
Electron-accepting π-conjugated species with 1,8-naphthalic anhydride or diketophosphanyl units†
Sergio
Sánchez
 a,
Alva Yuen Yiu
Woo
a and
Thomas
Baumgartner
*ab
a,
Alva Yuen Yiu
Woo
a and
Thomas
Baumgartner
*ab
aDepartment of Chemistry, Centre for Advanced Solar Materials, University of Calgary, 2500 University Drive NW, Calgary, AB T2N 1N4, Canada. E-mail: tbaumgar@yorku.ca
bDepartment of Chemistry, York University, 4700 Keele Street, Toronto, ON M3J 1P3, Canada
First published on 24th August 2017
Abstract
The synthesis and characterization of a series of 3- or 4-substituted 1,8-naphthalic anhydride compounds, and their organophosphorus analogues, bearing a cyclic diketophosphanyl moiety, are reported. The new anhydrides have been synthesized by Stille coupling with a variety of aryl groups with different donor strengths from the corresponding 3- and 4-bromo substituted anhydrides, respectively. The thienyl- and (dimethylamino)phenyl-substituted anhydrides have moreover been used as precursors for the synthesis of the corresponding diketophosphanyl derivatives by reaction with MesP(SiMe3)2. The photophysical and electrochemical properties of the new compounds have been studied. Density functional theory (DFT) and time-dependent DFT calculations have been carried out in order to rationalize their electronic structures and electronic absorption properties. The low-energy absorption bands exhibit mainly (π → π*) charge-transfer character from the aryl moieties to the anhydride or diketophosphanyl core, respectively. In the thiophene-containing diketophosphanyl compounds the low-energy absorption also shows some electron-transfer features from the phosphorus moiety (MesP) to the core. The charge-transfer nature of the transition has been confirmed by solvatochromic absorption and emission studies.
Introduction
1,8-Naphthalic anhydride (Fig. 1a) has been well-known for decades, mostly as an intermediate for the formation of different derivatives of 1,8-naphthalimide (Fig. 1b).1 1,8-Naphthalimide derivatives have attracted much attention, owing to their intriguing optical, thermal and chemical stabilities, as well as their high photoluminescence quantum efficiencies. Consequently, they have been widely used in high-tech fields such as laser diodes, solar-energy conversion, optical brighteners and liquid crystal displays.2–8 Moreover, 1,8-naphthalimide derivatives have shown success as anticancer agents and as potential imaging agents for cellular environments and markers for cancers.9–15 Surprisingly, far less attention has been given to the 1,8-naphthalic anhydride derivatives, mostly due to their hydrolyzation in water and in any other protic solvents.16 As a result, only very limited photophysical and electronic properties have been reported.17–19 The functionalization of 1,8-naphthalimide derivatives with electron-donating substituents produces a significant change in their photophysical and electrochemical properties.20–23 It is therefore expected that, due to the similar backbone between the 1,8-naphthalic anhydride and 1,8-naphthalimide, the synthesis and study of functionalized 1,8-naphthalic anhydrides would lead to a new series of compounds with desirable properties as well. Due to the electron-accepting characteristics of their core, these materials could be useful towards the advancement of organic electronics; anhydrides in general, have proven to be interesting materials for application in this field. Prominent examples are 3,4,9,10-perylenetetracarboxylicdianhydride (PTCDA),24,25 which is an archetypal crystalline molecular organic semiconductor, or 1,4,5,8-naphthalenetetracarboxylic dianhydride (NTCDA) which is a class of n-type organic semiconductor.26,27 Another class of anhydride-based materials that have been intensively investigated are the perylene monoimide dyes, where only one of the two anhydrides is converted to an imide, and they have been studied as sensitizers for solar cell applications.28,29 Great application potentials are revealed for these types of compounds, despite the lower solar cell efficiency relative to the traditional polypyridyl ruthenium complex-based cell.30,31 | ||
| Fig. 1 General structures of 1,8-naphthalic anhydride (a); 1,8-naphthalimide (b); 1,8-naphthalic diketophosphanyl (c). | ||
On the other hand, the development of organophosphorus π-conjugated systems has increasingly been attracting attention because of the intriguing physicochemical properties, induced by the incorporation of phosphorus functionalities into carbon rich π-conjugated frameworks.32–34 Within this field, aromatic-fused diketophosphanyl compounds, which can be seen as phosphorus analogues of aromatic-fused imide compounds such as phthalimides, have not attracted as much attention as other π-conjugated organophosphorus compounds.35 Cowley et al.36 reported the first diketophosphanyl analogue of 1,8-naphthalimide in 1987 (Fig. 1c). However, only NMR data was reported and photophysical and electronic properties were not investigated. The authors did, however, confirm the reactivity of the phosphorus lone pair in the adopted pyramidal geometry by preliminary reactivity studies. In 2013, our group reported the first diketophosphanyl derivatives and evaluating their utility for organic electronics by studying their physicochemical properties.37,38 Subsequently, Takeda, Minakata et al. reported a series of diketophosphanyl compounds as phosphorus analogues of aromatic diimides and pyrromelytic diimides that showed excellent electron-accepting abilities.35,39 The addition of the organophosphorus center gives rise not only to profound effects on the electronic properties of the materials, it also introduces a new site for functionalization on the phosphorus atom. The comparative study evaluating the replacement of the bridging oxygen atom of naphthalic anhydrides by a phosphanyl group, due to their larger electron-accepting abilities of the 2,3-naphthalene-fused diketophosphanyl compounds, could thus lead to a further understanding of the physicochemical properties of organophosphorus molecules oriented toward organic electronics applications.
Herein, we disclose the syntheses, characterization, and physicochemical properties (UV/vis absorption, luminescence profiles and electrochemical behavior) of functionalized 1,8-naphthalic anhydrides 1–10 as well as their P-functionalized congeners 11–14. Extension of the core has been achieved by functionalizing the 3- and 4-positions of 1,8-naphthalic anhydride with different π-donor/acceptor groups. DFT and TD-DFT calculations have been performed for all compounds in order to obtain a better understanding of their properties.
Experimental section
Materials and procedures
Reactions and manipulations were all carried out under dry nitrogen atmosphere employing standard Schlenk techniques. Solvents were dried via distillation and using appropriate drying agents or were used directly from an MBraun solvent purification system. All chemical reagents and NMR solvents were purchased from commercial sources and were, unless otherwise noted, used without further purification. Dry ice/acetone (−78 °C) and water/ice (0 °C) baths were used when low temperature reaction conditions were required, as well as for cooling receiving flasks during reagent addition. The compounds 3-bromo-1,8-napthalenedicarboxylic anhydride,40 4-bromo-1,8-napthalenedicarboxylic anhydride,41,42 3-phenyl-1,8-napthalenedicarboxylic anhydride (1),43 4-phenyl-1,8-napthalenedicarboxylic anhydride (6),43 MesP(SiMe3)239 and the stannylated precursor molecules44–52 were prepared according to the literature.General physical measurements
1H-NMR, 13C{1H}-NMR, and 31P{1H}-NMR were recorded on Bruker Avance (-II, -III) 400 MHz spectrometers. Chemical shifts were referenced to external 85% H3PO4 (31P), and external TMS (13C, 1H) through residual 1H and 13C resonance(s) of the employed solvents: CDCl3 (7.24 ppm) and CD2Cl2 (5.32 ppm) for 1H, and CDCl3 (77.23 ppm) and CD2Cl2 (53.5 ppm) for 13C spectra. Fluorescence and UV-vis experiments were recorded in dichloromethane (DCM) solution using a Jasco FP-6600 spectrofluorometer and a Cary 5000 UV-vis-NIR spectrophotometer, respectively. Atmospheric-pressure chemical ionization (APCI) and electrospray ionization (ESI) mass spectra were collected on a Bruker Esquire 3000 and on an Agilent 6520 Q-TOF instrument. APCI and ESI analyses were performed by Mr Wade White at the Department of Chemistry at the University of Calgary. Cyclic voltammetry was performed on an Autolab PGSTAT302 instrument, with a polished carbon electrode as the working electrode, a Pt wire as the counter electrode, and Ag/AgCl/KCl3M as the reference electrode, using ferrocene/ferrocenium (Fc/Fc+) as the internal standard. Cyclic voltammetry experiments were performed in DCM with tetrabutylammonium hexafluorophosphate, NBu4PF6 (0.1 M) as the supporting electrolyte.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) to yield the corresponding anhydride. The remaining powder was further subjected to multiple recrystallizations with acetic anhydride to afford a bright yellow powder (0.52 g, 52%). 1H NMR (400 MHz, CD2Cl2): δ = 8.88 (d, 4J = 2.0 Hz, 1H), 8.59 (dd, 4J = 1.2 Hz, 3J = 7.2 Hz, 1H), 8.53 (d, 4J = 2.0 Hz, 1H), 8.38 (ddd, 4J = 0.4 Hz, 4J = 1.2 Hz 3J = 8.4 Hz, 1H), 7.87 (dd, 3J = 7.2 Hz, 3J = 8.4 Hz, 1H), 7.67 (dd, 4J = 1.2 Hz 3J = 3.6 Hz 1H), 7.52 (dd, 4J = 1.2 Hz 3J = 5.2 Hz 1H), 7.25 (dd, 3J = 3.6 Hz 3J = 5.2 Hz 1H) ppm; 13C{1H} NMR (100 MHz, CD2Cl2): δ = 160.5 (s), 160.4 (s), 141.5 (s), 135.2 (s), 133.9 (s), 132.7 (s), 132.4 (s), 131.1 (s), 130.4 (s), 129.2 (s), 128.7 (s), 128.1 (s), 127.0 (s), 125.4 (s), 119.5 (s), 118.8 (s) ppm; HRMS (APCI): m/z calcd for C16H8O3S ([M + H]+): 281.02669; found: 281.02584.
:3) to yield the corresponding anhydride. The remaining powder was further subjected to multiple recrystallizations with acetic anhydride to afford bright fluffy greenish-golden needles (0.26 g, 52%). 1H NMR (400 MHz, CD2Cl2): δ = 8.81 (dd, 4J = 1.2 Hz, 3J = 8.4 Hz, 1H), 8.70 (dd, 4J = 1.2 Hz, 3J = 7.2 Hz, 1H), 8.65 (d, 3J = 7.6 Hz, 1H), 7.94 (d, 3J = 7.6 Hz, 1H), 7.89 (dd, 3J = 7.2 Hz, 3J = 8.4 Hz, 1H), 7.68 (dd, 4J = 1.2 Hz 3J = 5.2 Hz 1H), 7.47 (dd, 4J = 1.2 Hz, 3J = 3.6 Hz, 1H), 7.33 (dd, 3J = 3.6 Hz, 3J = 5.2 Hz 1H) ppm; 13C{1H} NMR (100 MHz, CDCl3): δ = 160.6 (s), 160.4 (s), 140.8 (s), 139.0 (s), 133.9 (s), 133.5 (s), 132.8 (s), 131.0 (s), 130.2 (s), 129.3 (s), 129.0 (s), 128.3 (s), 128.1 (s), 127.7 (s), 119.1 (s), 117.9 (s) ppm; HRMS (APCI): m/z calcd for C16H8O3S ([M + H]+): 281.02669; found: 281.02712.
:3) to yield the corresponding anhydride. The remaining powder was further subjected to multiple recrystallizations with acetic anhydride to afford a bright yellow powder (0.52 g, 52%). 1H NMR (400 MHz, CD2Cl2): δ = 8.88 (d, 4J = 2.0 Hz, 1H), 8.59 (dd, 4J = 1.2 Hz, 3J = 7.2 Hz, 1H), 8.53 (d, 4J = 2.0 Hz, 1H), 8.38 (ddd, 4J = 0.4 Hz, 4J = 1.2 Hz 3J = 8.4 Hz, 1H), 7.87 (dd, 3J = 7.2 Hz, 3J = 8.4 Hz, 1H), 7.67 (dd, 4J = 1.2 Hz 3J = 3.6 Hz 1H), 7.52 (dd, 4J = 1.2 Hz 3J = 5.2 Hz 1H), 7.25 (dd, 3J = 3.6 Hz 3J = 5.2 Hz 1H) ppm; 13C{1H} NMR (100 MHz, CD2Cl2): δ = 160.5 (s), 160.4 (s), 141.5 (s), 135.2 (s), 133.9 (s), 132.7 (s), 132.4 (s), 131.1 (s), 130.4 (s), 129.2 (s), 128.7 (s), 128.1 (s), 127.0 (s), 125.4 (s), 119.5 (s), 118.8 (s) ppm; HRMS (APCI): m/z calcd for C16H8O3S ([M + H]+): 281.02669; found: 281.02584.
:3) to yield the corresponding anhydride. The remaining powder was further subjected to multiple recrystallizations with acetic anhydride to afford bright fluffy greenish-golden needles (0.26 g, 52%). 1H NMR (400 MHz, CD2Cl2): δ = 8.81 (dd, 4J = 1.2 Hz, 3J = 8.4 Hz, 1H), 8.70 (dd, 4J = 1.2 Hz, 3J = 7.2 Hz, 1H), 8.65 (d, 3J = 7.6 Hz, 1H), 7.94 (d, 3J = 7.6 Hz, 1H), 7.89 (dd, 3J = 7.2 Hz, 3J = 8.4 Hz, 1H), 7.68 (dd, 4J = 1.2 Hz 3J = 5.2 Hz 1H), 7.47 (dd, 4J = 1.2 Hz, 3J = 3.6 Hz, 1H), 7.33 (dd, 3J = 3.6 Hz, 3J = 5.2 Hz 1H) ppm; 13C{1H} NMR (100 MHz, CDCl3): δ = 160.6 (s), 160.4 (s), 140.8 (s), 139.0 (s), 133.9 (s), 133.5 (s), 132.8 (s), 131.0 (s), 130.2 (s), 129.3 (s), 129.0 (s), 128.3 (s), 128.1 (s), 127.7 (s), 119.1 (s), 117.9 (s) ppm; HRMS (APCI): m/z calcd for C16H8O3S ([M + H]+): 281.02669; found: 281.02712.
Theoretical methods
Density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations were undertaken by using the Gaussian 09 software.53 Studies were performed on complexes 1–14 by using the functional B3LYP54 with the 6-31G(d) basis set.Results and discussion
Synthesis and characterization
The synthetic routes to the 1,8-naphthalic anhydrides 1–10 and the diketophosphanyl compounds 11–14 are illustrated in Schemes 1 and 2, respectively.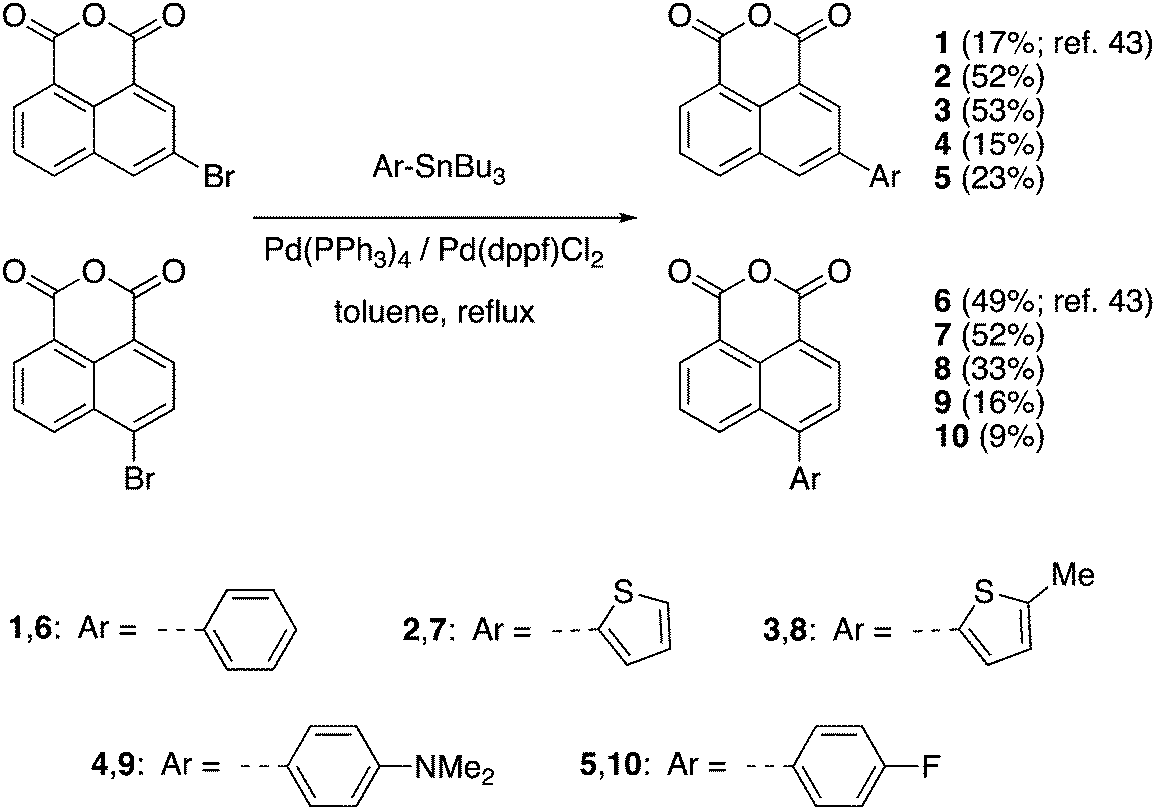 | ||
| Scheme 1 Synthesis of 3- and 4-π-extended 1,8-naphthalic anhydrides 1–10. | ||
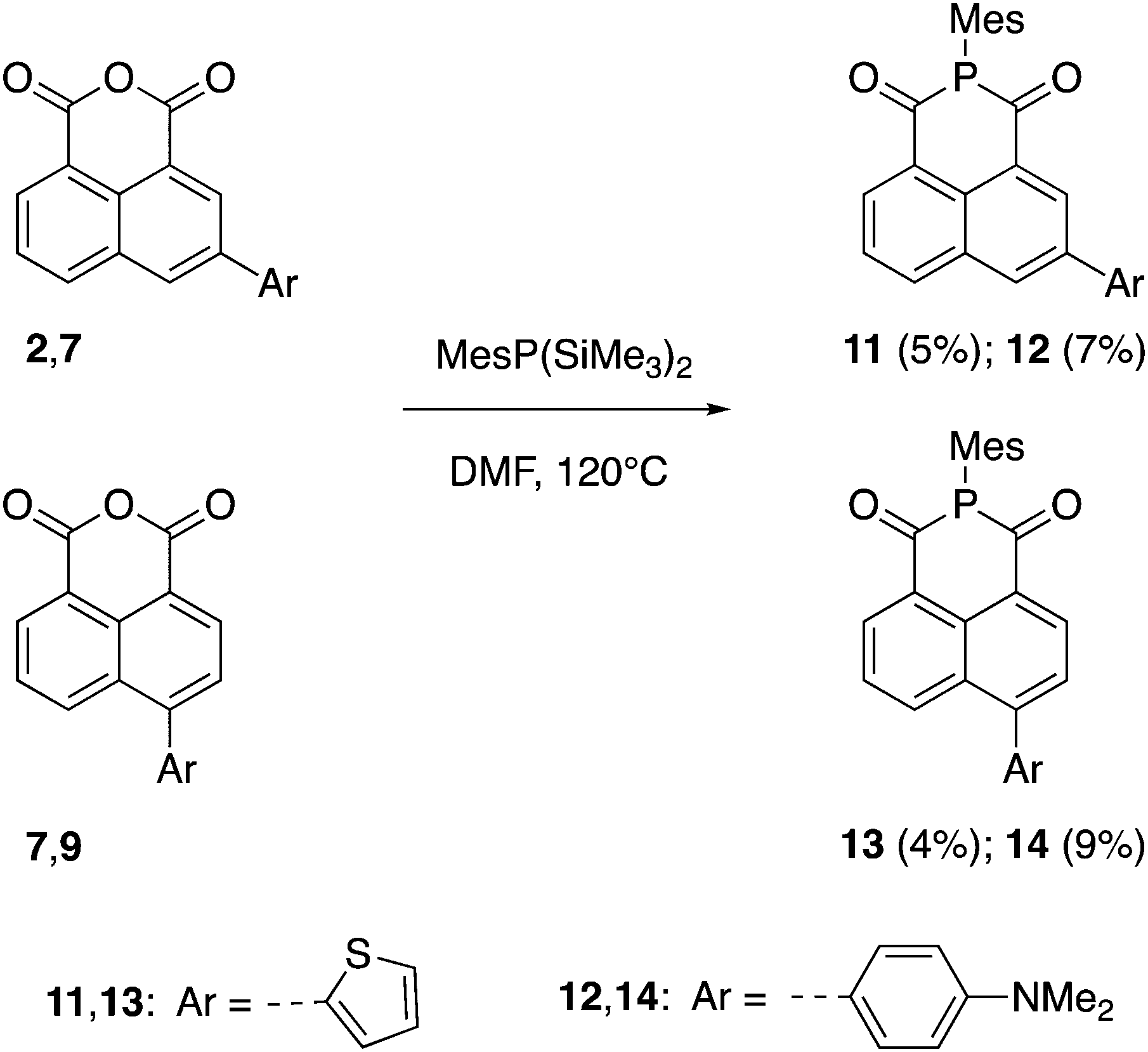 | ||
| Scheme 2 Synthesis of the diketophosphanyl species 11–14. | ||
It is important to note that compounds 1 and 6 have been reported before, but they have only been investigated for their potential to be incorporated into biological applications.43 The general synthesis towards π-extended 1,8-naphthalic anhydrides involved brominating the anhydride core at the corresponding 3- and 4-positions and, via subsequent Stille coupling, the desired extended products were obtained in low to moderate yields (9–53%). All synthesized functionalized 1,8-naphthalic anhydrides were purified differently, but ultimately via multiple recrystallizations from acetic anhydride due to their reduced solubility in many organic solvents. Diketophosphanyl compounds 11–14 were synthesized following the same procedure used by Takeda, Minakata et al.,35 by reaction of 1,8-naphthalic anhydrides with MesP(SiMe3)2, although the obtained yield was low for all compounds (4–9%).
The choice of mesityl group on the P center was due to the kinetic and thermodynamic stabilities of both of disilylphosphane reagent and resulting diketophosphanyl products against air and moisture, as established by Takeda, Minakata et al.35 In fact, when the reaction was attempted using PhP(SiMe3)2 instead of MesP(SiMe3)2 the desired product was observed by 31P and 1H NMR spectroscopy, however, together with many other side-products and could not be isolated. The air- and moisture-stable products were fully characterized by 1H (1–14), 13C{1H} (1–10,14) and 31P{1H} (11–14) NMR spectroscopy (Fig. S1–S25, ESI†), and high-resolution mass spectrometry (HRMS), except for 11–13, of which an insufficient quantity for the 13C{1H} characterization was available. All diketophosphanyl products showed a sharp 31P{1H} NMR singlet in CDCl3 at a similar region (δ = 24.5–26.5 ppm) and generally a slightly improved solubility over the corresponding anhydride precursors.
Photophysical properties
UV/vis absorption and photoluminescence spectra of CH2Cl2 solutions (10−5 M) of all compounds are depicted in Fig. 2 and 3 and the photophysical data are shown in Table 1.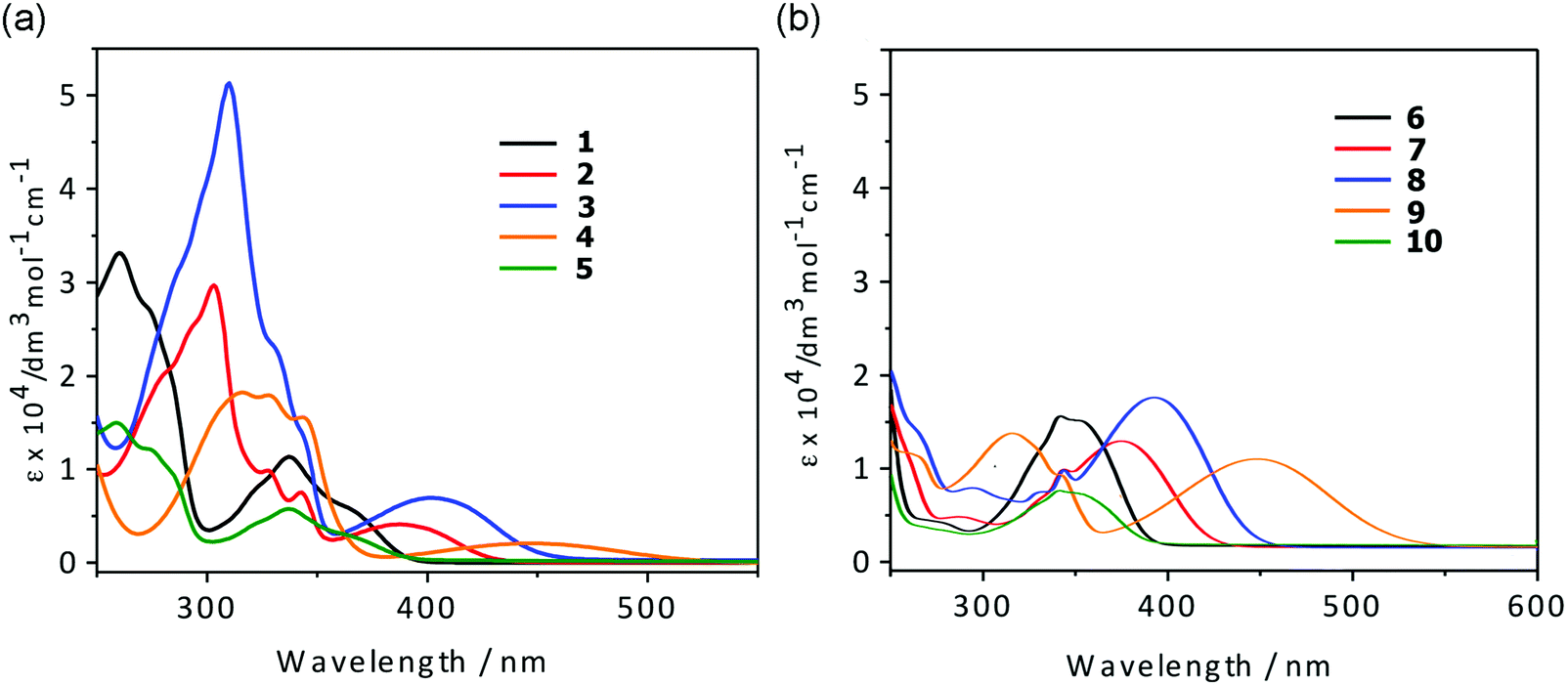 | ||
| Fig. 2 UV-vis spectra of compounds 1–10 in CH2Cl2 (c = 5.0 × 10−5 M). | ||
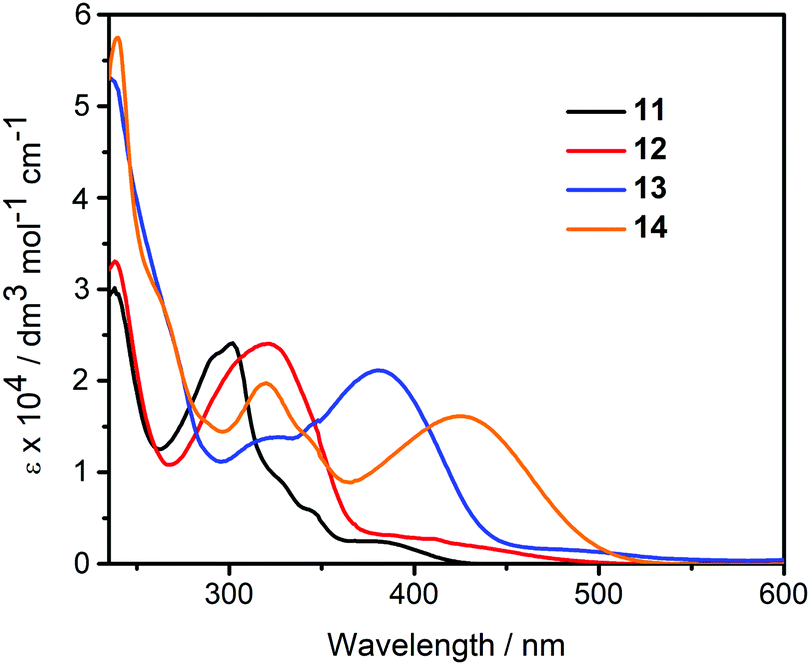 | ||
| Fig. 3 UV-vis spectra of compounds 11–14 in CH2Cl2 (c = 5.0 × 10−5 M). | ||
| Compound | λ abs [nm] (εmax [dm3 mol−1 cm−1])a | λ em [nm] | Φ PL |
|---|---|---|---|
| a Measured in CH2Cl2. b Photoluminescence quantum yields in CH2Cl2, relative to quinine sulfate. | |||
| 1 | 260 (33.1), 337 (11.3) | 415 | 0.17 |
| 2 | 303 (29.7), 388 (4.1) | 463 | 0.13 |
| 3 | 310 (51.3), 401 (6.9) | 495 | 0.20 |
| 4 | 316 (18.2), 328 (17.9), 343 (15.6), 446 (2.1) | 662 | 0.01 |
| 5 | 259 (15.0), 337 (5.8) | 418 | 0.15 |
| 6 | 342 (14.00) | 422 | 0.83 |
| 7 | 344 (8.3), 375 (11.3) | 468 | 0.46 |
| 8 | 344 (8.2), 393 (16.0) | 493 | 0.51 |
| 9 | 316 (12.2), 448 (9.4) | 638 | 0.28 |
| 10 | 341 (6.0) | 424 | 0.56 |
| 11 | 301 (35.9), 326 (14.1), 383 (3.7) | 465 | 0.09 |
| 12 | 238 (33.0), 319 (23.9), 409 (2.85) | 613 | 0.08 |
| 13 | 236 (53.2), 319 (13.8), 382 (21.1) | 496 | 0.35 |
| 14 | 239 (57.6), 319 (19.6), 425 (16.2) | 619 | 0.29 |
All compounds show a high-energy absorption band at ca. 240–340 nm and a low-energy band at ca. 340–450 nm. Comparison of the lower-energy absorption bands (λmax) of the anhydride derivatives 1–10 show the trend 4-(dimethylamino)phenyl (446, 448 nm) > 5-methylthienyl (401, 393 nm) > thienyl (388, 375 nm) > phenyl and 4-fluorophenyl (337–342 nm), which is consistent with the electron-donating abilities of the aromatic moieties.
To support the presence of a charge-transfer transition resulting from the donor–acceptor architecture within the molecules, solvatochromic absorption and emission studies in representative solvents of different polarity (hexane, CH2Cl2, CH3OH, and DMF) were carried out. As shown in Fig. S26–S33 and Table S1 (see ESI†), the low-energy absorption bands are shifted when changing the solvent, confirming their charge-transfer nature. The solvatochromic effect is not very pronounced (ca. 10–15 nm), indicating that both the ground and the excited states are not very polar. With regard to the effect of the substitution pattern on the intramolecular charge transfer, the 3-substituted compounds tend to exhibit a slightly stronger intramolecular charge transfer than the 4-substituted compounds.
The TD-DFT calculations performed for the anhydride derivatives (vide infra) support the low-energy absorption bands in dichloromethane mainly involving HOMO → LUMO (that is, π → π*) transitions, with some transference from the aryl moieties to the 1,8-naphthalic anhydride core. The energies of these transitions are within the same range of low-energy absorption bands of 1,8-naphthalimide derivatives (330–500 nm). This suggests that they are photophysically similar, likely due to having a similar electron-deficient core. The UV-vis spectroscopic data indicate that the 3- and 4-substitued compounds have comparable broadness in the low-energy absorption band, but the 3-substituted compounds have a much lower molar absorptivity.
The diketophosphanyl derivatives 11–14 show the same trend for the low-energy absorption bands 4-(dimethylamino)phenyl (409, 425 nm) > thienyl (382, 375 nm), with the aminophenyl derivatives showing the lowest-energy absorption. The incorporation of the phosphorus center produces a clear blue-shift when compared to their anhydride precursors in the aniline-containing compounds (Δλmax = 37 nm for 4vs.12 and 23 nm for 9vs.14), whereas in the thienyl-containing compounds there is a small blue-shift in the 3-substituted (Δλmax = 5 nm for 2vs.11) and a small red-shift in the 4-substituted species (Δλmax = 7 nm for 7vs.13). Notably, the extinction coefficients for the P-containing 4-substituted derivatives are twice as large and those of the corresponding anhydrides, whereas the 3-substituted derivatives show comparable values for all four species. However, both absorption peak values are red-shifted when compared to the non-substituted parent, reported by Takeda, Minakata et al. (λabs = 235 nm and 321 nm) as would be expected.35
According to TD-DFT, the low-energy transitions for 11–14 are mainly HOMO → LUMO (π → π*) transitions, that is, from the aryl moieties to the diketophosphanyl core. As shown in Table S1 (ESI†), the lowest-energy transition of 11 and 13 show also some transference from the MesP fragment to the core. In 11, there is a small contribution (7%) from HOMO-2 → LUMO, while there is a significant transference from HOMO−1 → LUMO (17%) in 13. This could explain the small blue shift and the red shift observed when compared to the low-energy absorption of their precursors, respectively.
Due to the low isolated yields obtained for most of the diketophospanyl compounds, solvatochromic absorption studies were performed only for the 4-substituted aminophenyl-containing derivative 14, as a proof-of-concept to confirm the nature of the low-energy band. The observed shift of the low-energy absorption band is more pronounced for 14 than for the anhydride precursor 9 (Table S1 and Fig. S34; ESI†), suggesting a higher polarity in the excited state, and a stronger intramolecular charge transfer.
The emission of the anhydride derivatives 1–10 in dichloromethane solutions (Fig. 4 and Table 1) show that the emission energies follow the trend consistent with the electron-donating abilities of the aromatic substituents 4-(dimethylamino)phenyl (662, 638 nm) > 5-methylthienyl (495, 493 nm) > thienyl (463, 468 nm) > phenyl and 4-fluorophenyl (415–424 nm). This indicates the decreased energy gap due to stronger electron-donating abilities, which is consistent with the theoretical calculations, as well as with the observations seen in the UV-vis absorption profiles mentioned above. Somewhat surprisingly, when the same aryl group is attached at different positions of the backbone, they emit at very similar wavelengths. However, this feature is supported by their calculated frontier molecular orbitals, exhibiting essentially similar LUMOs for all species and only slight differences in their HOMO–LUMO gaps for about 0.1 eV in favor of the 3-substituted family of derivatives (see ESI†).
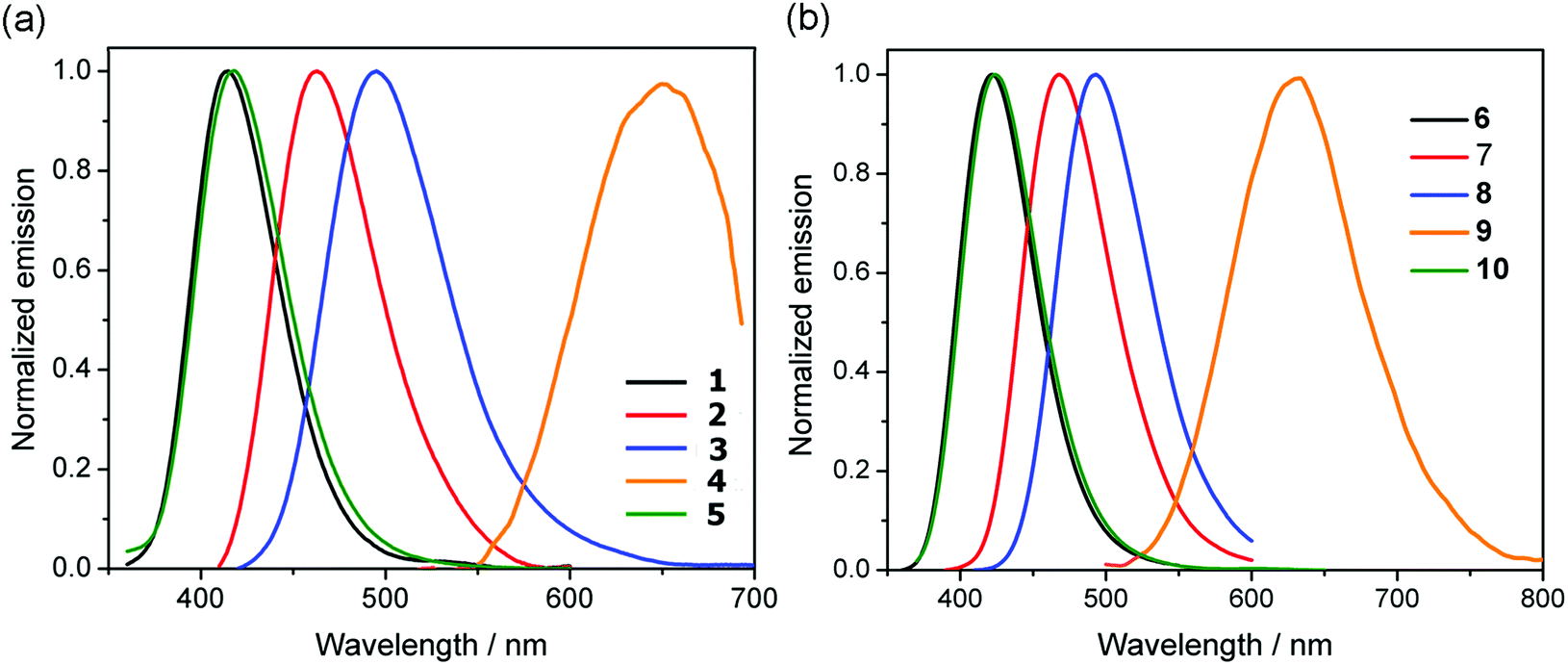 | ||
| Fig. 4 Emission spectra of compounds 1–10 in CH2Cl2 (c = 5.0 × 10−5 M). | ||
On the other hand, the quantum yields (Φ) of the 4-substituted compounds (6–10) have significantly higher values than the 3-substitued derivatives (1–5). The quantum yields for the 3-substituted range from 0.01 to 0.20 while for the 4-series range from 0.28 to 0.83. This observation was also seen in 3- and 4-substituted 1,8-naphthalimides, where 3-substituted naphthalimides have fluorescence quantum yields ranging from 0.03 to 0.24,55 and 4-substituted naphthalimides having quantum yields as high as 0.84.23,56,57 In the 1,8-naphthalimides, when the backbone is functionalized at the 4-position with electron-donating substituents, these compounds have decreased excited-state energy, red-shifted fluorescence spectrum and substantially increased florescence yields.20 These observations were attributed to the donors being in direct conjugation with the carbonyl groups of the molecule. Because 1,8-naphthalimides and 1,8-naphthalic anhydrides are quite similar, the same reasoning could be why 1,8-naphthalic anhydrides show different quantum yields depending on the position of the substituents.
Solvatochromic absorption and emission studies in representative solvents of different polarity (hexane, CH2Cl2, CH3OH, and DMF) were carried out for all anhydrides except the electron-acceptor 4-fluorophenyl substituted compounds 5 and 10. As shown in Table S1 (ESI†), the solvatochromism of all compounds is more pronounced in the emission wavelengths than in the lowest-energy absorption bands, supporting distinct differences in polarity for the various excited states. As can be seen in Fig. 5, the charge-transfer effect for the 1,8-naphthalic anhydride compounds is expectedly more efficient when strong donor groups are attached to the core with the largest red shift in the aminophenyl-substituted compounds (Δλem = 150 nm 4 and Δλem = 129 nm 9), leading to a distinctly charge-separated excited state.
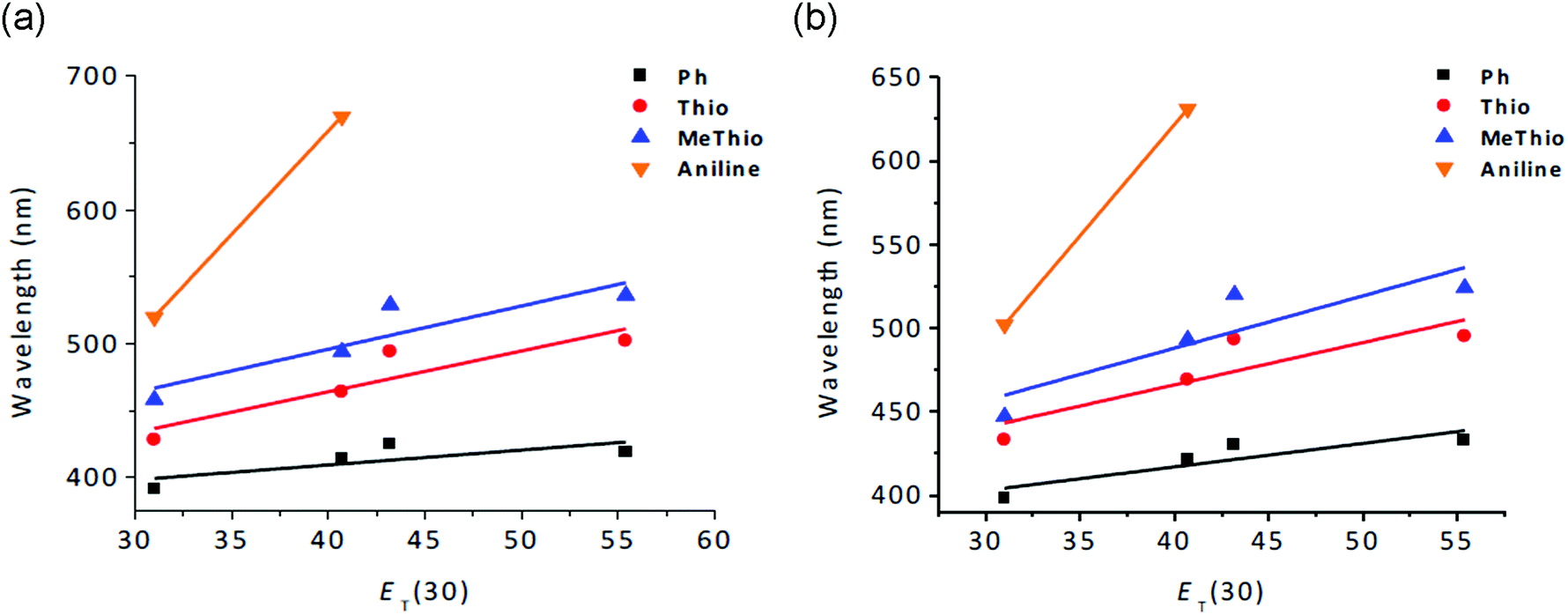 | ||
| Fig. 5 Solvatochromic shifts in the emission of 3-substituted (a) and 4-substituted (b) 1,8-naphthalic anhydrides at different ET(30) values: Hex: 31.0, DCM: 40.7, DMF: 43.2, and MeOH: 55.4. | ||
As shown in Fig. 6, the diketophosphanyl derivatives (11–14) show the same trend for the emission energies (4-(dimethylamino)phenyl (684, 619 nm) > thienyl (465, 496 nm)) as their anhydride precursors. Notably, the non-substituted parent species reported by Takeda and Minakata is non-fluorescent at room temperature, but becomes emissive at 77 K with a well-resolved spectrum showing clear vibronic fine structure (λem = 532 nm, 577 nm).35 The apparent wavelength inconsistencies with the new π-extended species can be attributed to incompatible experimental parameters (solvent, temperature), and/or different relaxation pathways.
 | ||
| Fig. 6 Emission spectra of compounds 11–14 in CH2Cl2 (c = 5.0 × 10−5 M). | ||
Again, similar emission wavelengths are observed in the isomeric derivatives with the same substituent, in agreement with the calculated frontier molecular orbitals (vide infra). As observed for the anhydrides, the quantum yields found for the 4-substituted derivatives are larger than for the 3-substitued relatives. Solvatochromic emission studies performed on 14 show a clear positive shift of the emission wavelength with increasing polarity of the solvent (Table S1, ESI†), however, they seem to be counter-balanced by the hydrogen-bonding ability of the solvent, ultimately leading to a blue shift in DMF and more so in MeOH, when compared to the emission in CH2Cl2. Interestingly however, when comparing the shift observed in the energy emission from hexane to dichloromethane with its precursor (Δλem = 142 nm for 14; Δλem = 129 nm for 9), a larger positive solvatochromism is observed in 14, supporting a larger charge reorganization from the donor to the acceptor unit in their excited state.
Theoretical calculations
In order to obtain a better understanding of the photophysical and electronic properties of the synthesized compounds, density functional theory (DFT) calculations (B3LYP/6-31G(d)) were performed and their frontier orbital energies were determined.53 The HOMO and LUMO energy levels and orbital diagrams of compounds 1–10 are shown in Fig. 7 and Fig. S35 (see ESI†), the HOMO–LUMO gaps (Table S2, ESI†) and other relevant frontier molecular orbitals are depicted in Fig. S36 and S37 (ESI†). While some of the theoretically calculated values do not exactly match the experimental values, both series show more or less the same trends with reasonable agreement. The HOMO is delocalized over the extended conjugated system in all compounds except for the aniline derivatives, where it mostly resides in the aminophenyl moiety. The LUMOs in all compounds mainly reside in the localized π* orbital of the 1,8-naphthalic anhydride core explaining why the calculated LUMO values were similar. Compared to the Ph-substituted compounds, all the other compounds have nearly identical LUMO levels; only the aminophenyl derivatives show very slight stabilization of the LUMO by 0.02–0.06 eV, respectively.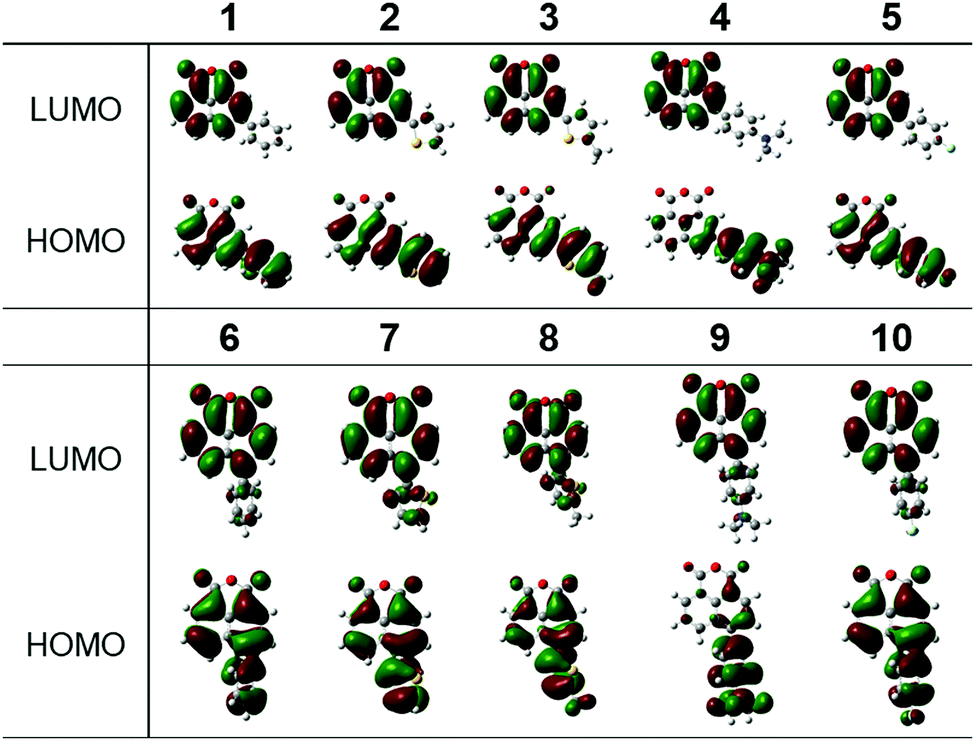 | ||
| Fig. 7 Frontier molecular orbital surfaces for compounds 1–10. | ||
Regarding the HOMO energy levels, all compounds have higher lying HOMOs compared to Ph-substituted derivatives, which suggests that most other substituents destabilize the HOMO, as expected. The absolute value of the shift of the HOMO level appears to be governed by the strength of the electronic coupling between the substituting group and the backbone. Overall, when compared to the Ph-substituted congeners, there is a decrease in the energy gap (Eg = HOMO–LUMO). Eg follows the trend 4-(dimethylamino)phenyl (2.63 eV, 2.76 eV) < 5-methylthienyl (3.24 eV, 3.35 eV) < thienyl (3.38 eV, 3.48 eV) < 4-fluorophenyl (3.65 eV, 3.74 eV) < phenyl (3.70 eV, 3.77 eV). The HOMO and LUMO energy levels and orbital diagrams of the diketophosphanyl derivatives (11–14) are shown in Fig. S38 and S39 (ESI†). A similar trend as for their anhydride precursors with regard to Eg is observed for these derivatives (4-(dimethylamino)phenyl: 3.06 eV, 3.24 eV < thienyl: 3.62 eV, 3.70 eV). The energy of the HOMOs in these compounds is similar to their precursors while the LUMOs are destabilized in the diketophosphanyl derivatives, therefore increasing the Eg. As shown in Fig. S39 (ESI†), the frontier orbitals also suggest possible intramolecular charge transfer since the HOMO resides within the donor substituents, while the LUMO is mainly localized in the electron-deficient naphthalic core, as a result of the pronounced donor–acceptor architecture.
To gain more insight into the optical absorption properties, time-dependent DFT (TD-DFT) calculations were carried out using a dichloromethane environment; the significant transitions are shown in Table S3 (ESI†). The low-energy absorption bands in the anhydride compounds 1–10 seem to mainly involve HOMO → LUMO (i.e., π → π*) transitions. Although the calculated lowest-energy transitions are red-shifted compared to the experimental UV-vis data, the energy of these bands follow the same trend (4-(dimethylamino)phenyl < 5-methylthienyl < thienyl < 4-fluorophenyl < phenyl) as observed experimentally. The high-energy absorption bands, on the other hand, involve the HOMO → LUMO+1/LUMO+2 transitions, composed of the π* orbital of the aryl moieties. The lowest-energy transitions for the diketophosphanyl compounds 11–14 are mainly composed of HOMO → LUMO (i.e., π → π*) transitions as well. As previously mentioned, in the thiophene-containing derivatives there is some transference from the MesP fragment to the core, that is, from HOMO−2 → LUMO in 11, and HOMO−1 → LUMO in 13. These calculated transitions are blue-shifted compared to their anhydride precursors, following the same trend observed in their UV-vis spectra.
Electrochemistry
The results of cyclic voltammetry (CV) measurements on compounds 1–14 are included in Table 2 and representative voltammograms are shown in Fig. 8 (and Fig. S40, ESI†).| Compound | E red,1/2 [V] | LUMOb [eV] | LUMOb [eV] | E red,1/2 [V] | Compound |
|---|---|---|---|---|---|
| a Potentials versus ferrocene/ferrocenium (Fc/Fc+). b Calculated using the ferrocene HOMO level at −(4.8 + Ered1/2) eV. | |||||
| 1 | E red1 = −1.66 | −3.14 | −3.14 | E red1 = −1.66 | 6 |
| E red2 = −2.29 | E red2 = −2.28 | ||||
| 2 | E red1 = −1.62 | −3.18 | −3.00 | E red1 = −1.80 | 7 |
| E red2 = −2.25 | E red2 = −2.51 | ||||
| 3 | E red1 = −1.63 | −3.17 | −3.19 | E red1 = −1.61 | 8 |
| E red2 = −2.25 | E red2 = −2.18 | ||||
| 4 | E red1 = −1.66 | −3.14 | −3.16 | E red1 = −1.64 | 9 |
| E red2 = −2.30 | E red2 = −2.30 | ||||
| E ox = 0.53 | E ox = 0.61 | ||||
| 5 | E red1 = −1.60 | −3.20 | −3.18 | E red1 = −1.62 | 10 |
| E red2 = −2.24 | E red2 = −2.25 | ||||
| 11 | E red1 = −1.65 | −3.15 | −3.20 | E red1 = −1.60 | 13 |
| E red2 = −2.20 | E red2 = −2.15 | ||||
| 12 | E red1 = −1.80 | −3.00 | −3.01 | E red1 = −1.79 | 14 |
| E ox = −2.20 | E red2 = −2.18 | ||||
| E ox = 0.47 | E ox = 0.55 | ||||
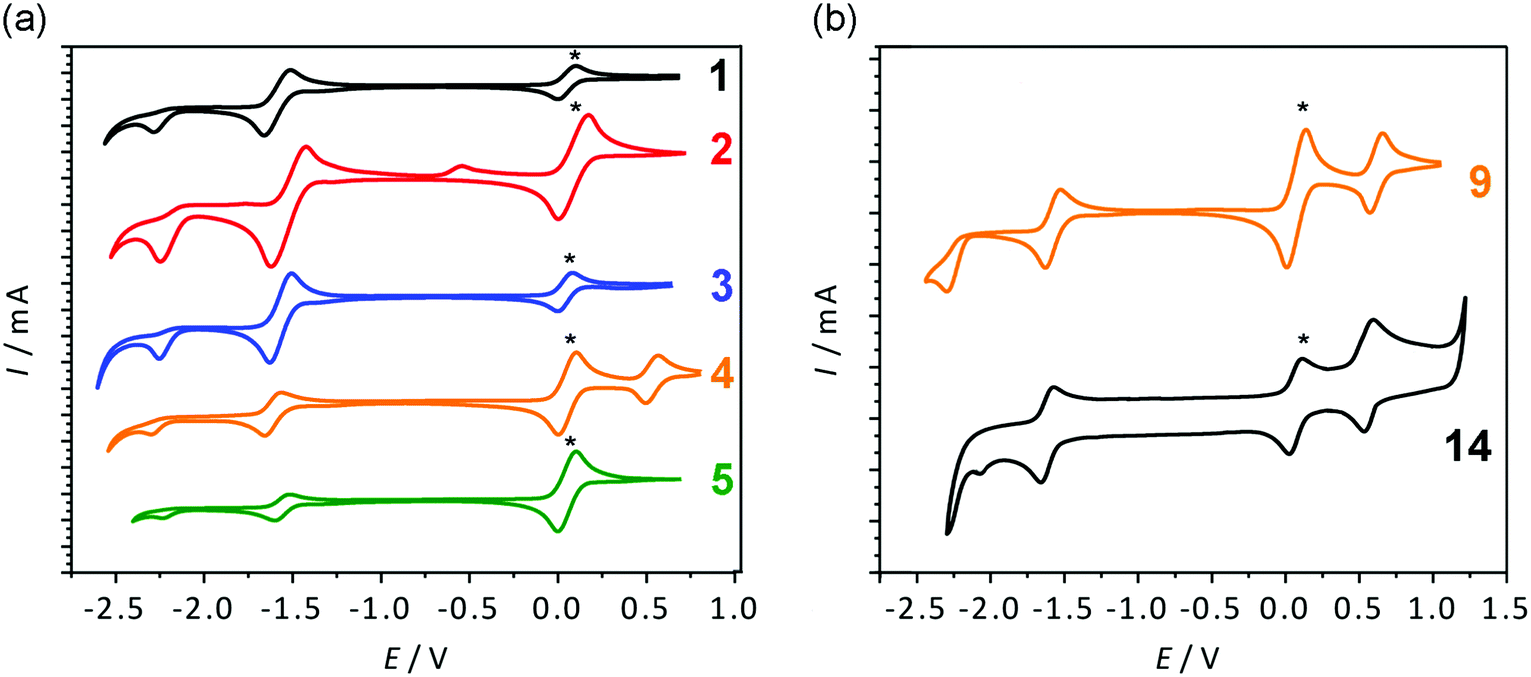 | ||
| Fig. 8 Cyclic voltammograms of compounds 1–5 (a) and 9 and 14 (b) in CH2Cl2 solution containing 0.1 M tetrabutylammonium hexafluorophosphate as the supporting electrolyte. Scan rate = 100 mV s−1. Ferrocene (*) was added as an internal standard and referenced to 0 V. | ||
The anhydride compounds 1–10 show a reversible and an irreversible reduction peak, respectively. Only the aminophenyl-containing compounds (4 and 9) show an oxidation peak attributed to the amine oxidation. From Table 2 it is evident that each corresponding pair of compounds has very similar Ered1 and Ered2 peaks, suggesting that their LUMO energy levels are also very similar, as supported by the theoretical calculations. The only exception is the thiophene-containing compounds 2 and 7, where the pair differs by 0.18 eV. A possible reason could be the thiophene being in a different orientation with respect to the core and thus causing a change in electronic communication from the donor to the acceptor group. Electronic communication could be more efficient in the 3-position and therefore have a lower LUMO energy level. This is supported by the theoretical calculations, where a smaller torsion angle is observed between the thiophene and the core in the 3-substituted compound (22.4°) compared to the 4-substituted relative (46.7°). Cyclic voltammetry shows that the position of substitution, whether in direct conjugation with the carbonyl group or not, has very little effect in the electrochemical properties (and the optical properties, vide supra) of these materials. Notwithstanding, all compounds have considerable electron-accepting character, as well as appreciable electrochemical stability.
The diketophosphanyl compounds 11–14 show a similar profile as the anhydride compounds, with a reversible and an irreversible reduction peak, as well as one oxidation peak for the aminophenyl-substituted derivatives. As previously observed with the anhydride precursors, the thiophene-containing derivatives 11 and 12 and the aminophenyl-substituted species 13 and 14 show similar values for their reduction peaks, as supported by the similar calculated LUMO energies. Direct comparison of the anhydride and diketophosphanyl derivatives with the same substituents reveals distinct differences for the phosphorus-analogues with regard to their electrochemical properties. The CV data suggest that most compounds have a lower LUMO than their precursors, and consequently should show increased electron-accepting abilities. The 4-substituted thiophene-containing 12, however, shows the opposite behavior, with a higher electrochemically determined LUMO than its precursor. This notable effect in the electrochemical properties contrasts with the similar redox behavior found in the related 1,8-naphthalimide and its P-analogue bearing a mesityl group on the N or P center, respectively (−1.84 V vs. Fc+/Fc and −1.85 V vs. Fc+/Fc).35
Conclusions
From the photophysical and electronic properties of the 1,8-naphthalic anhydride derivatives studied in this paper, they were found to exhibit relatively low-lying LUMO energy levels, good electron-accepting character, and good chemical stability. All compounds show emission at room temperature that can be tuned by the electron-donating abilities of the aromatic substituents. In addition, the observed photophysical properties were comparable to the better-known 1,8-naphthalimide class of compounds. However, the LUMO energy levels reported for the imide analogues in the literature tend to be lower (i.e., they are better electron acceptors) than the anhydride analogues in this study. On the other hand, the incorporation of an organophosphorus center that converts the backbone into a cyclic diketophosphanyl moiety, changes the electronic properties of the material. When compared to their anhydride precursors, the electrochemical data show a considerable effect on the LUMO energy upon incorporation of a P-center, suggesting that further research on new phosphorus-containing electron-accepting compounds could lead to promising materials towards organic electronics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canada Foundation for Innovation (CFI) is gratefully acknowledged. S. S. and A. Y. Y. W. thank the University of Calgary for an Eyes High postdoctoral fellowship and a QE-II graduate scholarship, respectively. We also thank Dr T. C. Sutherland for access to his instrumentation.Notes and references
- G. H. Zhang, L. J. Liu and W. L. Sun, Adv. Mater. Res., 2012, 393–395, 1359–1362 CAS.
- C. Thalacker, C. Röger and F. Würthner, J. Org. Chem., 2006, 71, 8098–8105 CrossRef CAS PubMed.
- C. Röger and F. Würthner, J. Org. Chem., 2007, 72, 8070–8075 CrossRef PubMed.
- W. W. Stewart, Cell, 1978, 14, 741–759 CrossRef CAS PubMed.
- G.-L. Tu, C.-Y. Mei, Q.-G. Zhou, Y.-X. Cheng, Y.-H. Geng, L.-X. Wang, D.-G. Ma, X.-B. Jing and F.-S. Wang, Adv. Funct. Mater., 2006, 16, 101–106 CrossRef CAS.
- I. Grabchev, I. Moneva, V. Bojinov and S. Guittonneau, J. Mater. Chem., 2000, 10, 1291–1296 RSC.
- D. Kolosov, V. Adamovich, P. Djurovich, M. E. Thompson and C. Adachi, J. Am. Chem. Soc., 2002, 124, 9945–9954 CrossRef CAS PubMed.
- I. Grabchev, I. Moneva, V. Bojinov and S. Guittonneau, J. Mater. Chem., 2000, 10, 1291–1296 RSC.
- B. Swagata, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC.
- P. Quintana-Espinoza, J. García-Luis, Á. Amesty, P. Martín-Rodríguez, I. Lorenzo-Castrillejo, A. G. Ravelo, L. Fernández-Pérez, F. Machín and A. Estévez-Braun, Bioorg. Med. Chem., 2013, 21, 6484–6495 CrossRef CAS PubMed.
- S. V. Bhosale, S. V. Bhosale and S. K. Bhargava, Org. Biomol. Chem., 2012, 10, 6455–6468 CAS.
- M. F. Braña and A. Ramos, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1, 237–255 CrossRef.
- M. Lv and H. Xu, Curr. Med. Chem., 2009, 16, 4797–4813 CrossRef CAS PubMed.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- G. J. Ryan, S. Quinn and T. Gunnlaugsson, Inorg. Chem., 2008, 47, 401–403 CrossRef CAS PubMed.
- T. C. Barros, S. Yunes, G. Menegon, F. Nome, H. Chaimovich, M. J. Politi, L. G. Dias and I. M. Cuccovia, J. Chem. Soc., Perkin Trans. 2, 2001, 2342–2350 RSC.
- S. Ghosh, S. Biswas, M. Mondal and S. Basu, J. Lumin., 2014, 145, 410–419 CrossRef CAS.
- E. Maltseva, S. Amirjalayer and W. J. Buma, Phys. Chem. Chem. Phys., 2017, 19, 5861–5869 RSC.
- A. Islam, C.-C. Cheng, S.-H. Chi, S. J. Lee, P. G. Hela, I.-C. Chen and C.-H. Cheng, J. Phys. Chem. B, 2005, 109, 5509–5517 CrossRef CAS PubMed.
- O. V. Prezhdo, B. V. Uspenskii, V. V. Prezhdo, W. Boszczyk and V. B. Distanov, Dyes Pigm., 2007, 72, 42–46 CrossRef.
- P. Kucheryavy, G. F. Li, S. Vyas, C. Hadad and K. D. Glusac, J. Phys. Chem. A, 2009, 113, 6453–6461 CrossRef CAS PubMed.
- V. Wintgens, P. Valat, J. Kossanyi, A. Demeter, L. Biczok and T. Berges, New J. Chem., 1996, 20, 1149–1158 CAS.
- M. S. Alexiou, V. Tychopoulos, S. Ghorbanian, J. H. P. Tyman, R. G. Brown and P. I. Brittain, J. Chem. Soc., Perkin Trans. 2, 1990, 837–842 RSC.
- S. R. Forrest, J. Phys.: Condens. Matter, 2003, 15, S2599–S2610 CrossRef CAS.
- D. Bellinger, J. Pflaum, C. Brüning, V. Engel. and B. Engels, Phys. Chem. Chem. Phys., 2017, 19, 2434–2448 RSC.
- A. Nollau, M. Pfeiffer, T. Fritz and K. Leo, J. Appl. Phys., 2000, 87, 4340–4343 CrossRef CAS.
- D. Qin, L. Chen, Y. Chen, J. Liu, G. Li, W. Quan, J. Zhang and D. Yan, Phys. Status Solidi A, 2012, 209, 790–794 CrossRef CAS.
- S. Ferrere, A. Zaban and B. A. Gregg, J. Phys. Chem. B, 1997, 101, 4490–4493 CrossRef CAS.
- G. Qi, R. Li, L. Wang and X. Li, J. Photochem. Photobiol., A, 2012, 239, 28–36 CrossRef CAS.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- H. Hoppe and N. S. Sariciftci, J. Mater. Res., 2004, 19, 1924–1945 CrossRef CAS.
- T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed.
- T. Baumgartner and C. Romero-Nieto, Synlett, 2013, 920–937 Search PubMed.
- A. Fukazawa and S. Yamaguchi, Chem. – Asian J., 2009, 4, 1386–1400 CrossRef CAS PubMed.
- Y. Takeda, T. Nishida, K. Hatanaka and S. Minakata, Chem. – Asian J., 2015, 21, 1666–1672 CAS.
- A. R. Barron, S. W. Hall and A. H. Cowley, J. Chem. Soc., Chem. Commun., 1987, 23, 1753–1754 RSC.
- X. He, J. A. Borau-Garcia, Y. Y. Woo, S. Trudel and T. Baumgartner, J. Am. Chem. Soc., 2013, 135, 1137–1147 CrossRef CAS PubMed.
- X. He and T. Baumgartner, Organometallics, 2013, 32, 7625–7628 CrossRef CAS.
- Y. Takeda, T. Nishida and S. Minakata, Chem. – Asian J., 2014, 20, 10266–10270 CAS.
- T. Kogiso, K. Yamamoto, H. Suemune and K. Usui, Org. Biomol. Chem., 2012, 10, 2934–2936 CAS.
- B. H. Rotstein, R. Mourtada, S. O. Kelley and A. K. Yudin, Chem. – Asian J., 2011, 17, 12257–12261 CAS.
- M. Shahid, P. Srivastava and A. Misra, New J. Chem., 2011, 35, 1690–1700 RSC.
- M. F. Braña, A. Gradillas, A. Gómez, N. Acero, F. Llinares, D. Muñoz-Mingarro, C. Abradelo, F. Rey-Stolle, M. Yuste, J. Campos, M. Á. Gallo and A. Espinosa, J. Med. Chem., 2004, 47, 2236–2242 CrossRef PubMed.
- S. Zeng, L. Yin, C. Ji, X. Jiang, K. Li, Y. Li and Y. Wang, Chem. Commun., 2012, 48, 10627–10629 RSC.
- A. V. Coelho, A. L. F. de Souza, P. G. de Lima, J. L. Wardell and O. A. C. Antunes, Appl. Organomet. Chem., 2008, 22, 39–42 CrossRef CAS.
- I. Palamà, F. Di Maria, I. Viola, E. Fabiano, G. Gigli, C. Bettini and G. Barbarella, J. Am. Chem. Soc., 2011, 133, 17777–17785 CrossRef PubMed.
- K. H. Kim, K. Watanabe, D. Menzel and H.-J. Freund, J. Am. Chem. Soc., 2009, 131, 1660–1661 CrossRef CAS PubMed.
- Y. Ren, Y. Dienes, S. Hettel, M. Parvez, B. Hoge and T. Baumgartner, Organometallics, 2009, 28, 734–740 CrossRef CAS.
- H. Jian and J. M. Tour, J. Org. Chem., 2003, 68, 5091–5103 CrossRef CAS PubMed.
- J. L. Sessler, M. Sathiosatham, K. Doerr, V. Lynch and K. A. Abboud, Angew. Chem., Int. Ed., 2000, 39, 1300–1303 CrossRef CAS PubMed.
- Z. Gu, L. Deng, H. Luo, X. Guo, H. Li, Z. Cao, X. Liu, X. Li, H. Huang, Y. Tan, Y. Pei and S. Tan, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3848–3858 CrossRef CAS.
- S. Pu, C. Zheng, Q. Sun, G. Liu and C. Fan, Chem. Commun., 2013, 49, 8036–8038 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. Pardo, E. Martin, J. M. L. Poyato, J. J. Camacho, M. F. Braña and J. M. Castellano, J. Photochem. Photobiol., A, 1987, 41, 69–78 CrossRef CAS.
- H. Shaki, K. Gharanjig, S. Rouhani and A. Khosravi, J. Photochem. Photobiol., A, 2010, 216, 44–50 CrossRef CAS.
- J. Kollár, P. Hrdlovič and Š. Chmela, J. Photochem. Photobiol., A, 2009, 204, 191–199 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Multinuclear NMR spectra, UV-vis and fluorescence spectra, solvatochromic data, cyclic voltammograms, DFT data (frontier orbitals, energy levels), and TD-DFT data (PDF); Cartesian coordinates for calculated structures (XYZ). See DOI: 10.1039/c7qm00336f |
| This journal is © the Partner Organisations 2017 |