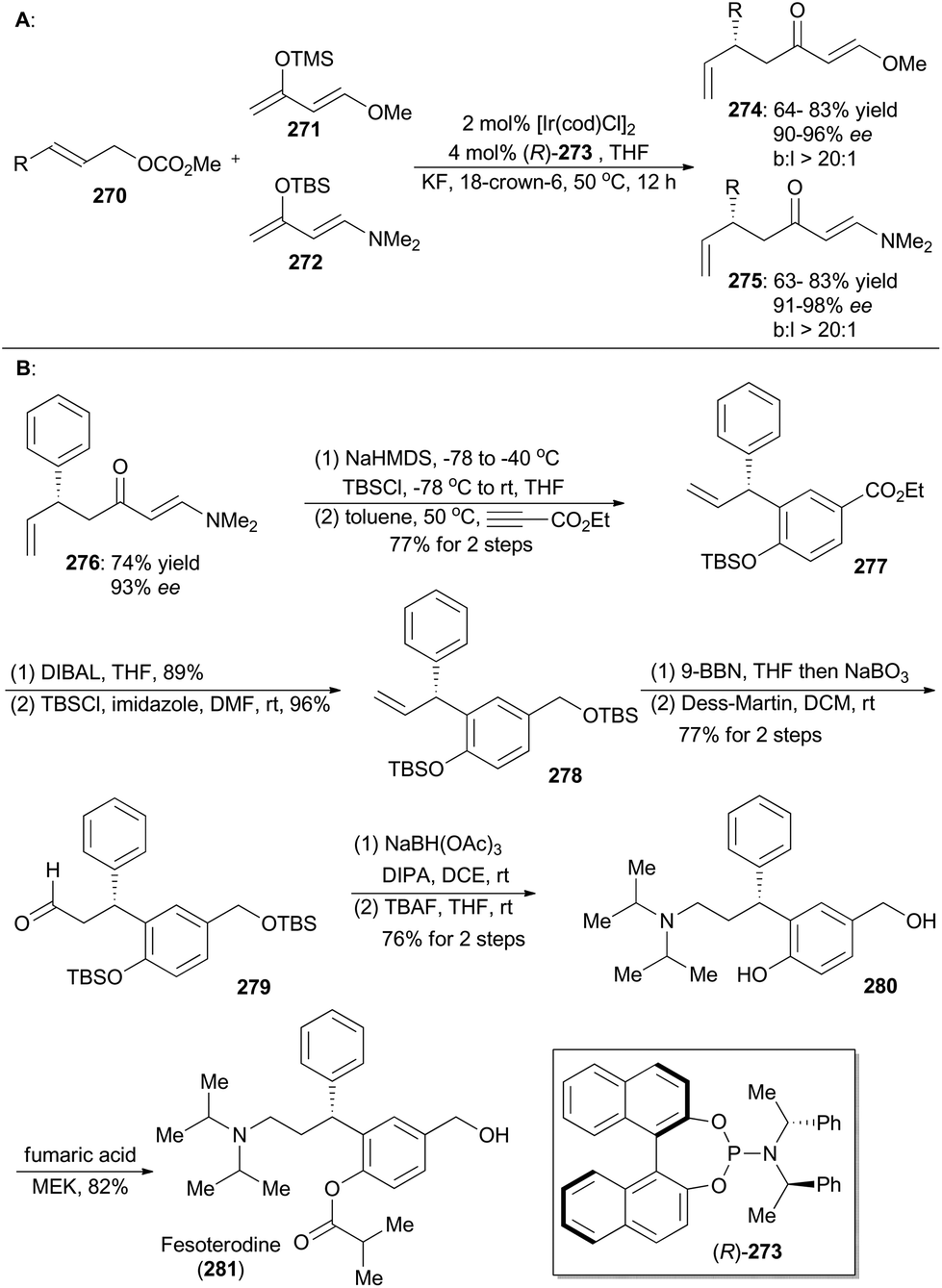 | ||
| Scheme 30 Hartwig's total synthesis of fesoterodine. | ||
5.5 Stark's synthesis of amauromine, novoamauromine and epi-amauromine
In 2016, Stark and coworkers reported an interesting regio- and diastereo-selective reverse prenylation of indole derivatives. N-Boc protected L- and D-tryptophan can lead to all four possible diastereomers (283a, 283b, 285a and 285b) through this iridium-catalyzed methodology under different conditions (Scheme 31A).151 In addition, N-Fmoc protected L-tryptophan (286) was proved to be another applicable substrate of this methodology to afford endo-287a and exo-287b (Scheme 31B). This method yielded a variety of building motifs, which may serve as synthons to reverse-prenylated hexahydropyrrolo[2,3]indole natural products,152 such as amauromine,153aepi-amauromine,153b and novoamauromine (Scheme 32).153c Accordingly, hydrolysis of methyl ester of exo-287b with CsOH·H2O facilitated the subsequent dimerization in the presence of HBTU to afford amauromine (288; Scheme 32). Novoamauromine (289) was synthesized as well similarly from endo-287a. A 1:1 mixture of diastereomeric exo-287b and endo-287a was subjected to the same dimerization conditions to afford epi-amauromine (290).
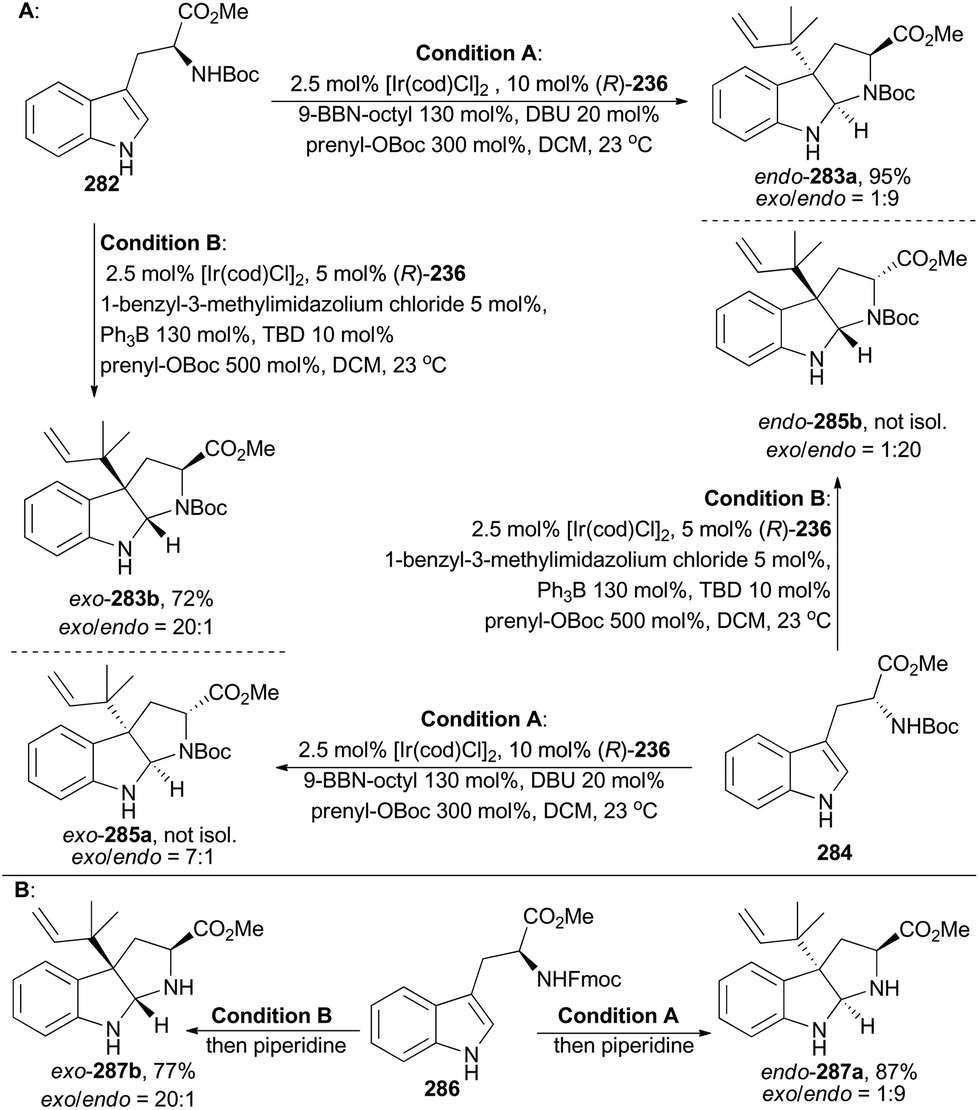 | ||
| Scheme 31 Stark's prenylation of indole derivatives. | ||
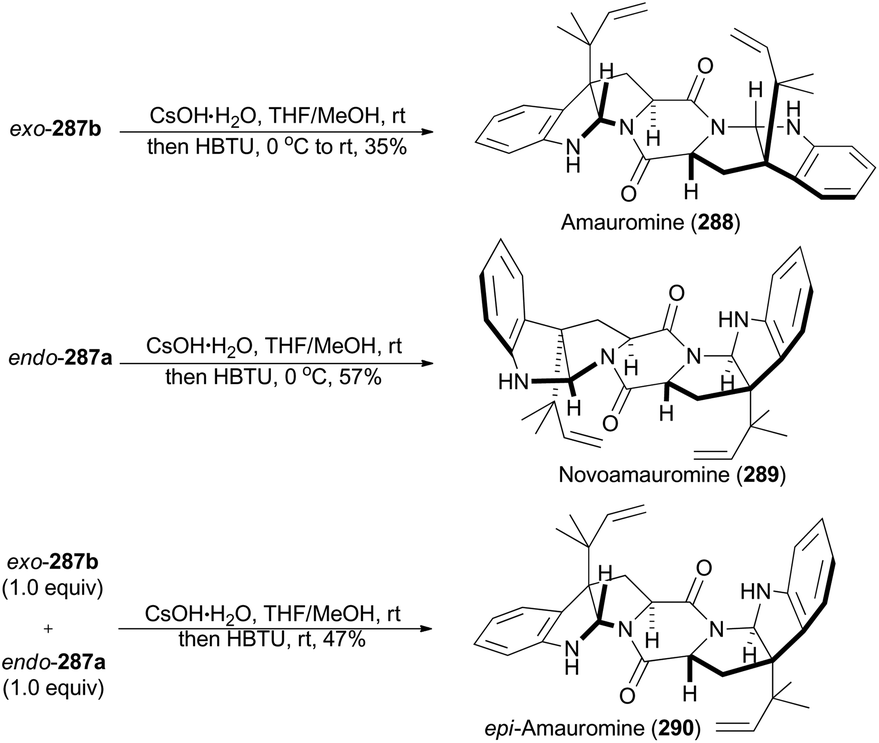 | ||
| Scheme 32 Stark's total synthesis of amauromine, novoamauromine and epi-amauromine. | ||
Generally, iridium-catalyzed asymmetric allylic substitution reactions have been well developed to generate a variety of chiral building blocks with versatile molecular skeletons and functionalities, which are useful for total synthesis of natural products and applicable for synthesis of pharmaceuticals and other bioactive compounds.117p,q,154
6 Polycyclization reaction
A cascade (domino) reaction is one of the most attractive strategies to promptly increase complexity of organic compounds from relatively simple starting materials.9,155 In this regard, polyene cyclization of unsaturated molecules is one of the most important methods for rapid construction of complex molecular structures in a single operation. Many types of polyene cyclization have been developed so far, including Brønsted acid-initiated cyclization, Lewis acid- and base-mediated cyclization, organocatalytic polyene cyclization and transition metal-mediated cyclization.9b Among them, transition metal catalysis offers an exceptional means to polyene cyclization in light of the mild reaction conditions and generally good selectivity, but its enantioselective versions are uncommon. In 2010, Toste and coworkers reported a gold(I)-catalyzed enantioselective polycyclization reaction potentially applicable in organic synthesis.156 Carreira and coworkers developed an iridium-catalyzed asymmetric polyene cyclization,9a,157 which has been significantly utilized in total synthesis of natural products.6.1 Carreira's synthesis of (+)-asperolide C
In 2012, Carreira and coworkers reported a highly enantioselective polycyclization method with iridium-catalyzed allylic substitution activated by Zn(OTf)2.9a Accordingly, a diverse set of carbocycles and heteropolycycles were obtained in good yields with excellent ee. Inspired by this methodology, Carreira and coworkers employed an allyl silane as the terminating group in compound 292, prepared from the known compound 291 in five steps, and thus triggered an Ir-catalyzed polyene cyclization to afford the trans-decalin 293 with excellent stereocontrol.157 It provided a practical synthetic entry into (+)-asperolide C (298) (Scheme 33), a tetranorlabdane-type diterpenoid isolated from Aspergillus wentii EN-48.158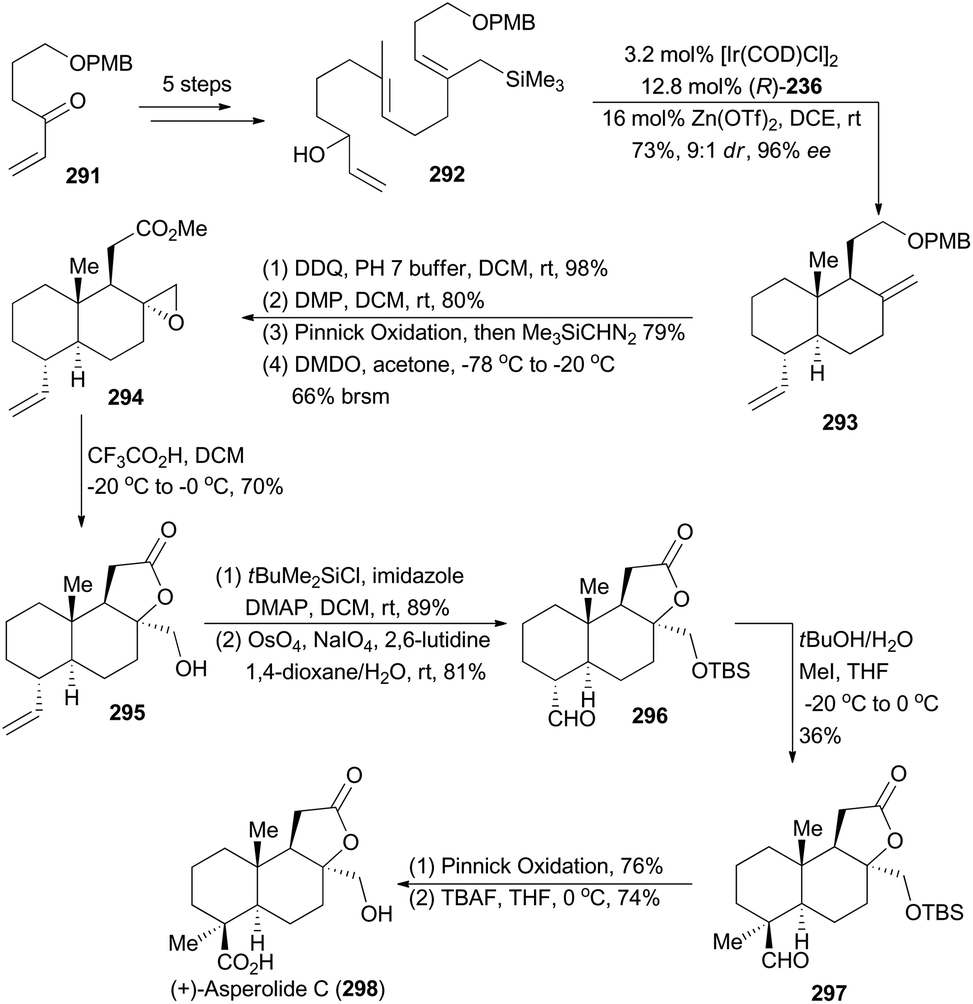 | ||
| Scheme 33 Carreira's synthesis of (+)-asperolide C. | ||
After deprotection of PMB ether, oxidation of the liberated primary hydroxy group to carboxylic acid, methyl esterification of the resulting carboxylic acid, and epoxidation of the exo-methylene with DMDO, compound 293 was transformed to the oxirane 294. It was then subjected to trifluoroacetic acid in DCM to furnish the tricyclic lactone 295, which underwent protection of the primary alcohol as a TBS ether and subsequent Lemieux–Johnson oxidation to provide the aldehyde 296. Treatment of 296 with tBuOK, followed by addition of iodomethane, delivered 297 to construct the quaternary chiral center. Finally, Pinnick oxidation of 297 and deprotection of the TBS group accomplished total synthesis of asperolide C (298).157
6.2 Li's synthesis of taiwaniadducts B, C, and D and (−)-mycoleptodiscin A
Taiwaniaquinoids are a class of terpenoids possessing significant biological activities and have been isolated from the endangered species Taiwania cryptomerioides in recent years.159 Taiwaniaquinoids B, C and D could be derived from a Diels–Alder reaction between two polycyclic structures, 305 and 311, based on a biosynthetic perspective (Schemes 34 and 35). Based on Carreira's Ir-catalyzed asymmetric polyene cyclization strategy,9a,157 Li and coworkers first synthetically achieved both 305b and 311, and finally accomplished total synthesis of taiwaniaquinoids B, C and D.160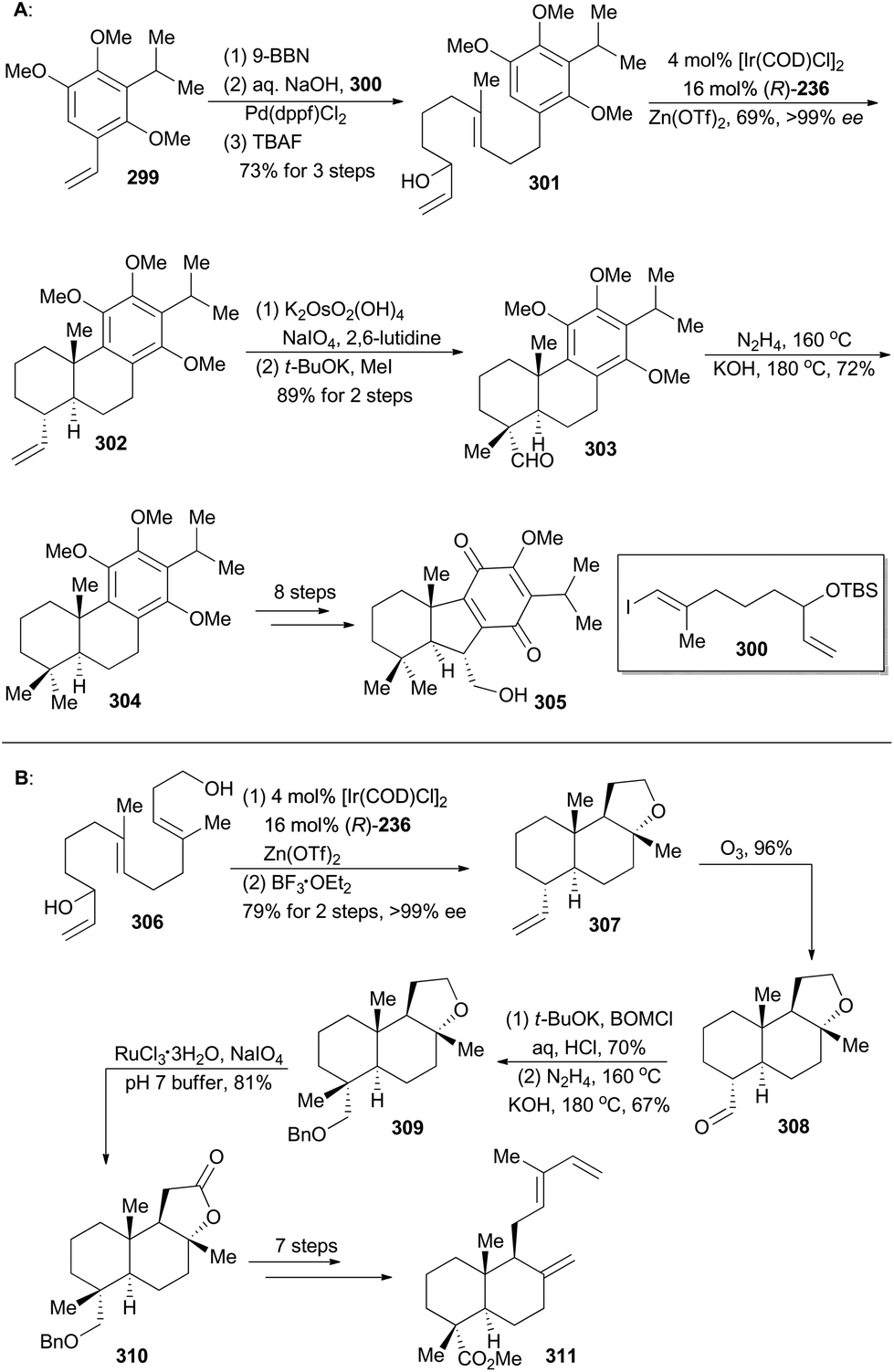 | ||
| Scheme 34 Li's synthesis of dienophile (305) and diene (311). | ||
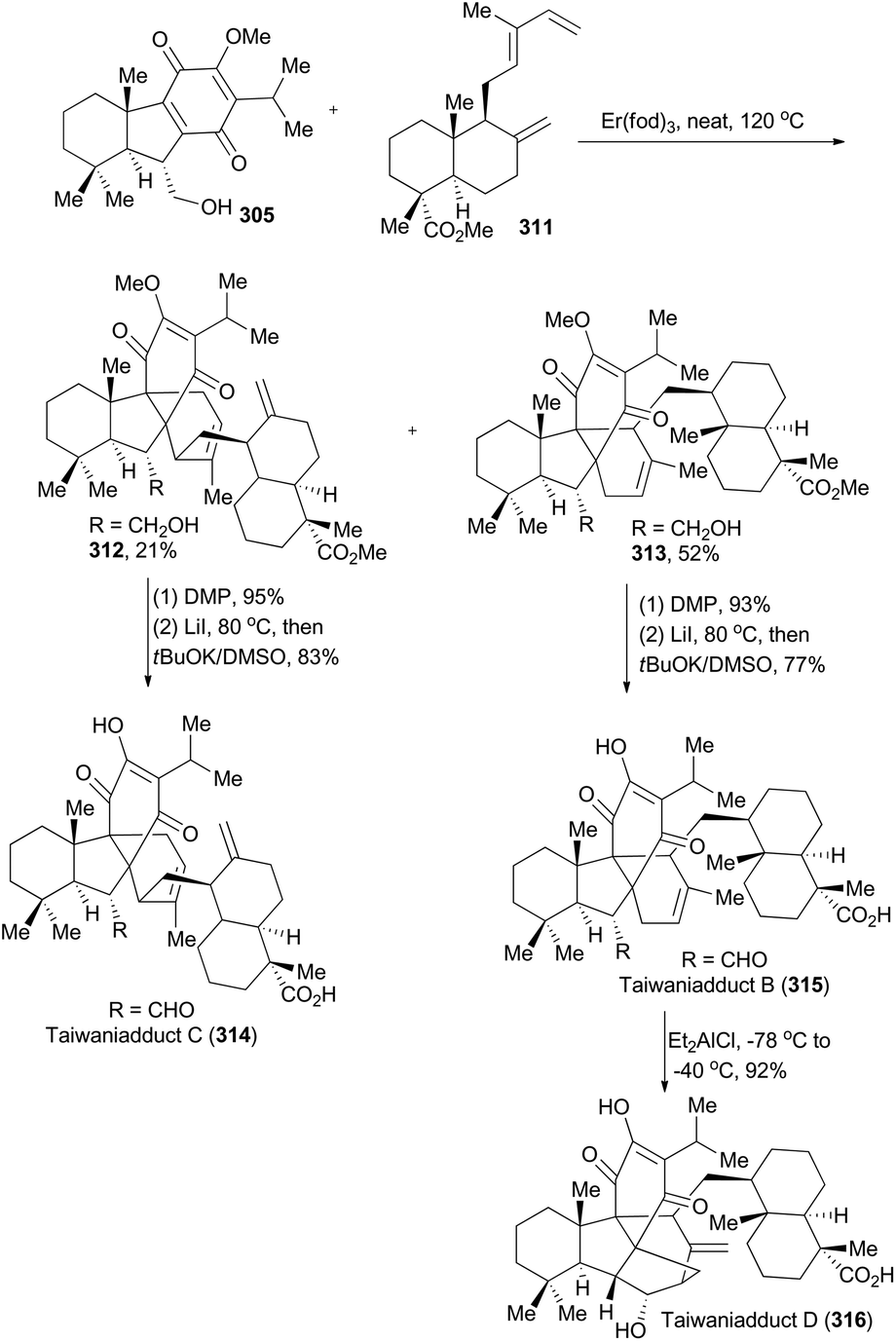 | ||
| Scheme 35 Li's synthesis of taiwaniadducts B, C, and D. | ||
Both 302 and 307 can be prepared with excellent enantioselectivity utilizing Carreira's iridium(I)/phosphoramidite catalytic system from compounds 301 and 306, respectively (Scheme 34). Notably, 302 could be prepared on a 15 g scale with consistent efficiency and selectivity, showing the robustness of this iridium catalysis methodology. On the one hand, cleavage of the terminal alkene in 302 and introduction of a methyl group furnished compound 303, which was subjected to Wolff–Kishner–Huang conditions to provide compound 304. It underwent a routine five-step sequence to afford the dienophile 305 (Scheme 34A).161 On the other hand, cleavage of 307 by ozonolysis afforded the aldehyde 308, which was converted into 309via alkylation with BOMCl and Wolff–Kishner–Huang reduction. Oxidation of 309 with RuCl3/NaIO4 resulted in formation of the lactone 310,162 which was converted into 311via further functionalization in 7 steps (Scheme 34B). Then the dienophile 305 and the diene 311 were combined and underwent Diels–Alder cycloaddition, promoted by Eu(fod)2 under neat conditions (Scheme 35), to produce the cycloadduct 312 (21% yield) and its regio-isomer 313 (52% yield). Both cycloadducts were subjected to oxidation of the primary alcohol and hydrolysis of the methyl ester to furnish taiwaniadducts C and B (314 and 315), respectively. Subsequently, carbonyl–ene cyclization of 315 promoted by Me2AlCl led to taiwaniadduct D (316, Scheme 35).
In 2015, the Li group reported their asymmetric total synthesis of (−)-mycoleptodiscin A (324, Scheme 36),163 a member of indolosesquiterpenoids, isolated by Cubilla-Rios and coworkers from the endophytic fungus Mycoleptodiscus sp.164 From a structural perspective, mycoleptodiscin A presents a unique synthetic challenge, due to its unusual indolosesquiterpenoid possessing an ortho-benzoquinone motif.
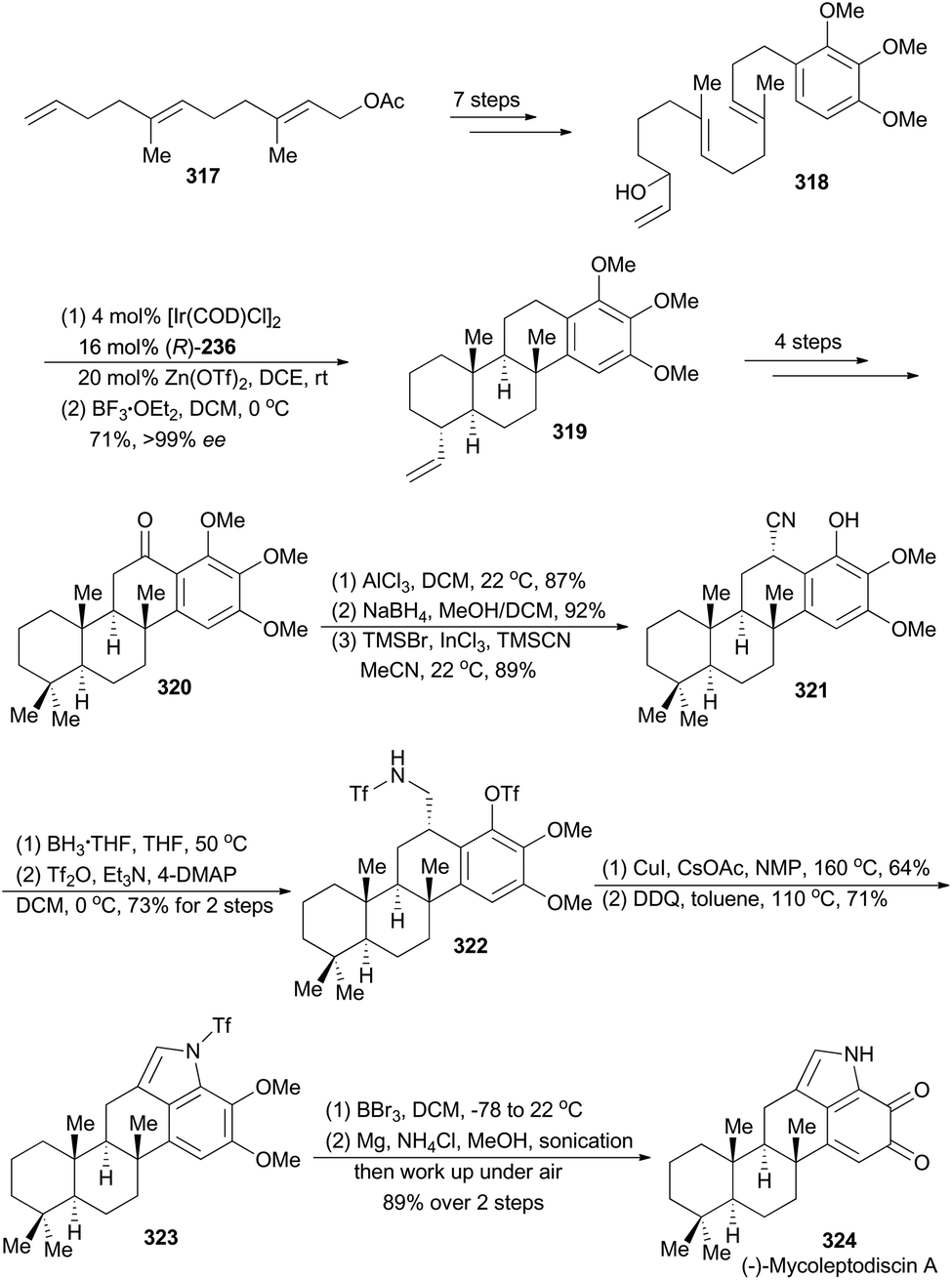 | ||
| Scheme 36 Li's synthesis of (−)-mycoleptodiscin A. | ||
Li's synthesis of (−)-mycoleptodiscin A commenced with a known compound 317 (Scheme 36),165 which produced the requisite linear precursor 318 in seven steps. Then the polyene 318 was subjected to Carreira's iridium-catalyzed polycyclization conditions to afford a mixture of the desired product 319 and the partial cyclization intermediates. Treatment of this mixture with BF3·OEt2 transformed these partial cyclization intermediates to 319. Thus the polycyclization product 319 was prepared in 71% overall yield with exceptionally high enantioselectivity (99% ee). Similar to the sequence for synthesis of compound 305, compound 320 was achieved in four steps. A three-step sequence, involving aluminum-promoted demethylation, ketone reduction and benzylic cyanation, delivered 321, which was subjected to reduction and protection of the resulting primary amine to afford compound 322. After a modified Buchwald–Hartwig amination by Fukuyama and coworkers,166 and oxidative formation of pyrrole with DDQ, compound 323 was obtained. Finally, total synthesis of (−)-mycoleptodiscin A (324) was accomplished from 323 after three steps.
7 Conclusions
In general, iridium complexes have been demonstrated to be active catalysts for asymmetric hydrogenation of alkenes, hydrogen transfer reaction, C–H functionalization, allylic substitution reaction and enantioselective polycyclization reaction. Recent accomplishments made in natural product total syntheses with an iridium catalyst are highlighted as above and other elegant examples of total synthesis of complex natural products are illustrated in Fig. 2, in which iridium-catalyzed methodologies, such as allylic amination,167 silane reduction of amide,168 alkene isomerization,169 oxidative desymmetrization,170 and photoredox catalysis,171 have been exploited. Undoubtedly, iridium catalysis must continue to serve as an attractive synthetic tool for accessing natural products with significant bioactivity and pharmaceutically active compounds, especially in an atom-economical and environmentally benign fashion.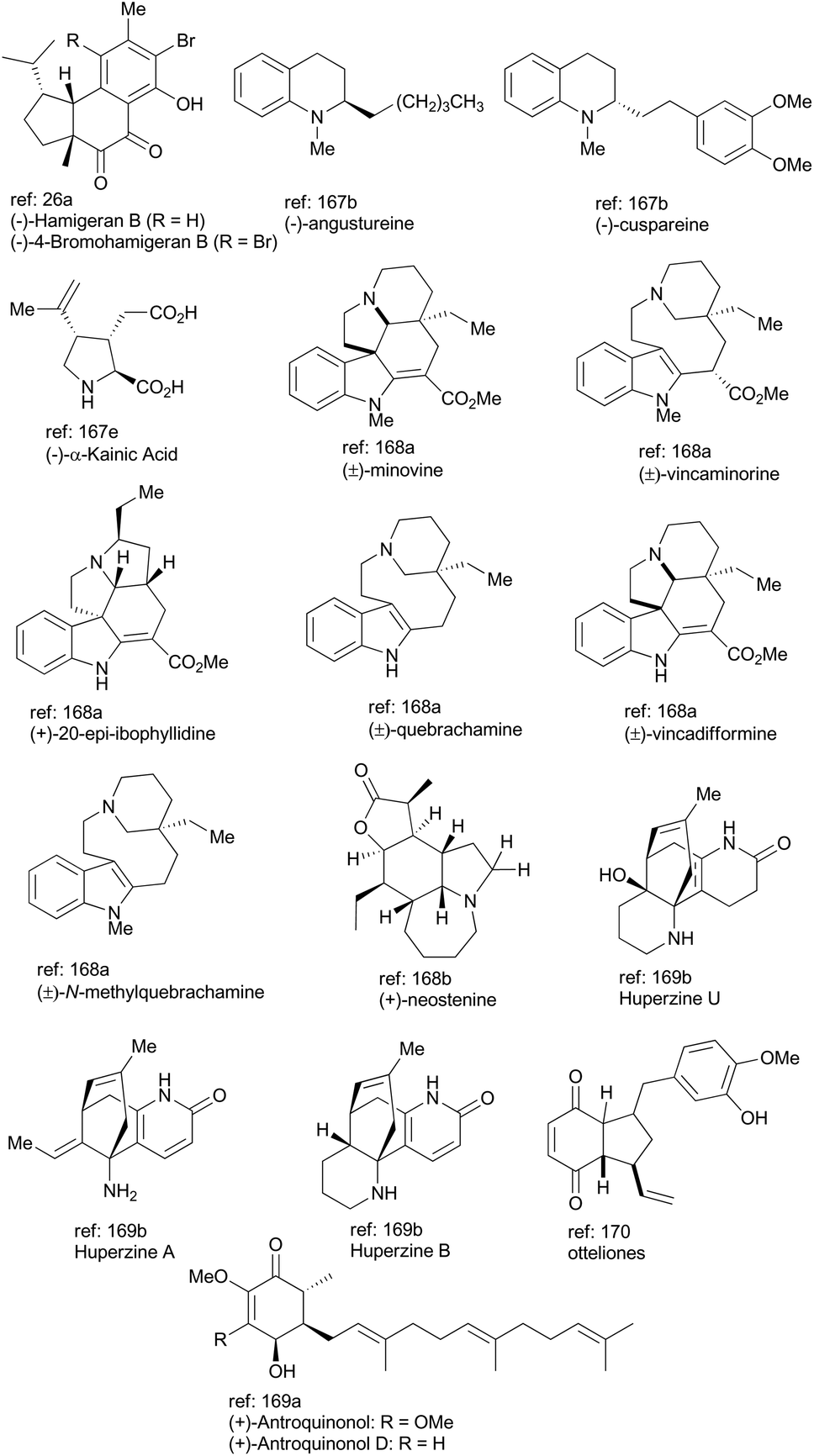 | ||
| Fig. 2 More examples of natural products synthesized with an iridium catalyst; the newly formed bonds via iridium catalysis are highlighted in red color. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the NSFC (21702192, 21672153 and 21290180).Notes and references
- Q.-L. Zhou, Angew. Chem., Int. Ed., 2016, 55, 5352 CrossRef CAS PubMed.
- R. H. Crabtree, Top. Organomet. Chem., 2011, 34, 1 CrossRef CAS.
- L. Vaska, Acc. Chem. Res., 1968, 1, 335 CrossRef CAS.
- For select references, see: (a) M. Diéguez, O. Pàmies and C. Claver, Top. Organomet. Chem., 2011, 34, 11 CrossRef; (b) D. H. Woodmansee and A. Pfaltz, Top. Organomet. Chem., 2011, 34, 31 CrossRef CAS; (c) J. Zhang, D. Yang and Y. Long, Chin. J. Org. Chem., 2009, 29, 835 CrossRef CAS; (d) S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402 CrossRef CAS PubMed; (e) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2017, 50, 988 CrossRef CAS PubMed; (f) T. Zhou, B. Peters, M. F. Maldonado, T. Govender and P. G. Andersson, J. Am. Chem. Soc., 2012, 134, 13592 CrossRef CAS PubMed.
- (a) O. Saidi and J. M. J. Williams, Top. Organomet. Chem., 2011, 34, 77 CrossRef CAS; (b) Q. Tu and Q. Ding, Chin. J. Org. Chem., 2010, 30, 1164 CAS; (c) N. Donati, M. Königsmann, D. Stein, L. Udino and H. Grützmacher, C. R. Chim., 2007, 10, 721 CrossRef CAS; (d) F. Foubelo, C. Nájera and M. Yus, Tetrahedron: Asymmetry, 2015, 26, 769 CrossRef CAS.
- (a) J. Choi and A. S. Goldman, Top. Organomet. Chem., 2011, 34, 139 CrossRef CAS; (b) S. Miyamura, M. Araki, T. Suzuki, J. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2014, 53, 1 CrossRef; (c) H. He, W.-B. Liu, L.-X. Dai and S.-L. You, J. Am. Chem. Soc., 2009, 131, 8346 CrossRef CAS PubMed; (d) Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433 CrossRef CAS PubMed.
- (a) J. F. Hartwig and M. J. Pouy, Top. Organomet. Chem., 2011, 34, 169 CrossRef CAS; (b) S. L. Rössler, S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 3603 CrossRef PubMed; (c) X. Zhang, W.-B. Liu, Q. Cheng and S.-L. You, Organometallics, 2016, 35, 2467 CrossRef CAS; (d) J. Y. Hamilton, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 125, 7680 CrossRef; (e) J. Y. Hamilton, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2014, 136, 3006 CrossRef CAS PubMed; (f) S. Breitler and E. M. Carreira, J. Am. Chem. Soc., 2015, 137, 5296 CrossRef CAS PubMed; (g) J. Y. Hamilton, N. Hauser, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2014, 126, 10935 CrossRef.
- (a) D. Carmona and L. A. Oro, Top. Organomet. Chem., 2011, 34, 209 CrossRef CAS; (b) D. Carmona, J. Ferrer, N. García, P. Ramıŕez, F. J. Lahoz, P. García-Ordunña and L. A. Oro, Organometallics, 2013, 32, 1609 CrossRef CAS; (c) H. Bian and D. Yang, Chin. J. Org. Chem., 2010, 30, 506 CAS.
- (a) M. A. Schafroth, D. Sarlah, S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2012, 134, 20276 CrossRef CAS PubMed; (b) C. N. Ungarean, E. H. Southgate and D. Sarlah, Org. Biomol. Chem., 2016, 14, 5454 RSC.
- (a) D. Staveness, I. Bosque and C. R. J. Stephenson, Acc. Chem. Res., 2016, 49, 2295 CrossRef CAS PubMed; (b) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (c) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875 CrossRef CAS PubMed; (d) J. W. Tucker and C. R. J. Stephenson, Org. Lett., 2011, 20, 5468 CrossRef PubMed; (e) M. H. Larraufie, R. Pellet, L. Fensterbank, J.-P. Goddard, E. Lacôte, M. Malacria and C. Ollivier, Angew. Chem., Int. Ed., 2011, 50, 4463 CrossRef CAS PubMed; (f) J. W. Tucker, J. M. R. Narayanam, P. S. Shah and C. R. J. Stephenson, Chem. Commun., 2011, 47, 5040 RSC; (g) J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74 CrossRef CAS PubMed; (h) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87 CrossRef CAS PubMed; (i) D. Hager and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 16986 CrossRef CAS PubMed; (j) J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2014, 53, 1 CrossRef.
- (a) L. Mantilli and C. Mazet, Tetrahedron Lett., 2009, 50, 4141 CrossRef CAS; (b) L. Mantilli, D. Gérard, S. Torche, C. Besnard and C. Mazet, Angew. Chem., Int. Ed., 2009, 121, 5245 CrossRef; (c) A. W. Gregory, A. Chambers, A. Hawkins, P. Jakubec and D. J. Dixon, Chem. – Eur. J., 2015, 21, 111 CrossRef CAS PubMed.
- (a) X.-W. Li and B. Nay, Nat. Prod. Rep., 2014, 31, 533 RSC; (b) R. Long, J. Huang, J. Gong and Z. Yang, Nat. Prod. Rep., 2015, 32, 1584 RSC; (c) G. Mao, Q. Huang and C. Wang, Eur. J. Org. Chem., 2017, 3549 CrossRef CAS.
- G.-H. Hou, J.-H. Xie, P.-C. Yan and Q.-L. Zhou, J. Am. Chem. Soc., 2009, 131, 1366 CrossRef CAS PubMed.
- (a) T. R. Wu and J. M. Chong, J. Am. Chem. Soc., 2006, 128, 9646 CrossRef CAS PubMed; (b) J. Szawkało, S. J. Czarnocki, A. Zawadzka, K. Wojtasiewicz, A. Leniewski, J. K. Maurin, Z. Czarnockia and J. Drabowicz, Tetrahedron: Asymmetry, 2007, 18, 406 CrossRef.
- (a) Q. Zhang, G. Tu, Y. Zhao and T. Cheng, Tetrahedron, 2002, 58, 6795 CrossRef CAS; (b) H.-J. Knölker and S. Agarwal, Tetrahedron Lett., 2005, 46, 1173 CrossRef.
- (a) Y.-C. Chang and M. G. Nair, J. Nat. Prod., 1995, 58, 1892 CrossRef CAS PubMed; (b) K. Morito, T. Hirose, J. Kinjo, T. Hirakawa, M. Okawa, T. Nohara, S. Ogawa, S. Inoue, M. Muramatsu and Y. Masamune, Biol. Pharm. Bull., 2001, 24, 351 CrossRef CAS PubMed; (c) K. D. R. Setchell, N. M. Brown and E. Lydeking-Olsen, J. Nutr., 2002, 132, 3577 CrossRef CAS PubMed.
- S. Yang, S.-F. Zhu, C.-M. Zhang, S. Song, Y.-B. Yu, S. Li and Q.-L. Zhou, Tetrahedron, 2012, 68, 5172 CrossRef CAS.
- Y. Dai, L. Harinantenaina, P. J. Brodie, M. Goetz, Y. Shen, K. TenDyke and D. G. Kingston, J. Nat. Prod., 2013, 76, 865 CrossRef CAS PubMed.
- G. O′Donnell, F. Bucar and S. Gibbons, Phytochemistry, 2006, 67, 178 CrossRef PubMed.
- T. Poisson, V. Gembus, V. Dalla, S. Oudeyer and V. Levacher, J. Org. Chem., 2010, 75, 7704 CrossRef CAS PubMed.
- Z.-Y. Li, S. Song, S.-F. Zhu, N. Guo, L.-X. Wang and Q.-L. Zhou, Chin. J. Chem., 2014, 32, 783 CrossRef CAS.
- S. Song, S.-F. Zhu, S. Yang, S. Li and Q.-L. Zhou, Angew. Chem., Int. Ed., 2012, 51, 2708 CrossRef CAS PubMed.
- T.-S. Wu, Y.-L. Tsai, A. G. Damu, P.-C. Kuo and P.-L. Wu, J. Nat. Prod., 2002, 65, 1522 CrossRef CAS.
- V. K. Honwad and A. S. Rao, Tetrahedron, 1965, 21, 2593 CrossRef CAS.
- H. Rimpler, R. Haensel and L. Kochendoerfer, Z. Naturforsch., 1970, 25, 995 CAS.
- (a) H. Lin, L.-J. Xiao, M.-J. Zhou, H.-M. Yu, J.-H. Xie and Q.-L. Zhou, Org. Lett., 2016, 18, 1434 CrossRef CAS PubMed; (b) S. Yang, S.-F. Zhu, N. Guo, S. Song and Q.-L. Zhou, Org. Biomol. Chem., 2014, 12, 2049 RSC; (c) S. Song, S.-F. Zhu, L.-Y. Pu and Q.-L. Zhou, Angew. Chem., Int. Ed., 2013, 52, 6072 CrossRef CAS PubMed; (d) Q.-Q. Zhang, J.-H. Xie, X.-H. Yang, J.-B. Xie and Q.-L. Zhou, Org. Lett., 2012, 14, 6158 CrossRef CAS PubMed.
- M. G. Schrems and A. Pfaltz, Chem. Commun., 2009, 6210 RSC.
- F. Bohlmann, C. Zdero, H. Robinson and R. M. King, Phyrochemistry, 1979, 18, 1675 CrossRef CAS.
- (a) L. F. Tietze and T. Raschke, Liebigs Ann., 1996, 1981 CrossRef CAS; (b) L. F. Tietze and T. Raschke, Synlett, 1995, 597 CrossRef CAS; (c) K. C. Nicolaou, R. Reingruber, D. Sarlah and S. Bräse, J. Am. Chem. Soc., 2009, 131, 2086 CrossRef CAS PubMed; (d) H. G. Schmalz, O. Kiehl, U. Korell and J. Lex, Synthesis, 2003, 1851 CrossRef CAS.
- F. Bohlmann, C. Zdero and N. Le Van, Phytochemistry, 1979, 18, 99 CrossRef CAS.
- G. G. Bianco, H. M. C. Ferraz, A. M. Costa, L. V. Costa-Lotufo, C. Pessoa, M. O. de Moraes, M. G. Schrems, A. Pfaltz and L. F. Silva Jr., J. Org. Chem., 2009, 74, 2561 CrossRef CAS PubMed.
- B. D. Morris, R. R. Smyth, S. P. Foster, M. P. Hoffmann, W. L. Roelofs, S. Franke and W. Francke, J. Nat. Prod., 2005, 68, 26 CrossRef CAS PubMed.
- (a) C. F. Weise, M. Pischl, A. Pfaltz and C. Schneider, Chem. Commun., 2011, 47, 3248 RSC; (b) C. F. Weise, M. Pischl, A. Pfaltz and C. Schneider, J. Org. Chem., 2012, 77, 1477 CrossRef CAS PubMed.
- B. A. Baker, Z. V. Boškovic and B. H. Lipshutz, Org. Lett., 2008, 10, 289 CrossRef CAS PubMed.
- A. Grassia, L. Bruno, C. Debitus, S. Marzocco, A. Pinto, L. Gomez-Paloma and R. Riccio, Tetrahedron, 2001, 57, 6257 CrossRef CAS.
- (a) T. Brodmann, M. Lorenz, R. Schaeckel, S. Simsek and M. Kalesse, Synlett, 2009, 174 CAS; (b) B. Schetter and R. Mahrwald, Angew. Chem., Int. Ed., 2006, 45, 7506 CrossRef CAS PubMed; (c) C. Palomo, M. Oiarbide and J. M. Garcia, Chem. Soc. Rev., 2004, 33, 65 RSC.
- (a) B. Horst, B. L. Feringa and A. J. Minnaard, Chem. Commun., 2010, 46, 2535 RSC; (b) S. Hanessian, S. Giroux and V. Mascitti, Synthesis, 2006, 1057 CrossRef CAS.
- Y. Zhu, A. Loudet and K. Burgess, Org. Lett., 2010, 12, 4392 CrossRef CAS PubMed.
- M. Isaka, P. Chinthanom, S. Supothina, P. Tobwor and N. L. Hywel-Jones, J. Nat. Prod., 2010, 73, 2057 CrossRef CAS PubMed.
- H. J. Jessen, A. Schumacher, F. Schmid, A. Pfaltz and K. Gademann, Org. Lett., 2011, 13, 4368 CrossRef CAS PubMed.
- H. J. Jessen, A. Schumacher, T. Shaw, A. Pfaltz and K. Gademann, Angew. Chem., Int. Ed., 2011, 50, 4222 CrossRef CAS PubMed.
- (a) W. Lolsberg, S. Werle, J.-M. Neudorfl and H.-G. Schmalz, Org. Lett., 2012, 14, 5996 CrossRef PubMed; (b) X. Wei, B. Qu, X. Zeng, J. Savoie, K. R. Fandrick, J.-N. Desrosiers, S. Tcyrulnikov, M. A. Marsini, F. G. Buono, Z. Li, B.-S. Yang, W. Tang, N. Haddad, O. Gutierrez, J. Wang, H. Lee, S. Ma, S. Campbell, J. C. Lorenz, M. Eckhardt, F. Himmelsbach, S. Peters, N. D. Patel, Z. Tan, N. K. Yee, J. J. Song, F. Roschangar, M. C. Kozlowski and C. H. Senanayake, J. Am. Chem. Soc., 2016, 138, 15473 CrossRef CAS PubMed; (c) B. K. Peters, J. Liu, C. Margarita and P. G. Andersson, Chem. – Eur. J., 2015, 21, 7292 CrossRef CAS PubMed; (d) F. Schmid, M. Bernasconi, H. J. Jessen, A. Pfaltz and K. Gademann, Synthesis, 2014, 864 Search PubMed; (e) P.-Y. Yang and Y.-G. Zhou, Tetrahedron: Asymmetry, 2004, 15, 1145 CrossRef CAS.
- C. Tejel and M. A. Ciriano, Top. Organomet. Chem., 2007, 22, 97 CrossRef CAS.
- (a) J. F. Bower, I. S. Kim, R. L. Patman and M. J. Krische, Angew. Chem., Int. Ed., 2009, 48, 34 CrossRef CAS PubMed; (b) T. Suzuki, Chem. Rev., 2011, 111, 1825 CrossRef CAS PubMed; (c) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681 CrossRef CAS PubMed; (d) J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761 CrossRef CAS PubMed; (e) D. Bezier and M. Brookhart, ACS Catal., 2014, 4, 3411 CrossRef CAS.
- (a) S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787 CrossRef CAS; (b) M. P. Joseph, Nat. Prod. Rep., 2008, 25, 166 RSC.
- J. Zhou and J. Fang, J. Org. Chem., 2011, 76, 7730 CrossRef CAS PubMed.
- J. Fang and J. Zhou, Org. Biomol. Chem., 2012, 10, 2389 Search PubMed.
- B. K. Joshi, J. B. Gloer, D. T. Wicklow and P. F. Dowd, J. Nat. Prod., 1999, 62, 650 CrossRef CAS PubMed.
- Z. Z. Ma, Y. Hano, T. Nomura and Y. J. Chen, Heterocycles, 1999, 51, 1883 CrossRef CAS.
- (a) A. H. Amin and D. R. Mehta, Nature, 1959, 183, 1317 CrossRef; (b) D. R. Mehta, J. S. Naravane and R. M. Desai, J. Org. Chem., 1963, 28, 445 CrossRef CAS; (c) M. P. Jain, S. K. Koul, K. L. Dhar and C. K. Atal, Phytochemistry, 1980, 19, 1880 CrossRef CAS; (d) A. Al-Shamma, S. Drake, D. L. Flynn, L. A. Mitscher, Y. H. Park, G. S. R. Rao, A. Simpson, J. K. Swayze, T. Veysoglu and S. T. S. Wu, J. Nat. Prod., 1981, 44, 745 CrossRef CAS PubMed.
- (a) S. R. Johns and J. A. Lamberton, Chem. Commun., 1965, 267 RSC; (b) J. S. Fitzgerald, S. R. Johns, J. A. Lamberton and A. H. Redcliffe, Aust. J. Chem., 1966, 19, 151 CrossRef CAS.
- J. Kokosi, I. Hermecz, G. Szasz and Z. Meszaros, Tetrahedron Lett., 1981, 22, 4861 CrossRef CAS.
- J. Kokosi, G. Szasz and I. Hermecz, Tetrahedron Lett., 1992, 33, 2995 CrossRef.
- (a) J. F. Bower and M. J. Krische, Top. Organomet. Chem., 2011, 34, 107 CrossRef CAS PubMed; (b) A. Hassan and M. J. Krische, Org. Process Res. Dev., 2011, 15, 1236 CrossRef CAS PubMed; (c) J. Moran and M. J. Krische, Pure Appl. Chem., 2012, 84, 1729 CrossRef CAS PubMed.
- X. Gao, H. Han and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 12795 CrossRef CAS PubMed.
- (a) M. Ishibashi, R. E. Moore and G. M. L. Patterson, J. Org. Chem., 1986, 51, 5300 CrossRef CAS; (b) R. E. Moore, G. M. L. Patterson, J. S. Mynderse, J. Barchi, T. R. Norton, E. Furusawa and S. Furusawa, Pure Appl. Chem., 1986, 58, 263 CrossRef CAS.
- (a) K. C. Nicolaou, K. Ajito, A. P. Patron, H. Khatuya, P. K. Richter and P. Bertinato, J. Am. Chem. Soc., 1996, 118, 3059–3060 CrossRef CAS; (b) K. C. Nicolaou, A. P. Patron, K. Ajito, P. K. Richter, H. Khatuya, P. Bertinato, R. A. Miller and M. J. Tomaszewski, Chem. – Eur. J., 1996, 2, 847 CrossRef CAS.
- (a) H. Naqaoka, G. Schmid, H. Iio and Y. Kishi, Tetrahedron Lett., 1981, 22, 899 CrossRef; (b) H. Nagaoka, W. Rutsch, G. S. H. Iio, M. R. Johnson and Y. Kishi, J. Am. Chem. Soc., 1980, 102, 7962 CrossRef CAS; (c) E. J. Corey and D. A. Clark, Tetrahedron Lett., 1980, 2045 CrossRef CAS.
- X. Gao, S. K. Woo and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 4223 CrossRef CAS PubMed.
- (a) Z. A. Kasun, X. Gao, R. M. Lipinski and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 8900 CrossRef CAS PubMed; (b) F. Perez, A. R. Waldeck and M. J. Krische, Angew. Chem., Int. Ed., 2016, 55, 5049 CrossRef CAS PubMed; (c) S. B. Han, A. Hassan, I. S. Kim and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 15559 CrossRef CAS PubMed; (d) A. R. Waldeck and M. J. Krische, Angew. Chem., Int. Ed., 2013, 52, 4470 CrossRef CAS PubMed; (e) Y. Feng, X. Jiang and J. K. De Brabander, J. Am. Chem. Soc., 2012, 134, 17083 CrossRef CAS PubMed; (f) J. Willwacher and A. Fürstner, Angew. Chem., Int. Ed., 2014, 53, 4217 CrossRef CAS PubMed; (g) J. Willwacher, B. Heggen, C. Wirtz, W. Thiel and A. Fürstner, Chem. – Eur. J., 2015, 21, 10416 CrossRef CAS PubMed; (h) S. Thiede, P. R. Wosniok, D. Herkommer, T. Debnar, M. Tian, T. Wang, M. Schrempp and D. Menche, Chem. – Eur. J., 2017, 23, 3300 CrossRef CAS PubMed.
- (a) S. Masamune, M. Hirama, S. Mori, S. A. Ali and D. S. Garvey, J. Am. Chem. Soc., 1981, 103, 1568 CrossRef CAS; (b) D. C. Myles and S. J. Danishefsky, J. Org. Chem., 1990, 55, 1636 CrossRef CAS; (c) D. A. Evans and A. S. Kim, Tetrahedron Lett., 1997, 38, 53 CrossRef CAS; (d) D. A. Evans, A. S. Kim, R. Metternich and V. J. Novack, J. Am. Chem. Soc., 1998, 120, 5921 CrossRef CAS; (e) E. M. Stang and M. C. White, Nat. Chem., 2009, 1, 547 CrossRef CAS PubMed; (f) M. T. Crimmins and D. J. Slade, Org. Lett., 2006, 8, 2191 CrossRef CAS PubMed.
- (a) H. A. Brooks, D. Gardner and J. P. Poyser, J. Antibiot., 1984, XXXVII, 1501 CrossRef; (b) U. Grafe, W. Schade and M. Roth, J. Antibiot., 1984, XXXVII, 836 CrossRef.
- E. Tonew, M. Tonew, U. Graefe and P. Zopel, Pharmazie, 1988, 43, 717 CAS.
- Z. Song, A. G. Lohse and R. P. Hsung, Nat. Prod. Rep., 2009, 26, 560 RSC.
- J. Feng, V. J. Garza and M. J. Krische, J. Am. Chem. Soc., 2014, 136, 8911 CrossRef CAS PubMed.
- K. Takada, H. Kajiwara and N. Imamura, J. Nat. Prod., 2010, 73, 698 CrossRef CAS PubMed.
- (a) Y. Takaishi, K. Shishido, N. Wariishi, M. Shibuya, K. Goto, M. Kido, Y. Ono and M. Taka, Tetrahedron Lett., 1992, 33, 7177 CrossRef CAS; (b) K. Shishido, K. Nakano, N. Wariishi, H. Tateishi, T. Omodani, M. Shibuya, K. Goto, Y. Ono and Y. Takaishi, Phytochemistry, 1994, 35, 731 CrossRef CAS; (c) H. Q. Duan, H. Y. Takaishi, H. Momota, Y. Ohmoto, T. Taki, Y. F. Jia and D. Li, J. Nat. Prod., 1999, 62, 1522 CrossRef CAS PubMed.
- (a) P. Crabbé, S. Burstein and C. Djerassi, Bull. Soc. Chim. Belg., 1958, 67, 632 CrossRef; (b) E. Bratoeff, M. C. Perez-Amador and E. Ramirez, Phyton, 1996, 58, 119 CAS.
- J. Feng, F. Noack and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 12364 CrossRef CAS PubMed.
- (a) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333 CrossRef CAS PubMed; (b) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS PubMed; (c) F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345, 1077 CrossRef CAS; (d) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (e) H. Davies and J. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (f) X. Chen, K. Engle, D. Wang and J. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (g) T. Lyons and M. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (h) C. Sun, B. Li and Z. Shi, Chem. Commun., 2010, 46, 677 RSC; (i) C. Yeung and V. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (j) S. Cho, J. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC.
- (a) C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 3375 CrossRef CAS PubMed; (b) X. Liao, L. M. Stanley and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2088 CrossRef CAS PubMed; (c) J. M. Murphy, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 15434 CrossRef CAS PubMed.
- Y. Ebe and T. Nishimura, J. Am. Chem. Soc., 2015, 137, 5899 CrossRef CAS PubMed.
- (a) T. Ishiyama, K. Sato, Y. Nishio, T. Saiki and N. Miyaura, Chem. Commun., 2005, 5065 RSC; (b) B. Lu and J. R. Falck, Angew. Chem., Int. Ed., 2008, 47, 7508 CrossRef CAS PubMed.
- J. Kobayashi, Y. Hirasawa, N. Yoshida and H. Morita, Tetrahedron Lett., 2000, 41, 9069 CrossRef CAS.
- (a) K. Ishiuchi, T. Kubota, T. Hoshino, Y. Obara, N. Nakahata and J. Kobayashi, Bioorg. Med. Chem., 2006, 14, 5995 CrossRef CAS PubMed; (b) H. Morita, K. Ishiuchi, A. Haganuma, T. Hoshino, Y. Obara, N. Nakahata and J. Kobayashi, Tetrahedron, 2005, 61, 1955 CrossRef CAS.
- (a) D. F. Fischer and R. Sarpong, J. Am. Chem. Soc., 2010, 132, 5926 CrossRef CAS PubMed; (b) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390 CrossRef CAS PubMed.
- (a) For compound 121 see: M. N. Kopylovich, V. Y. Kukushikin, M. Haukka, J. J. R. Frausto da Silva and A. J. L. Pombiero, Inorg. Chem., 2002, 41, 4798 CrossRef CAS PubMed; (b) For compound 122 see: E. Schuster, G. Jas and D. Schumann, Org. Prep. Proced. Int., 1992, 24, 670 CrossRef CAS; (c) K.-M. Liu, C.-M. Chau and C.-K. Sha, Chem. Commun., 2008, 91 RSC.
- (a) G. Majetich and J. M. Shimkus, J. Nat. Prod., 2010, 73, 284 CrossRef CAS PubMed; (b) W. H. Lin, J. M. Fang and Y. S. Cheng, Phytochemistry, 1995, 40, 871 CrossRef CAS; (c) W. H. Lin, J. M. Fang and Y. S. Cheng, Phytochemistry, 1996, 42, 1657 CrossRef CAS; (d) K. Kawazoe, M. Yamamoto, Y. Takaishi, G. Honda, T. Fujita, E. Sezik and E. Yesilada, Phytochemistry, 1999, 50, 493 CrossRef CAS; (e) H. Ohtsu, M. Iwamoto, H. Ohishi, S. Matsunaga and R. Tanaka, Tetrahedron Lett., 1999, 40, 6419 CrossRef CAS; (f) C. I. Chang, S. C. Chien, S. M. Lee and Y. H. Kuo, Chem. Pharm. Bull., 2003, 51, 1420 CrossRef CAS PubMed; (g) C. I. Chang, J. Y. Chang, C. C. Kuo, W. Y. Pan and Y. H. Kuo, Planta Med., 2005, 71, 72 CrossRef CAS PubMed.
- G. X. Liang, Y. Xu, I. B. Seiple and D. Trauner, J. Am. Chem. Soc., 2006, 128, 11022 CrossRef CAS PubMed.
- E. Fillion and D. Fishlock, J. Am. Chem. Soc., 2005, 127, 13144 CrossRef CAS PubMed.
- (a) R. J. Cole, J. W. Kirksey, J. H. Moore, B. R. Blankenship, U. L. Diener and N. D. Davis , Appl. Microbiol., 1972, 24, 248 CAS; (b) R. J. Cole and J. W. Kirksey, J. Agric. Food Chem., 1973, 21, 927 CrossRef CAS PubMed; (c) J. Fayos, D. Lokensgard, J. Clardy, R. J. Cole and J. W. Kirksey, J. Am. Chem. Soc., 1974, 96, 6785 CrossRef CAS PubMed; (d) R. J. Cole, J. W. Kirksey, R. H. Cox and J. Clardy, J. Agric. Food Chem., 1975, 23, 1015 CrossRef CAS PubMed; (e) T. Yoshizawa, N. Morooka, Y. Sawada and S. Udagawa, Appl. Environ. Microbiol., 1976, 32, 441 CAS.
- (a) M. Yamazaki, S. Suzuki and K. Miyaki, Chem. Pharm. Bull., 1971, 19, 1739 CrossRef CAS PubMed; (b) M. Yamazaki, H. Fujimoto and T. Kawasaki, Chem. Pharm. Bull., 1980, 28, 245 CrossRef CAS PubMed.
- (a) S. K. Rabindran, D. D. Ross, L. A. Doyle, W. Yang and L. M. Greenberger, Cancer Res., 2000, 60, 47 CAS; (b) J. D. Allen, A. van Loevezijn, J. M. Lakhai, M. van der Valk, O. van Tellingen, G. Reid, J. H. M. Schellens, G.-J. Koomen and A. H. Schinkel, Mol. Cancer Ther., 2002, 1, 417 CrossRef CAS PubMed.
- X. Wang, T. Furukawa, T. Nitanda, M. Okamoto, Y. Sugimoto, S.-L. Akiyama and M. Baba, Mol. Pharmacol., 2003, 63, 65 CrossRef CAS PubMed.
- Y. Feng, D. Holte, J. Zoller, S. Umemiya, L. R. Simke and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 10160 CrossRef CAS PubMed.
- R. E. Shade, A. M. Hyde, J.-C. Olsen and C. A. Merlic, J. Am. Chem. Soc., 2010, 132, 1202 CrossRef CAS PubMed.
- L. Randriambola, J.-C. Quirion, C. Kan-Fan and H.-P. Husson, Tetrahedron Lett., 1987, 28, 2123 CrossRef CAS.
- (a) S. Takano, T. Sato, K. Inomata and K. Ogasawara, J. Chem. Soc., Chem. Commun., 1991, 462 RSC; (b) C. L. Morales and B. L. Pagenkopf, Org. Lett., 2008, 10, 157 CrossRef CAS PubMed; (c) F. D. Simone, J. Gertsch and J. Waser, Angew. Chem., Int. Ed., 2010, 49, 5767 CrossRef PubMed; (d) M. Mizutani, F. Inagaki, T. Nakanishi, C. Yanagihara, I. Tamai and C. Mukai, Org. Lett., 2011, 13, 1796 CrossRef CAS PubMed; (e) L. Jiao, E. Herdtweck and T. Bach, J. Am. Chem. Soc., 2012, 131, 14563 CrossRef PubMed; (f) Z. Xu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2013, 52, 3272 CrossRef CAS PubMed; (g) G. Lewin, G. Bernadat, G. Aubert and T. Cresteil, Tetrahedron, 2013, 69, 1622 CrossRef CAS.
- S. Zhou and Y. Jia, Org. Lett., 2014, 16, 3416 CrossRef CAS PubMed.
- T. Ueki, M. Doe, Y. Morimoto, T. Kinoshita, R. Tanaka and K. Yoshihara, J. Heterocycl. Chem., 2001, 38, 165 CrossRef CAS.
- C. F. Huebner and S. R. Ames, J. Am. Chem. Soc., 1946, 68, 1621 CrossRef CAS PubMed.
- G. Stork and K. Zhao, Tetrahedron Lett., 1989, 30, 2173 CrossRef CAS.
- (a) T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed., 2002, 41, 3056 CrossRef CAS; (b) T. Ishiyama, Y. Nobuta, J. F. Hartwig and N. Miyaura, Chem. Commun., 2003, 2924 RSC; (c) T. Ishiyama, J. Takagi, Y. Yonekawa, J. F. Hartwig and N. Miyaura, Adv. Synth. Catal., 2003, 345, 1103 CrossRef CAS; (d) H. Y. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995 CrossRef CAS PubMed; (e) J. Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305 CrossRef CAS PubMed.
- (a) H. Seow, P. G. Penketh, R. P. Baumann and A. C. Sartorelli, Methods Enzymol., 2004, 382, 221 CAS; (b) F. J. Alcain and J. M. Villalba, Expert Opin. Ther. Pat., 2007, 17, 649 CrossRef CAS.
- A. Jain, R. M. Phillips, A. J. Scally, G. Lenaz, M. Beer and R. Puri, Urology, 2009, 73, 1083 CrossRef PubMed.
- H. Oka, T. Yoshinari, T. Murai, K. Kuwamura, F. Satoh, K. Funaishi, A. Okura, H. Suda, M. Okanishi and Y. Shizuri, J. Antibiot., 1991, 44, 486 CrossRef CAS PubMed.
- Y. Fukuyama, C. Iwatsuki, M. Kodama, M. Ochi, K. Kataoka and K. Shibata, Tetrahedron, 1998, 54, 10007 CrossRef CAS.
- (a) C. Wang and J. Sperry, J. Org. Chem., 2012, 77, 2584 CrossRef CAS PubMed; (b) S. H. Kim and J. Sperry, Tetrahedron Lett., 2015, 56, 5914 CrossRef CAS.
- (a) J. Nobuhiro, M. Hirayama, T. Choshi, K. Kamoshita, S. Maruyama, Y. Sukenaga, T. Ishizu, H. Fujioka and S. Hibino, Heterocycles, 2006, 70, 491 CrossRef CAS; (b) M. Inman and C. J. Moody, J. Org. Chem., 2010, 75, 6023 CrossRef CAS PubMed; (c) J. M. Saa, C. Marti and A. Raso-García, J. Org. Chem., 1992, 57, 589 CrossRef CAS.
- D. W. Robbins, T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 4068 CrossRef CAS PubMed.
- P. Sulanga, G. A. Chotana, D. Holmes, R. C. Reichle, R. E. Maleczka Jr. and M. R. Smith III, J. Am. Chem. Soc., 2006, 128, 15552 CrossRef PubMed.
- M. A. Larsen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4287 CrossRef CAS PubMed.
- Y.-J. Lee, D.-G. Lee, H. S. Rho, V. B. Krasokhin, H. J. Shin, J. S. Lee and H.-S. Lee, J. Heterocycl. Chem., 2013, 50, 1400 CrossRef CAS.
- M. Chakraborty, T. Kundu and Y. Harigaya, Synth. Commun., 2006, 36, 2069 CrossRef.
- P. Hume, D. P. Furkert and M. A. Brimble, Tetrahedron Lett., 2012, 53, 3771 CrossRef CAS.
- K. Schunemann, D. P. Furkert, S. Connelly, J. D. Fraser, J. Sperry and M. A. Brimble, Synlett, 2014, 556 CAS.
- S. B. Singh, M. G. Cordingley, R. G. Ball, J. L. Smith, A. W. Dombrowski and M. A. Goetz, Tetrahedron Lett., 1991, 32, 5279 CrossRef CAS.
- K. Schünemann, D. P. Furkert, S. Connelly, J. D. Fraser, J. Sperry and M. A. Brimble, Eur. J. Org. Chem., 2014, 122 CrossRef.
- T. Ishiyama, Y. Nobuta, J. F. Hartwig and N. Miyaura, Chem. Commun., 2003, 2924 RSC.
- Z. Xu, Q. Li, L. Zhang and Y. Jia, J. Org. Chem., 2009, 74, 6859 CrossRef CAS PubMed.
- (a) Q. Liu and Y. Jia, Org. Lett., 2011, 13, 4810 CrossRef CAS PubMed; (b) Z. Xu, F. Zhang, L. Zhang and Y. Jia, Org. Biomol. Chem., 2011, 9, 2512 RSC; (c) Y. Zhang, Q. Liu, C. Wang and Y. Jia, Org. Lett., 2013, 15, 3662 CrossRef CAS PubMed; (d) Z. Xu, W. Hu, Q. Liu, L. Zhang and Y. Jia, J. Org. Chem., 2010, 75, 7626 CrossRef CAS PubMed.
- (a) J. Ku, B. Jeong, S. Jew and H. Park, J. Org. Chem., 2007, 72, 8115 CrossRef CAS PubMed; (b) J. Li, T. Mei and J. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452 CrossRef CAS PubMed; (c) Q. Liu, Q. Li, Y. Ma and Y. Jia, Org. Lett., 2013, 15, 4528 CrossRef CAS PubMed.
- Y.-K. Tahara, M. Ito, K. S. Kanyiva and T. Shibata, Chem. – Eur. J., 2015, 21, 11340 CrossRef CAS PubMed.
- (a) J. Robbers and H. Floss, Tetrahedron Lett., 1969, 10, 1857 CrossRef; (b) G. King, P. Mantle, C. Szczyrbak and E. Waight, Tetrahedron Lett., 1973, 14, 215 CrossRef; (c) G. S. King, E. Waight, P. Mantle and C. Szczyrbak, J. Chem. Soc., Perkin Trans. 1, 1977, 2099 RSC; (d) J. Robbers, H. Otsuka, H. Floss, E. Arnold and J. Clardy, J. Org. Chem., 1980, 45, 1117 CrossRef CAS.
- (a) R. Bartholomaus, F. Dommershausen, M. Thiele, N. S. Karanjule, K. Harms and U. Koert, Chem. – Eur. J., 2013, 19, 7423 CrossRef PubMed; (b) J. Schwaben, J. Cordes, K. Harms and U. Koert, Synthesis, 2011, 2929 CAS.
- Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, in Topics in Organometallic Chemistry, ed. U. Kazmaier, Springer, Heidelberg, 2012, vol. 38 Search PubMed.
- (a) R. Takeuchi and M. Kashio, Angew. Chem., Int. Ed. Engl., 1997, 36, 263 CrossRef CAS; (b) J. P. Janssen and G. Helmchen, Tetrahedron Lett., 1997, 38, 8025 CrossRef CAS; (c) T. Ohmura and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 15164 CrossRef CAS PubMed; (d) F. Giacomina, D. Riat and A. Alexakis, Org. Lett., 2010, 12, 1156 CrossRef CAS PubMed; (e) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461 CrossRef CAS PubMed; (f) M. Grtner, S. Mader, K. Seehafer and G. Helmchen, J. Am. Chem. Soc., 2011, 133, 2072 CrossRef PubMed; (g) K.-Y. Ye, H. He, W.-B. Liu, L.-X. Dai, G. Helmchen and S.-L. You, J. Am. Chem. Soc., 2011, 133, 19006 CrossRef CAS PubMed; (h) W. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 15249 CrossRef CAS PubMed; (i) W.-B. Liu, C. M. Reeves, S. C. Virgil and B. M. Stoltz, J. Am. Chem. Soc., 2013, 135, 10626 CrossRef CAS PubMed; (j) W.-B. Liu, C. M. Reeves and B. M. Stoltz, J. Am. Chem. Soc., 2013, 135, 17298 CrossRef CAS PubMed; (k) W. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 377 CrossRef CAS PubMed; (l) M. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 2014, 53, 12172 CrossRef CAS PubMed; (m) W. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2068 CrossRef CAS PubMed; (n) C.-X. Zhuo, C. Zheng and S.-L. You, Acc. Chem. Res., 2014, 47, 2558 CrossRef CAS PubMed; (o) X. Zhang, Z.-P. Yang, L. Huang and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 1873 CrossRef CAS PubMed; (p) J. Y. Hamilton, D. Sarlah and E. M. Carreir, Angew. Chem., Int. Ed., 2015, 54, 7644 CrossRef CAS PubMed; (q) P. Tosatti, A. Nelson and S. P. Marsden, Org. Biomol. Chem., 2012, 10, 3147 RSC.
- J. Ruchti and E. M. Carreira, J. Am. Chem. Soc., 2014, 136, 16756 CrossRef CAS PubMed.
- M. Kusano, G. Sotoma, H. Koshino, J. Uzawa, M. Chijimatsu, S. Fujioka, T. Kawano and Y. J. Kimura, J. Chem. Soc., Perkin Trans. 1, 1998, 2823 RSC.
- Y. Kimura, T. Hamasaki, H. Nakajima and A. Isogai, Tetrahedron Lett., 1982, 23, 225 CrossRef CAS.
- C. J. Barrow and H. H. Sun, J. Nat. Prod., 1994, 57, 471 CrossRef CAS.
- K. Matsumura and T. Kitahara, Heterocycles, 2001, 54, 727 CrossRef CAS.
- C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed.
- (a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (c) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258 CrossRef CAS PubMed; (d) J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS PubMed.
- (a) Z.-P. Yang, Q.-F. Wu and S.-L. You, Angew. Chem., Int. Ed., 2014, 53, 6986 CrossRef CAS PubMed; (b) Z.-P. Yang, Q.-F. Wu, W. Shao and S.-L. You, J. Am. Chem. Soc., 2015, 137, 15899 CrossRef CAS PubMed; (c) X. Zhang, L. Han and S.-L. You, Chem. Sci., 2014, 5, 1059 RSC.
- Y. Ito, E. Nakajo, M. Nakatsuka and T. Saegusa, Tetrahedron Lett., 1983, 24, 2881 CrossRef CAS.
- J. W. Daly, B. Witkop, T. Tokuyama, T. Nishikawa and I. L. Karle, Helv. Chim. Acta, 1977, 60, 1128 CrossRef CAS PubMed.
- (a) J. W. Daly, Fortschr. Chem. Org. Naturst., 1982, 41, 205 CrossRef CAS PubMed; (b) C. Souccar, W. A. Varanda, J. W. Daly and E. X. Albuquerque, Mol. Pharmacol., 1984, 25, 384 CAS; (c) C. Souccar, W. A. Varanda, R. S. Aronstam, J. W. Daly and E. X. Albuquerque, Mol. Pharmacol., 1984, 25, 395 CAS.
- D. H. R. Barton and M. Samadi, Tetrahedron, 1992, 48, 7083 CrossRef CAS.
- (a) M. D. Hill, Chem. – Eur. J., 2010, 16, 12052 CrossRef CAS PubMed; (b) Y. Nakao, Synthesis, 2011, 3209 CrossRef CAS; (c) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642 CrossRef CAS PubMed; (d) D. Best and H. W. Lam, J. Org. Chem., 2014, 79, 831 CrossRef CAS PubMed.
- X.-J. Liu and S.-L. You, Angew. Chem., Int. Ed., 2017, 56, 4002 CrossRef CAS PubMed.
- S. Fukumoto, E. Imamiya, K. J. Kusumoto, S. Fujiwara, T. Watanabe and M. Shiraishi, J. Med. Chem., 2002, 45, 3009 CrossRef CAS PubMed.
- K. Ishiuchi, T. Kubota, H. Morita and J. Kobayashi, Tetrahedron Lett., 2006, 47, 3287 CrossRef CAS.
- T. Lei, H. Zhang and Y.-R. Yang, Tetrahedron Lett., 2015, 56, 5933 CrossRef CAS.
- A. G. Kozlovskii, T. F. Soloveva, V. G. Sahkarovskii and V. M. Adanin, Dokl. Akad. Nauk SSSR, 1981, 260, 230 CAS.
- (a) S. Krishnan, J. T. Bagdanoff, D. C. Ebner, Y. K. Ramtohul, U. K. Tambar and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 13745 CrossRef CAS PubMed; (b) D. C. Behenna, S. Krishnan and B. M. Stoltz, Tetrahedron Lett., 2011, 52, 2152 CrossRef CAS.
- K. C. Nicolaou, S. A. Snyder, X. H. Huang, K. B. Simonsen, A. E. Koumbis and A. Bigot, J. Am. Chem. Soc., 2004, 126, 10162 CrossRef CAS PubMed.
- S. Suetsugu, H. Nishiguchi, C. Tsukano and Y. Takemoto, Org. Lett., 2014, 16, 996 CrossRef CAS PubMed.
- X.-W. Yang, C.-P. Yang, L.-P. Jiang, X.-J. Qin, Y.-P. Liu, Q.-S. Shen, Y.-B. Chen and X.-D. Luo, Org. Lett., 2014, 16, 5808 CrossRef CAS PubMed.
- X. Liang, S.-Z. Jiang, K. Wei and Y.-R. Yang, J. Am. Chem. Soc., 2016, 138, 2560–2562 CrossRef CAS PubMed.
- (a) J. Y. Hamilton, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2013, 135, 994 CrossRef CAS PubMed; (b) O. F. Jeker, A. G. Kravina and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 12166 CrossRef CAS PubMed; (c) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065 CrossRef CAS PubMed; (d) J. Y. Hamilton, N. Hauser, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2014, 53, 10759 CrossRef CAS PubMed; (e) C. Defieber, H. Grützmacher and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4482 CrossRef CAS PubMed; (f) C. Defieber, M. A. Ariger, P. Moriel and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 3139 CrossRef CAS PubMed.
- G. Subramaniam, O. Hiraku, M. Hayashi, T. Koyano, K. Komiyama and T.-S. Kam, J. Nat. Prod., 2007, 70, 1783 CrossRef CAS PubMed.
- S.-Z. Jiang, X.-Y. Zeng, X. Liang, T. Lei, K. Wei and Y.-R. Yang, Angew. Chem., Int. Ed., 2016, 55, 4044 CrossRef CAS PubMed.
- V. Van Rheenen, Tetrahedron Lett., 1969, 10, 985 CrossRef.
- M. Teng, W. Zi and D. Ma, Angew. Chem., Int. Ed., 2014, 53, 1814 CrossRef CAS PubMed.
- (a) A. Numata, C. Takahashi, Y. Ito, T. Takada, K. Kawai, Y. Usami, E. Matsumura, M. Imachi, T. Ito and T. Hasegawa, Tetrahedron Lett., 1993, 34, 2355 CrossRef CAS; (b) R. Jadulco, R. A. Edrada, R. Ebel, A. Berg, K. Schaumann, V. Wray, K. Steube and P. Proksch, J. Nat. Prod., 2004, 67, 78 CrossRef CAS PubMed; (c) H. Hayashi, H. Matsumoto and K. Akiyama, Biosci., Biotechnol., Biochem., 2004, 68, 753 CrossRef CAS PubMed; (d) B. Andersen, J. Smedsgaard and J. C. Frisvad, J. Agric. Food Chem., 2004, 52, 2421 CrossRef CAS PubMed; (e) P. W. Dalsgaard, J. W. Blunt, M. H. G. Munro, J. C. Frisvad and C. Christophersen, J. Nat. Prod., 2005, 68, 258 CrossRef CAS PubMed.
- (a) J. Yang, H. Wu, L. Shen and Y. Qin, J. Am. Chem. Soc., 2007, 129, 13794 CrossRef CAS PubMed; (b) P. Liu, J. H. Seo and S. M. Weinreb, Angew. Chem., Int. Ed., 2010, 49, 2000 CrossRef CAS PubMed; (c) J. H. Seo, P. Liu and S. M. Weinreb, J. Org. Chem., 2010, 75, 2667 CrossRef CAS PubMed; (d) Z. Zuo, W. Xie and D. Ma, J. Am. Chem. Soc., 2010, 132, 13226 CrossRef CAS PubMed; (e) Z. Zuo and D. Ma, Angew. Chem., Int. Ed., 2011, 50, 12008 CrossRef CAS PubMed; (f) J. Belmar and R. L. Funk, J. Am. Chem. Soc., 2012, 134, 16941 CrossRef CAS PubMed; (g) S. P. Lathrop, M. Pompeo, W.-T. T. Chang and M. Movassaghi, J. Am. Chem. Soc., 2016, 138, 7763 CrossRef CAS PubMed; (h) S.-J. Han, F. Vogt, S. Krishnan, J. A. May, M. Gatti, S. C. Virgil and B. M. Stoltz, Org. Lett., 2014, 16, 3316 CrossRef CAS PubMed.
- X. Liang, T.-Y. Zhang, X.-Y. Zeng, Y. Zheng, K. Wei and Y.-R. Yang, J. Am. Chem. Soc., 2017, 139, 3364 CrossRef CAS PubMed.
- M. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 13972 CrossRef CAS PubMed.
- (a) O. Dirat, A. J. Bibb, A. C. M. Burns, G. D. Checksfield, B. R. Dillon, S. E. Field, S. J. Fussell, S. P. Green, C. Mason, J. Mathew, S. Mathew, I. B. Moses, P. I. Nikiforov, A. J. Pettman and F. Susanne, Org. Process Res. Dev., 2011, 15, 1010 CrossRef CAS; (b) S. J. Desai, K. M. Patel, A. M. Shah, R. S. Pada and H. M. Makwana, PCT Int. Appl, WO2011141932A2, 2011 Search PubMed; (c) S. Mantegazza, P. Allegrini and E. Attolino, Eur. Pat. Appl, EP2316817A1, 2011 Search PubMed; (d) A. Lorente Bonde-Larsen, F. J. Gallo Nieto, J. J. Ferreiro Gil and P. Martin Pascual, PCT Int. Appl, WO2013113946A2, 2013 Search PubMed; (e) O. Piccolo, E. Giannini, L. Bigini, E. Gianolli and D. Vigo, PCT Int. Appl, WO2014012832A1, 2014 Search PubMed.
- J. M. Müller and C. B. W. Stark, Angew. Chem., Int. Ed., 2016, 55, 4798 CrossRef PubMed.
- (a) R. M. Williams, E. M. Stocking and J. F. Sanz-Cervera, Top. Curr. Chem., 2000, 209, 97 CrossRef CAS; (b) D. Crich and A. Banerjee, Acc. Chem. Res., 2007, 40, 151 CrossRef CAS PubMed; (c) M. A. Schmidt and M. Movassaghi, Synlett, 2008, 313 CAS; (d) S.-M. Li, Nat. Prod. Rep., 2010, 27, 57 RSC; (e) T. Lindel, N. Marsch and S. K. Adla, Top. Curr. Chem., 2011, 309, 67 CrossRef PubMed.
- (a) S. Takase, M. Iwami, T. Ando, M. Okamoto, K. Yoshida, H. Horiai, M. Kohsaka II, H. Aoki and H. Imanaka, J. Antibiot., 1984, 37, 1320 CrossRef CAS PubMed; (b) F. S. de Guzmanj, J. B. Gloer, D. T. Wicklow and P. F. Dowd, J. Nat. Prod., 1992, 55, 931 CrossRef; (c) K. Ishikawa, T. Hosoe, T. Itabashi, D. Wakana, K. Takizawa, T. Yaguchi and K. Kawai, Chem. Pharm. Bull., 2010, 58, 717 CrossRef CAS PubMed.
- (a) B. N. Reddy and C. V. Ramana, Tetrahedron, 2017, 73, 888 CrossRef CAS; (b) G. Franck, K. Broedner and G. Helmchen, Org. Lett., 2010, 12, 3886 CrossRef CAS PubMed; (c) G. Satyanarayana, D. Pflaesterer and G. Helmchen, Eur. J. Org. Chem., 2011, 6877 CrossRef CAS and references therein.
- (a) R. A. Yoder and J. N. Johnston, Chem. Rev., 2005, 105, 4730 CrossRef CAS PubMed; (b) K. U. Wendt, G. E. Schulz, E. J. Corey and D. R. Liu, Angew. Chem., Int. Ed., 2000, 39, 2812 CrossRef CAS; (c) R. Ardkhean, D. F. J. Caputo, S. M. Morrow, H. Shi, Y. Xiong and E. A. Anderson, Chem. Soc. Rev., 2016, 45, 1557 RSC.
- S. G. Sethofer, T. Mayer and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8276 CrossRef CAS PubMed.
- O. F. Jeker, A. G. Kravina and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 12166 CrossRef CAS PubMed.
- H.-F. Sun, X.-M. Li, L. Meng, C.-M. Cui, S.-S. Gao, C.-S. Li, C.-G. Huang and B.-G. Wang, J. Nat. Prod., 2012, 75, 148 CrossRef CAS PubMed.
- G. Majetich and J. M. Shimkus, J. Nat. Prod., 2010, 73, 284 CrossRef CAS PubMed.
- J. Deng, S. Zhou, W. Zhang, J. Li, R. Li and A. Li, J. Am. Chem. Soc., 2014, 136, 8185 CrossRef CAS PubMed.
- J. Deng, R. F. Li, Y. J. Luo, J. Li, S. P. Zhou, Y. J. Li, J. Y. Hu and A. Li, Org. Lett., 2013, 15, 2022 CrossRef CAS PubMed.
- P. H. J. Carlsen, T. Katsuki, V. S. Martin and K. B. Sharpless, J. Org. Chem., 1981, 46, 3936 CrossRef CAS.
- S. Zhou, H. Chen, Y. Luo, W. Zhang and A. Li, Angew. Chem., Int. Ed., 2015, 54, 6878 CrossRef CAS PubMed.
- H. E. Ortega, P. R. Graupner, Y. Asai, K. Tendyke, D. Qiu, Y. Shen, N. Rios, A. E. Arnold, P. D. Coley, T. A. Kursar, W. H. Gerwick and L. Cubilla-Rios, J. Nat. Prod., 2013, 76, 741 CrossRef CAS PubMed.
- S. Yildizhan, J. van Loon, A. Sramkova, M. Ayasse, C. Arsene, C. Ten Broeke and S. Schulz, ChemBioChem, 2009, 10, 1666 CrossRef CAS PubMed.
- (a) K. Yamada, T. Kubo, H. Tokuyama and T. Fukuyama, Synlett, 2002, 231 CrossRef CAS; (b) M. Pineschi, F. Bertolini, P. Crotti and F. Macchia, Org. Lett., 2006, 8, 2627 CrossRef CAS PubMed.
- For application of iridium-catalyzed allylic amination in natural product synthesis, see: (a) J. Hoecker, G. C. Ruldolf, F. Baechle, S. Fleischer, B. D. Lindner and G. Helmchen, Eur. J. Org. Chem., 2013, 5149 CrossRef CAS; (b) G. Satyanarayana, D. Pflaesterer and G. Helmchen, Eur. J. Org. Chem., 2011, 6877 CrossRef CAS; (c) M. Gaertner, R. Weihofen and G. Helmchen, Chem. – Eur. J., 2011, 17, 7605 CrossRef CAS PubMed; (d) C. Gnamm, K. Broedner, C. M. Krauter and G. Helmchen, Chem. – Eur. J., 2009, 15, 10514 CrossRef CAS PubMed; (e) A. Farwick and G. Helmchen, Org. Lett., 2010, 12, 1108 CrossRef CAS PubMed.
- (a) P. W. Tan, J. Seayad and D. J. Dixon, Angew. Chem., Int. Ed., 2016, 55, 13436 CrossRef CAS PubMed; (b) Y. Nakayama, Y. Maeda, M. Kotatsu, R. Sekeya, M. Ichiki, T. Sato and N. Chida, Chem. – Eur. J., 2016, 22, 3300 CrossRef CAS PubMed.
- (a) R. S. Sulake and C. Chen, Org. Lett., 2016, 17, 1138 CrossRef PubMed; (b) R. Ding, J.-G. Fu, G.-Q. Xu, B.-F. Sun and G.-Q. Lin, J. Org. Chem., 2014, 79, 240 CrossRef CAS PubMed; (c) N. Tanaka, T. Suzuki, T. Matsumura, Y. Hosoya and M. Nakada, Angew. Chem., Int. Ed., 2009, 48, 2580 CrossRef CAS PubMed; (d) J. Jauch, C. Wallner and E. Herdtweck, Eur. J. Org. Chem., 2003, 3060 CrossRef CAS; (e) I. R. Baxendale, A.-L. Lee and S. V. Ley, J. Chem. Soc., Perkin Trans. 1, 2002, 1850 RSC.
- T. Suzuki, K. Ghozati, D.-Y. Zhou, T. Katoh and H. Sasai, Tetrahedron, 2010, 66, 7562 CrossRef CAS.
- Photoredox catalysis and its application have been well summarized in elegant reviews: (a) M. D. Karkas, J. A. Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683 CrossRef CAS PubMed; (b) T. P. Nicholls, D. Leonori and A. C. Bissember, Nat. Prod. Rep., 2016, 33, 1248 RSC; (c) F. Tan and W. Xiao, Acta Chim. Sin., 2015, 73, 85 CrossRef CAS; (d) T. M. Monos and C. R. J. Stephenson, Iridium(III) in Optoelectronic and Photonics Applications, John Wiley & Sons Ltd, 1st edn, 2017, p. 541 Search PubMed. For elegant work in natural product total synthesis with photoredox catalysis, see: (e) S. Inuki, K. Sato, T. Fukuyama, I. Ryu and Y. Fujimoto, J. Org. Chem., 2017, 82, 1248 CrossRef CAS PubMed; (f) S. Hwang, I. Baek and C. Lee, Org. Lett., 2016, 18, 2154 CrossRef CAS PubMed; (g) J. W. Beatty and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 10270 CrossRef CAS PubMed; (h) Z. Ma, X. i. Wang, X. Wang, R. A. Rodriguez, C. E. Moore, S. Gao, X. Tan, Y. Ma, A. L. Rheingold, P. S. Baran and C. Chen, Science, 2014, 346, 219 CrossRef CAS PubMed; (i) Y. Sun, R. Li, W. Zhang and A. Li, Angew. Chem., Int. Ed., 2013, 52, 9201 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2018 |