
Direct correlation of PNIPAM thermal transition and magnetic resonance relaxation of iron oxide nanoparticles†
Nurul
Izza Taib
 ac,
Vipul
Agarwal
*ab,
Nicole M.
Smith
a,
Robert C.
Woodward
c,
Timothy G.
St. Pierre
c and
K. Swaminathan
Iyer
*a
ac,
Vipul
Agarwal
*ab,
Nicole M.
Smith
a,
Robert C.
Woodward
c,
Timothy G.
St. Pierre
c and
K. Swaminathan
Iyer
*a
aSchool of Molecular Sciences, The University of Western Australia, Crawley WA 6009, Australia. E-mail: swaminatha.iyer@uwa.edu.au
bDepartment of Materials Engineering, Indian Institute of Science, Bangalore, Karnataka 560012, India. E-mail: agarwalvipul84@gmail.com
cSchool of Physics, The University of Western Australia, Crawley WA 6009, Australia
First published on 1st August 2017
Abstract
Poly(N-isopropylacrylamide) (PNIPAM), which undergoes a temperature dependent transition from hydrophilic to hydrophobic, has played a crucial role in the development of stimuli-responsive multifunctional nanoparticles. In particular, iron oxide nanoparticles coated with PNIPAM have been effectively developed to enable stimuli responsive drug delivery and imaging agents. However, the PNIPAM transition from hydrophilic to hydrophobic at physiologically relevant temperatures renders colloidal nanoparticles unstable resulting in aggregation and precipitation from solution. Consequently, a direct correlative analysis of the effect of the thermally induced phase transition of PNIPAM on the magnetic resonance properties of nanoparticles has not been possible as the changes in proton relaxivity have been dominated by the colloidal agglomeration of the nanoparticles. Herein, we report colloidally stable thermoresponsive PNIPAM-grafted-PGMA coated magnetite core/shell nanoparticles (PNIPAM–PGMA–NPs) that enable the direct analysis of the effect of PNIPAM phase changes in solution on the overall magnetic resonance relaxivity of nanoparticles in suspension.
Magnetically responsive iron oxide nanomaterials have been a topic of intense research for applications ranging from sensing, to diagnostics, drug delivery and therapy.1–3 The ability to manipulate the system remotely using an external magnetic field offers a non-invasive trigger for on-demand drug release applications.4,5 Of the various nanomaterial composites that have been explored, polymer/iron oxide nanocomposites have been most widely exploited to engineer magnetic responsive materials that exhibit a high amplitude magneto-response.6 In particular, poly(N-isopropylacrylamide) (PNIPAM) based systems have been widely explored owing to their lower critical solution temperature (LCST) (∼32 °C), above which the polymer chains in aqueous solution undergo a phase transition from an expanded coil to a collapsed state, as a result of loss of hydration.7,8 Indeed, this property of the polymer in combination with the interesting property of magnetic materials to produce heat when exposed to an alternating magnetic field (AMF) has resulted in several core–shell (magnetic core/polymer shell) on-demand drug release nanomaterials.9–11 However, one of the major problems that is often encountered in PNIPAM coated colloidal systems is that the nanoparticles undergo aggregation above the LCST because of the loss of steric stabilization.12 This colloidal aggregation is accompanied by substantial shortening of the proton transverse relaxation times of aqueous suspensions of the iron oxide nanoparticles.12 As a consequence of the poor colloidal stability of the PNIPAM coated iron-oxide nanoparticles, a direct correlative analysis between the thermally induced transition of the polymer from extended chain to collapsed chains and its corresponding effect on the proton relaxivity has been poorly understood. In this communication, we report the use of iron oxide nanoparticles encapsulated within poly(glycidyl methacrylate) (PGMA) shells as a reactive core for the grafting of high density PNIPAM chains to form a colloidally stable system to decipher the direct consequence of thermally induced phase transition of PNIPAM on the magnetic resonance properties of iron oxide nanoparticles in solution. Importantly, the use of a functional polymer like PGMA as an anchoring platform to graft PNIPAM chains and the traditional method of attaching to the surface of an iron oxide nanoparticle lies in the mobility of the epoxide functional groups13–17 located in the loops and tails of the core macromolecule. The mobility of the reactive loops of PGMA ensures greater access to anchoring, resulting in a 2–3 fold greater grafting density when compared to a monolayer of epoxy groups on an iron oxide nanoparticle surface of similar dimension.18 We demonstrate that the PNIPAM–PGMA–NP composite core offers a highly stable colloidal platform to directly correlate the PNIPAM thermo-transition using magnetic resonance relaxometry measurements. Herein, we fabricated two types of nanoparticles, namely thermoresponsive PNIPAM–PGMA–NPs and as a control, PGMA–NPs (PGMA/iron oxide nanoparticles). The PNIPAM–PGMA copolymer was synthesized by end grafting PNIPAM–COOH with PGMA via a ring-opening reaction with the epoxy group (see the ESI† for synthesis details).
The synthesized copolymer was characterized using 1H NMR and FTIR. 1H NMR confirmed the PNIPAM contribution with peaks evident at 1.13 ppm (–CH3) and 3.99 ppm (–CH2) while the PGMA contribution was observed with peaks at 2.5–2.9 ppm (–CH2) and 3.22 ppm (–CH) (see the ESI,† Fig. S1). Subsequent FTIR analysis validated PNIPAM grafting over PGMA as observed in the NMR analysis (Fig. 1E). The characteristic peaks from a PGMA segment (1726 cm−1 for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl vibration and 1147 cm−1 for C–O stretching from the ester group of glycidyl methacrylate (GMA)) and a PNIPAM segment (1642 cm−1 for amide I (CO/C–N), 1536 cm−1 for amide II (N–H/C–H), and 1387 and 1367 cm−1 for two methyl groups on isopropyl groups (–CH(CH3)2)) can be clearly observed and are consistent with previously published reports.19,20
O carbonyl vibration and 1147 cm−1 for C–O stretching from the ester group of glycidyl methacrylate (GMA)) and a PNIPAM segment (1642 cm−1 for amide I (CO/C–N), 1536 cm−1 for amide II (N–H/C–H), and 1387 and 1367 cm−1 for two methyl groups on isopropyl groups (–CH(CH3)2)) can be clearly observed and are consistent with previously published reports.19,20
 | ||
| Fig. 1 TEM micrograph of the nanoparticles: (A) magnetite, (B) PNIPAM–PGMA–NPs and (C) PGMA–NPs; (the insets in (B) and (C) show higher resolution images of a single nanoparticle), (D) DLS analysis showing the particle size distribution of PGMA–NPs and PNIPAM–PGMA–NPs at 25 °C in Pluronic F-108, and (E) FTIR spectrum of PNIPAM–PGMA–NPs showing the relevant contribution from both PNIPAM and PGMA. Scale bars: (A) 50 nm, (B) and (C) 200 nm (insets: 20 nm). | ||
Subsequently, polymer nanoparticles were fabricated using an oil-in-water emulsion method using the synthesised PGMA grafted PNIPAM copolymer encapsulating magnetite (Fe3O4) nanoparticles (PNIPAM–PGMA–NPs) while PGMA encapsulating magnetite nanoparticles (PGMA–NPs) were synthesised as a control (see the ESI† for synthesis details). The synthesised magnetite nanoparticles were approximately 7 ± 0.7 nm (mean ± standard deviation) as quantified using TEM (Fig. 1A). TEM analysis confirmed the formation of the spherical morphology of the nanoparticles with particle size distributions of 100 ± 10.8 nm and 110 ± 15.3 nm for the PNIPAM–PGMA–NPs and PGMA–NPs, respectively (Fig. 1B and C). Although TEM analysis is sufficient to confidently gauge the size of the nanoparticles, it is not the best technique to study the solution properties of the nanoparticle formulation. For example, it is not clear if the PGMA–PNIPAM–NPs are aggregated in Fig. 1B or if it is the result of the drying effect during the sample preparation for TEM analysis.
To ascertain the effect of temperature on particle size, DLS measurements were carried out using a temperature sweep from 25 °C to 45 °C at 1 °C step intervals. Interestingly, PNIPAM–PGMA–NPs yielded the mean hydrodynamic radius of 150.2 ± 1.3 nm (mean ± standard deviation) in pluronic F-108 at 25 °C (T < LCST of PNIPAM) whereas PGMA–NPs were observed to be 172.6 ± 2.4 nm under the same conditions (Fig. 2). Notably, we observed a significant reduction in the PNIPAM–PGMA–NP mean hydrodynamic radius from 150.2 nm to 82.8 nm with increasing temperature from 32 to 45 °C.
 | ||
| Fig. 2 DLS data showing the mean hydrodynamic radius for PGMA–NPs (blue squares) and PNIPAM–PGMA–NPs (orange dots) as a function of temperature. Mean hydrodynamic radius values were taken as Z-average of 6 measurements. Data presented as average ± SD. | ||
This observation indicated that PNIPAM–PGMA–NPs have a typical negative thermoresponsive volume phase transition characteristic.19,21 DLS data validated the lower critical solution temperature to be 32 °C. The plausible explanation for this is due to the complex dissociation of the interactions between PNIPAM and water molecules mediated by the breakage of hydrogen bonds.22 Therefore, when the temperature was increased above the LCST (T > 32 °C of PNIPAM), the thermodynamic size of the nanoparticles decreased significantly because the PNIPAM segment gradually becomes hydrophobic and shrinks in water (Fig. 2). Decrease in temperature below the LCST (T < 32 °C) favours the formation of hydrogen bonding between PNIPAM and water resulting in the expansion of the PNIPAM–PGMA chains. The control experiments, using the PGMA–NPs showed no shift in the particle size as a function of temperature (Fig. 2). This observation indicated that the size changes observed in the composite polymer nanoparticles were contributed by the PNIPAM component. DLS analysis confirmed the non-aggregation behaviour of PNIPAM–PGMA–NPs and PGMA–NPs in pluronic F-108 solution. However, both PNIPAM–PGMA–NPs and PGMA–NPs were observed to be unstable in water.
The aggregation behaviour of the magnetic nanoparticles is governed by their surface properties and their interaction with the solvent molecules. Furthermore, it is critical for any nanoparticle formulation to be colloidally stable for their end use applications especially in tissue engineering, drug delivery and as an MRI contrast agent. Magnetic resonance relaxation has found significant importance in the field of medical diagnostics especially in MRI and related techniques.23 Despite the higher spatial resolution, detection of small tissue continues to be a significant limitation with the use of MRI. Magnetic nanoparticles have been employed as contrast agents to circumvent this limitation. Magnetic nanoparticles accelerate the relaxation rates (longitudinal or traverse) thereby improving the contrast between the pathological legion and normal tissue. The magnetic nanoparticle induced relaxation rates are strongly correlated with the physicochemical parameters such as size, composition and surface coating of the magnetic nanoparticles.23 The size of the magnetic nanoparticles has a significant effect on MRI contrast. In order to elucidate the effect of a nanoparticle size on traverse relaxation time (T2) contrast enhancement, magnetite nanoparticles of different sizes (4, 6, 9 and 12 nm) were synthesised. The nanoparticle size was found to have a positive influence on the magnetic saturation which significantly increased the T2 value mediated MRI contrast.23,24 In an another approach, the controlled encapsulation of 7.4 nm magnetic nanoparticle clusters within the amphiphilic poly(ethylene oxide-co-lactide) micelles has been shown to produce significant contrast (r2) compared to the commercially used Feridex®. Encapsulation of magnetic nanoparticles within the pH responsive poly(NIPAM-AA) hydrogel has been shown to significantly enhance the transverse relaxation rates corresponding to the increase in the pH of the solution reaching the maximum value of 505 mM−1 s−1 at pH 7 which was theorised to be caused by the lowering of the diffusion coefficient of water near the particle surface with the increasing pH.25 Interestingly, linear alignment of polyelectrolyte stabilised magnetic nanoparticles has been shown to facilitate their entry into the brain tissue through the circulatory system and therefore can be used to image the brain capillary network. As little as 0.4 mL of colloidal linearly aligned nanoparticles was shown to provide significant improvement in the longitudinal weighted image contrast (T1) within 13.6 s post injection.26T1 contrast effects are also dependent on the nanoparticle size where with the reduction in the particle size enhances contrast which has been attributed to the magnetically disordered spin layers on the nanoparticle surface which makes the dominant contrast effects of the magnetic nanoparticle.23 Extremely small-sized iron oxide nanoparticles with 3 nm diameter were shown to exhibit a significantly higher T1 contrast effect as compared to the 12 nm particles, which has been attributed to the large number of surface Fe3+ ions with 5 unpaired valence electrons.27In vivo MRI imaging revealed that extremely small-sized iron oxide nanoparticles had longer circulation time than the commercial gadolinium complex-based contrast agent facilitating high resolution imaging of blood vessels with sizes down to 0.2 mm.27 The composition of magnetic nanoparticles is another factor with a significant influence on MRI contrast. The metal ferrite nanoparticles using transition metal dopants such as Mn, Ni and Co were fabricated by substituting Fe2+ in the octahedral sites of 12 nm nanoparticles.23,28 Mn doped iron oxide nanoparticles demonstrated the highest r2 values resulting in significantly superior contrast in a small tumor xenograft model (50 mg) as compared to other doped-nanoparticles.28 Subsequently, the incorporation of Zn29 as a dopant in the metal ferrite nanoparticles or rare-earth metal like Gd-embedded iron oxide nanoparticles30 has been shown to significantly enhance T2 and T1 values, respectively compared to the iron oxide nanoparticles of the same size. Despite the advantage in terms of improvement in MRI contrast, the use of dopants, especially rare earth metals, raises significant safety concerns and therefore, needs to be considered before translation into the clinic.
In order to decipher the mechanism behind the anomalous DLS data observed between the two solvent systems, zeta potential measurements were carried out to study the surface potential of the nanoparticles. The zeta potential measurements were carried out in 1 °C step intervals from 25 °C to 45 °C (see the ESI† for a method). The PNIPAM–PGMA–NPs and the control PGMA–NPs had nearly neutral zeta potentials of (+3.9 to +5.6 mV) and (−1.4 to −1.9 mV), respectively (see the ESI,† Fig. S2). These near neutral surface charges indicate that electrostatic repulsion is not a major mechanism behind the colloidal stability of these materials. Given that the particles without pluronic surfactant are colloidally unstable it seems that the presence of the surfactant, sterically stabilizes the particles against aggregation.
Magnetization properties of the two nanoparticle systems were elucidated using SQUID magnetometry (Superconducting Quantum Interference Device). Both PNIPAM–PGMA–NPs and PGMA–NPs were superparamagnetic at room temperature with the zero-field-cooled curves showing a maximum blocking temperature of approximately 150 K and 50 K, respectively (Fig. 3). The specific saturation Ms was determined to be 66.5 and 63.6 emu g−1 of Fe3O4 for the PNIPAM–PGMA–NPs and PGMA-NPs respectively. The magnetite content was quantified using ICP analysis and found to be 0.93 mg and 0.43 mg of Fe for the PNIPAM–PGMA–NPs and PGMA–NPs, respectively. The difference in the blocking temperature could be attributed to the amount of Fe content and the shielding effect of the polymer coating which disrupts the inter-particle and particle-field interactions. This could cause a particle spin canting effect at the polymer coating layer in addition to the broken symmetry due to crystalline disorder, reduced coordination, and broken exchange at the particle surface.12
 | ||
| Fig. 3 SQUID magnetometry of PNIPAM–PGMA–NPs (red circles) and PGMA–NPs (blue dots) reveals superparamagnetic behaviour. (A) Hysteresis loop at 5 K (inset), (B) the magnetization curve at 300 K displays no hysteresis and shows a specific saturation magnetization of 66.5 emu g−1 and 63.6 emu g−1 for PNIPAM–PGMA–NPs and PGMA-NPs, respectively. Zero-field-cooled (red circles) and field-cooled (blue dots) curves are coincident above ca. 150 K and 50 K for (C) PNIPAM–PGMA-NPs and (D) PGMA–NPs, respectively. | ||
As the temperature of a suspension of PNIPAM–PGMA–NPs in pluronic solution is increased, the mean hydrodynamic radius of the PNIPAM–PGMA–NPs started to decrease at approximately 32 °C. The rate of mean hydrodynamic radius decrease with temperature reaches its maximum value at approximately 35 °C (Fig. 2). As the temperature is increased further, the rate of mean hydrodynamic radius decrease slows. By 45 °C, the mean hydrodynamic radius reaches a mean value that is about 60% lower than that observed at 32 °C. The range of temperatures over which the change in mean hydrodynamic radius is observed (32 °C to 45 °C) most likely represents the range of temperatures between the LCST and the UCST (upper critical solution temperature). Measurements of the proton transverse relaxivity (r2) of a suspension of PNIPAM–PGMA–NPs in aqueous pluronic solution as the temperature is increased from 25 °C to 40 °C show a steady decrease (Fig. 4). No change in the rate of change of relaxivity is observed below 40 °C. This observation indicates that the significant decrease in mean hydrodynamic radius observed between 32 °C and 40 °C has no significant effect on the proton transverse relaxivity suggesting that any collapse of the PNIPAM is not restricting diffusive access of water molecules to the magnetic core of the particle. Between 40 °C and 45 °C, a significant drop in proton transverse relaxivity occurs at a rate significantly higher than the steady rate observed between 25 °C and 40 °C suggesting that between 25 °C and 40 °C there is a significant reduction of diffusive access of water molecules to the magnetic core of the particles. Above 45 °C, the proton relaxivity continues to decline steadily with increasing temperature at a slower rate. Taken together, these data suggest that the collapse of the PNIPAM polymer chains into a hydrophobic shell as the temperature is increased is a gradual process that occurs over a temperature span from approximately 32 °C to 45 °C. The fact that no change in the rate of change of proton transverse relaxivity is observed below 40 °C suggests that no particles have reached full collapse by 40 °C. This observation suggests that the observed decrease in mean hydrodynamic radius is a result of all particles partially and continuously collapsing over the temperature range rather than a result of an increase in the fraction of collapsed particles in a mixed population of extended and collapsed particles. At 40 °C, the mean hydrodynamic radius of the PNIPAM–PGMA–NPs is approximately 75% of its value below 32 °C and yet there is no significant restriction of diffusive access of water to the magnetic core of the particle. Somewhere between 40 °C and 45 °C, the remaining collapse of the PNIPAM polymer into a hydrophobic shell with a hydrodynamic radius 65% of its original value appears to have the effect of abruptly shutting off the diffusive access of water to the magnetic core of the nanoparticle. Since PGMA–NPs showed no change in the rate of change of hydrodynamic size or r2 values with change in temperature, it can be confirmed that the observed temperature induced effects in the PNIPAM–PGMA–NP suspensions have been contributed solely by the grafted PNIPAM moiety.
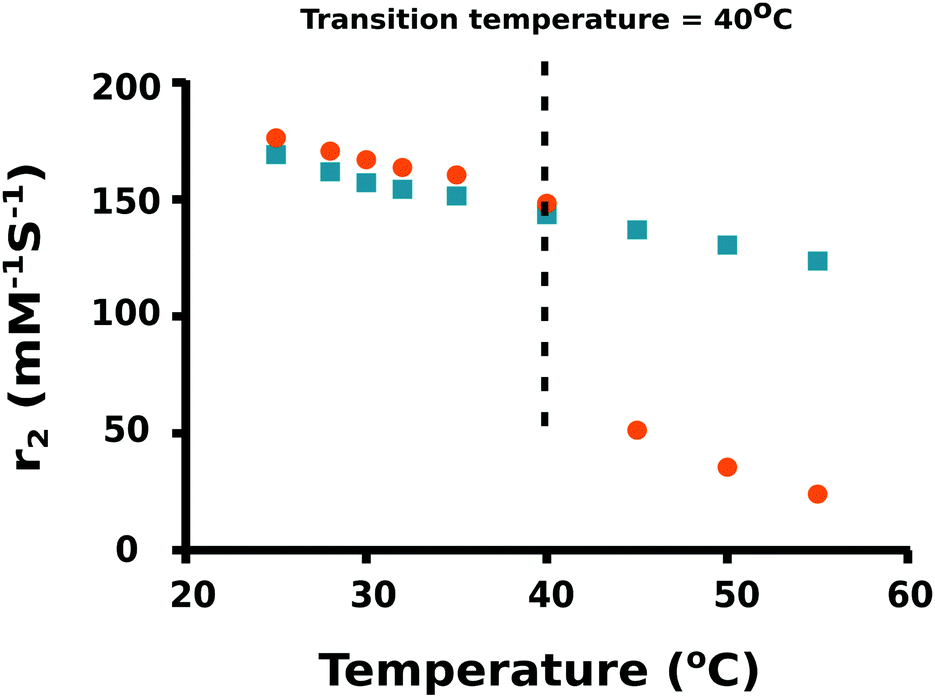 | ||
| Fig. 4 Relaxivity coefficient (r2) of PNIPAM–PGMA–NPs (orange dots) and PGMA–NPs (blue squares) at different temperatures. | ||
Conclusions
We have synthesised a thermoresponsive magnetic nanoparticle system comprising 7 nm magnetic iron oxide (magnetite) nanoparticles embedded in a PNIPAM-g-PGMA shell. The PNIPAM–PGMA–NPs show colloidal stability in pluronic F-108 solution. DLS and proton relaxometry measurements as a function of temperature indicate that the PNIPAM is extended in a hydrophilic form below 32 °C, and gradually collapses to a hydrophobic shell with increasing temperature between 32 °C and 45 °C. Solvent water molecules have diffusive access through the PNIPAM to the PGMA core up to temperatures of 40 °C. Between 40 and 45 °C, diffusive access of water through the PNIPAM to the PGMA core is abruptly shut off so that above 45 °C a collapsed PNIPAM hydrophobic shell around the PGMA core restricts diffusive access of water molecules to the PGMA core.Acknowledgements
This work was supported by funds from the Australian Research Council and The University of Western Australia. NIT acknowledges the postgraduate scholarship from the University of Technology MARA (UiTM) changed to Universiti Teknologi MARA (UiTM) and Ministry of Higher Education, Malaysia. VA would like to acknowledge the Department of Science and Technology (SERB-DST), India for the National Postdoctoral Fellowship (PDF/2016/002953). The microscopy analysis was carried out using facilities at the Centre for Microscopy, Characterisation and Analysis, The University of Western Australia, which are supported by University, State and Federal Government funding.References
- L. Hajba and A. Guttman, Biotechnol. Adv., 2016, 34, 354–361 CrossRef CAS PubMed.
- X. Li, J. Wei, K. E. Aifantis, Y. Fan, Q. Feng, F. Z. Cui and F. Watari, J. Biomed. Mater. Res., Part A, 2016, 104, 1285–1296 CrossRef CAS PubMed.
- B. Sierra-Martin and A. Fernandez-Barbero, Adv. Colloid Interface Sci., 2016, 233, 25–37 CrossRef CAS PubMed.
- A. M. Derfus, G. von Maltzahn, T. J. Harris, T. Duza, K. S. Vecchio, E. Ruoslahti and S. N. Bhatia, Adv. Mater., 2007, 19, 3932–3936 CrossRef CAS.
- J. H. Lee, K. J. Chen, S. H. Noh, M. A. Garcia, H. Wang, W. Y. Lin, H. Jeong, B. J. Kong, D. B. Stout and J. Cheon, Angew. Chem., Int. Ed., 2013, 125, 4480–4484 CrossRef.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- B. Ray, Y. Okamoto, M. Kamigaito, M. Sawamoto, K.-I. Seno, S. Kanaoka and S. Aoshima, Polym. J., 2005, 37, 234 CrossRef CAS.
- S. Patra, E. Roy, P. Karfa, S. Kumar, R. Madhuri and P. K. Sharma, ACS Appl. Mater. Interfaces, 2015, 7, 9235–9246 CAS.
- H. Kakwere, M. Pernia Leal, M.-E. Materia, A. Curcio, P. Guardia, D. Niculaes, R. Marotta, A. Falqui and T. Pellegrino, ACS Appl. Mater. Interfaces, 2015, 7, 10132–10145 CAS.
- A. Chiu-Lam and C. Rinaldi, Adv. Funct. Mater., 2016, 26, 3933–3941 CrossRef CAS.
- S. Balasubramaniam, N. Pothayee, Y. Lin, M. House, R. C. Woodward, T. G. St. Pierre, R. M. Davis and J. Riffle, Chem. Mater., 2011, 23, 3348–3356 CrossRef CAS.
- R. Frenzel, S. Höhne, C. Hanzelmann, T. Schmidt, R. Winkler, A. Drechsler, E. Bittrich, K.-J. Eichhorn and P. Uhlmann, ACS Appl. Mater. Interfaces, 2015, 7, 12355–12366 CAS.
- B. Zdyrko and I. Luzinov, Macromol. Rapid Commun., 2011, 32, 859–869 CrossRef CAS PubMed.
- A. W. Eckert, D. Gröbe and U. Rothe, Biomaterials, 2000, 21, 441–447 CrossRef CAS PubMed.
- B. Zdyrko, K. S. Iyer and I. Luzinov, Polymer, 2006, 47, 272–279 CrossRef CAS.
- V. Agarwal, D. Ho, D. Ho, Y. Galabura, F. Yasin, P. Gong, W. Ye, R. Singh, A. Munshi and M. Saunders, ACS Appl. Mater. Interfaces, 2016, 8, 4934–4939 CAS.
- K. S. Iyer, B. Zdyrko, H. Malz, J. Pionteck and I. Luzinov, Macromolecules, 2003, 36, 6519–6526 CrossRef CAS.
- S. Shi, Q. Wang, T. Wang, S. Ren, Y. Gao and N. Wang, J. Phys. Chem. B, 2014, 118, 7177–7186 CrossRef CAS PubMed.
- B. Jiang, L. Zhang, J. Yan, Q. Huang, B. Liao and H. Pang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2442–2453 CrossRef CAS.
- X. C. Xiao, L. Y. Chu, W. M. Chen, S. Wang and Y. Li, Adv. Funct. Mater., 2003, 13, 847–852 CrossRef CAS.
- Z. Zhou, S. Zhu and D. Zhang, J. Mater. Chem., 2007, 17, 2428–2433 RSC.
- T.-H. Shin, Y. Choi, S. Kim and J. Cheon, Chem. Soc. Rev., 2015, 44, 4501–4516 RSC.
- Y.-w. Jun, Y.-M. Huh, J.-s. Choi, J.-H. Lee, H.-T. Song, S. Kim, S. Kim, S. Yoon, K.-S. Kim and J.-S. Shin, J. Am. Chem. Soc., 2005, 127, 5732–5733 CrossRef CAS PubMed.
- C. Paquet, H. W. de Haan, D. M. Leek, H.-Y. Lin, B. Xiang, G. Tian, A. Kell and B. Simard, ACS Nano, 2011, 5, 3104–3112 CrossRef CAS PubMed.
- S. A. Corr, S. J. Byrne, R. Tekoriute, C. J. Meledandri, D. F. Brougham, M. Lynch, C. Kerskens, L. O'Dwyer and Y. K. Gun'ko, J. Am. Chem. Soc., 2008, 130, 4214–4215 CrossRef CAS PubMed.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon and H. B. Na, J. Am. Chem. Soc., 2011, 133, 12624–12631 CrossRef CAS PubMed.
- J.-H. Lee, Y.-M. Huh, Y.-w. Jun, J.-w. Seo, J.-t. Jang, H.-T. Song, S. Kim, E.-J. Cho, H.-G. Yoon and J.-S. Suh, Nat. Med., 2007, 13, 95–99 CrossRef CAS PubMed.
- J. t. Jang, H. Nah, J. H. Lee, S. H. Moon, M. G. Kim and J. Cheon, Angew. Chem., Int. Ed., 2009, 121, 1260–1264 CrossRef.
- Z. Zhou, L. Wang, X. Chi, J. Bao, L. Yang, W. Zhao, Z. Chen, X. Wang, X. Chen and J. Gao, ACS Nano, 2013, 7, 3287–3296 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, 1H NMR spectra and zeta potentials. See DOI: 10.1039/c7qm00202e |
| This journal is © the Partner Organisations 2017 |