
Late transition metal catalyzed α-olefin polymerization and copolymerization with polar monomers
Lihua
Guo
a,
Wenjing
Liu
a and
Changle
Chen
 *b
*b
aSchool of Chemistry and Chemical Engineering, Qufu Normal University, Qufu, 273165, China
bCAS Key Laboratory of Soft Matter Chemistry, Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei, 230026, China. E-mail: changle@ustc.edu.cn
First published on 29th August 2017
Abstract
In this review, recent developments on late transition metal catalyzed α-olefin polymerization and copolymerization with polar comonomers are described. First, the polymerization mechanisms of early and late transition metal catalyzed α-olefin polymerization are compared. Second, cationic catalysts bearing α-diimine and related ligands as well as neutral catalysts bearing anionic ligands for α-olefin homopolymerization are discussed in detail. Third, late transition metal catalyzed α-olefin copolymerization with polar functionalized comonomers is summarized. Special attention is paid to the regio- and stereo-selectivity induced by late transition metal catalysts in these polymerization and copolymerization reactions.
 Changle Chen | Changle Chen obtained his BS degree from the University of Science and Technology of China in 2005. After receiving his PhD from the University of Chicago in 2010, he moved to Northwestern University to start his postdoctoral studies. After some time at Celanese Corporation, he started his independent career as a professor at USTC in 2013. His current research focuses on development of organometallic complexes and new strategies for olefin polymerization and copolymerization. His notable awards include the Chinese Chemical Society Award for Outstanding Young Chemists, American Chemical Society DIC Young Investigator Award and IUPAC Prizes-Honorable Mention. |
1 Introduction
Polyolefins have enormous annual production and diverse applications as plastics, fibers and elastomers. These materials are derived from ethylene and α-olefins, which are mainly produced by the petroleum industry. Since Ziegler's discovery of transition metal catalyzed ethylene polymerization and Natta's discovery of stereoselective propylene polymerization,1,2 intense research efforts have been made in this field.3–14 Hogan and Banks's seminal contribution should also be noted.15 In particular, transition metal catalyzed ethylene polymerization has emerged as a major strategy in the production of polyethylenes. In contrast to polyethylene, transition metal catalyzed α-olefin polymerization is the predominant method in industry for the production of α-olefin polymers and copolymers.16 Specifically, various early transition metal based catalysts are widely used for the production of poly(α-olefins). Their study has greatly advanced our understanding of the mechanistic aspects, including the regio- and stereo-chemistry of the (co)polymerization processes.The non-polar nature of polyolefins represents one of their biggest limitations. The introduction of some polar functional groups could greatly improve their many properties and further expand their applications.17 As the most direct and economic strategy, transition metal catalyzed olefin copolymerization with polar monomers is highly fascinating but also highly challenging. The industrially relevant early transition metal catalysts are incompetent for this task because they are easily poisoned by polar functional groups. Because of their low oxophilicity, late transition metal catalysts have achieved great success in the area of ethylene copolymerization with polar monomers. However, most of these catalysts are not able to realize regio- or stereo-controlled α-olefin polymerization. Moreover, very few efforts have been made in developing efficient catalytic systems for α-olefin/polar monomer copolymerization. As a result, the synthetic approach for functional polypropylenes and poly(α-olefins) is generally limited to multi-step techniques such as post-polymerization functionalization or the masking/protection of polar monomers. The direct synthesis of polar functionalized polypropylenes from the transition metal catalyzed copolymerization of propylene and polar monomers remains a big challenge in this field.
In the 1990s, Brookhart et al. reported that α-diimine Pd(II) complexes could copolymerize olefin with polar monomers.18–21 A key insight from these early studies is that increasing steric bulk on the metal axial position could efficiently retard chain transfer and lead to high molecular weight polymers. Since then, tremendous progress has been made and a huge amount of high performance late transition metal catalysts have been designed and synthesized.22–24 The use of late transition metal catalysts for the coordination–insertion polymerization of ethylene, cycloolefins, dienes, trienes, and norbonene type monomers and copolymerization with polar monomers has been extensively studied3–15 and is beyond the scope of this review. In contrast, only a small fraction of these late transition metal catalysts have been investigated in α-olefin polymerization or copolymerization reactions. This review will focus on recent investigations on late transition metal catalyzed propylene and higher α-olefin homopolymerization and copolymerization with polar monomers. These catalysts can be categorized into two groups based on the nature of the active species during polymerization: cationic catalysts and neutral catalysts.
In this review, we will first discuss the mechanism of transition metal catalyzed ethylene and α-olefin polymerization. Subsequently, the cationic catalysts based on diimine ligands and other ligands, as well as neutral catalysts bearing phosphine–sulfonate and derivative ligands for α-olefin homopolymerization and copolymerization with polar monomers will be discussed. Specifically, the control of polymer microstructures including branching structure, regioselectivity and stereoselectivity will be focused on.
2 Polymerization mechanism
Ethylene polymerization by early transition metal catalysts usually affords highly linear polyethylene (Scheme 1a). The chain growth involves repeated insertion of ethylene into the metal–alkyl chain. Chain transfer processes such as β-hydride elimination limit the molecular weight of the polymer. As such, the polyethylene product possesses a highly linear microstructure. However, highly branched even branch-on-branch structures can be produced using late transition metal catalysts such as α-diimine Ni(II) and Pd(II) catalysts through a process known as chain walking polymerization. Detailed studies revealed the polymerization mechanism shown in Scheme 1b. The key step is that the 1° alkyl agostic complex can undergo β-hydride elimination, and reinsertion into the olefin hydride intermediate to give either the same 1° alkyl agostic complex or a new 2° alkyl agostic complex with opposite regio-chemistry. Ethylene trapping of the 2° alkyl species followed by further insertion results in the formation of a methyl branch. Subsequent chain walking along the polymer chain leads to the formation of longer branches.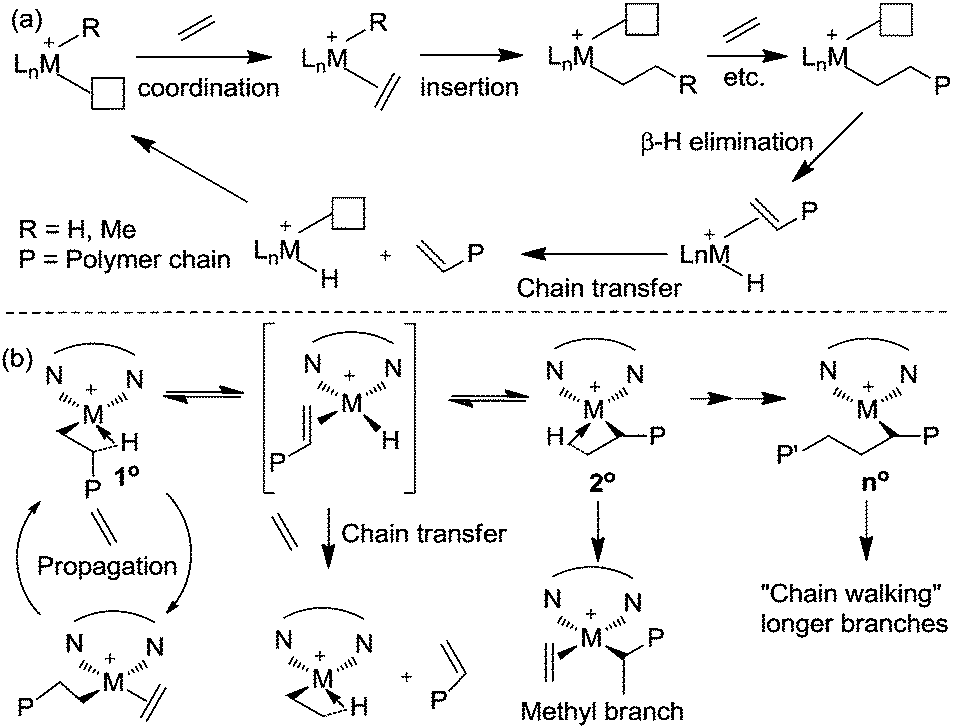 | ||
| Scheme 1 Early and late transition metal-catalyzed polymerization of ethylene. | ||
α-Olefin polymerization catalyzed by early transition metal catalysts usually exhibits highly selective 1,2-insertion of the monomer, in which the terminal or “1” carbon of the α-olefin is directly bound to the metal center and the neighboring “2” carbon is attached to the incipient polymer chain, resulting in poly(α-olefins) with n-alkyl side chains (Scheme 2a).25–27 The occasional 2,1-insertion can lead to a 2,1-enchainment or 1,ω-enchainment, which has been observed as isolated regioerrors.28
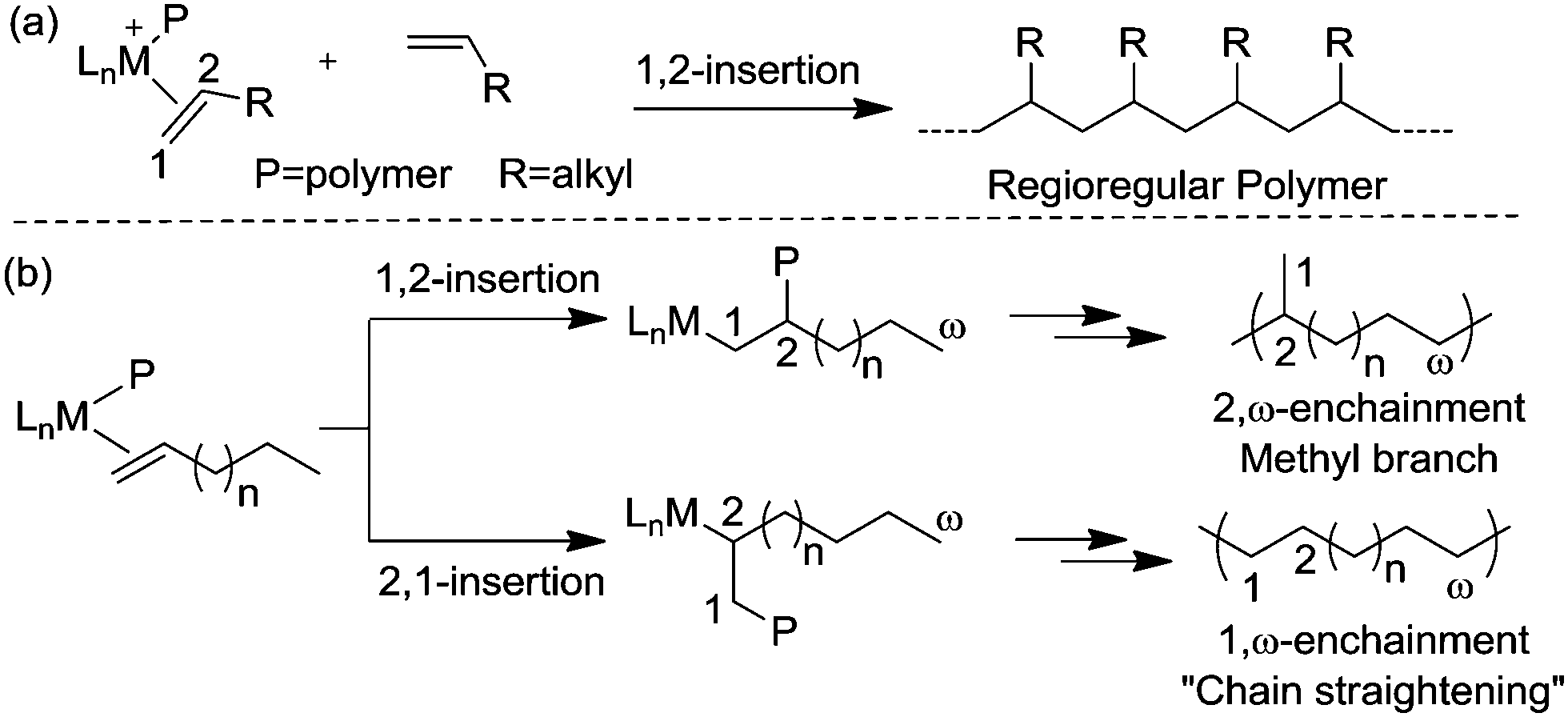 | ||
| Scheme 2 Early and late transition metal-catalyzed polymerization of α-olefins. | ||
In contrast, polymerization of α-olefins using cationic α-diimine Ni(II) and Pd(II) catalysts occurs through a chain straightening mechanism to give polymers that contain fewer branches per 1000 carbons than predicted from simple 1,2-monomer enchainment (Scheme 2b).21,29,30 The monomer undergoes 1,2- as well as 2,1-insertion into the M–C bond and the metal usually walks to the end of the branch before the next insertion happens. When 1,2-insertion takes place, the subsequent β-hydride elimination followed by metal migration to the primary carbon atom leads to 2,ω-enchainment to form a polymer chain with a methyl branch. On the other hand, 2,1-insertion followed by chain walking results in 1,ω-enchainment to give repeating units with a linear structure.
3 Catalysts for α-olefin homopolymerization
Late transition metal catalysts for α-olefin homopolymerization can be categorized into two groups: (1) cationic catalysts bearing neutral ligands (Fig. 1); (2) neutral catalysts bearing anionic ligands (Fig. 2). The influence of the catalyst structure and polymerization conditions on the polymerization results is summarized in Table 1.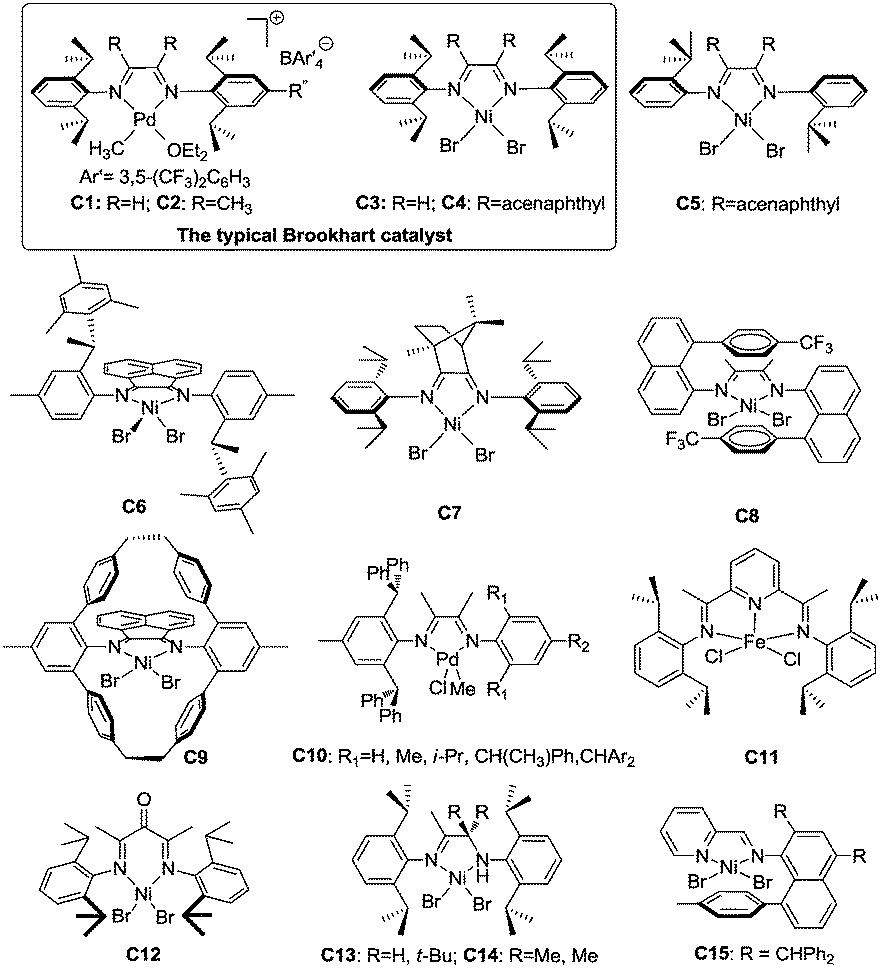 | ||
| Fig. 1 Cationic catalysts with neutral ligands for α-olefin polymerization. | ||
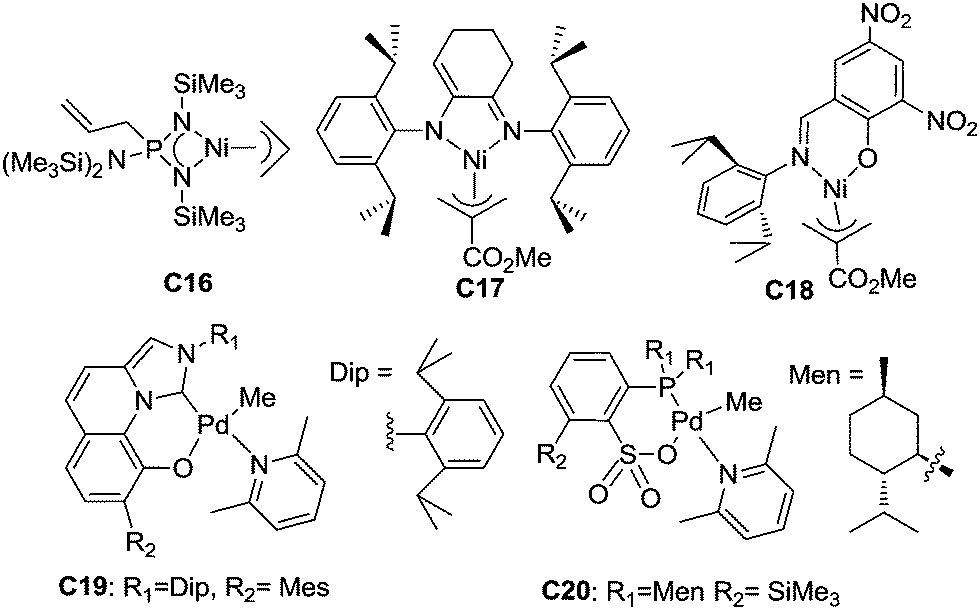 | ||
| Fig. 2 Neutral late transition metal catalysts bearing anionic ligands for α-olefin polymerization. | ||
| Catalyst | T (°C) | Time (h) | Olefin (conc.)a | TOFb (h−1) | M n (×10−3) | PDIc | B /1000C | Thermal analysise (°C) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a P = propylene, H = 1-hexene, D = 1-decene. b Turnover frequency, which is calculated as the moles of monomer consumed per mole of catalyst per hour. c Determined by Gel Permeation Chromatography (GPC). d Branching density, branches per 1000 carbon atoms, determined by 1H NMR. nd: not determined. e Determined by Differential Scanning Calorimetry (DSC). f Broad and weak endotherms, amorphous. | |||||||||
| C2 | 25 | 16 | P (1 atm) | 579 | 15 | 4.30 | 213 | −43 (Tg) | 18 |
| C4 | 0 | 2 | P (1 atm) | 3005 | 150 | 1.60 | 300 | −20, −78 (Tg) | 18 |
| C4 | 0 | 1.5 | H (0.8 M) | 907 | 74 | 1.15 | 83 | nd | 29 |
| C5 | 0 | 2 | H (0.8 M) | 528 | 74 | 1.19 | 98 | nd | 29 |
| C6 | −78 | 96 | P (15 g) | 1 | 5.7 | 1.37 | nd | −0.5 (Tg), 137.3 (Tm) | 37 |
| C6 | 0 | 2 | P (5 g) | 385 | 48.2 | 1.08 | nd | −54.4 (Tg) | 37 |
| C7 | 20 | 1 | P (1.2 atm) | 380 | 53.8 | 1.13 | 141 | 30.1 (Tm) | 33 |
| C8 | 22 | 24 | D (0.1 M) | 0.9 | 10 | 1.30 | nd | 109 (Tm) | 43 |
| C9 | 75 | 3 | H (2.66 M) | 1822 | 529 | 1.17 | 52 | −47 (Tg), 58 (Tm) | 44 |
| C9 | 50 | 1 | P (1 atm) | 2032 | 133 | 1.13 | 105 | −55 (Tg) | 44 |
| C11 | −20 | 2 | P (1 atm) | 12900 | 6.5 | 2.1 | nd | nd | 51 |
| C12 | −60 | 8 | P (40 mL) | 13 | 6.4 | 1.30 | nd | 134 (Tm) | 53 |
| C12 | −20 | 3 | P (20 mL) | 170 | 42 | 1.10 | nd | 76 (Tm) | 53 |
| C13 | 25 | 8 | H (1.06 M) | 26.4 | 31.8 | 1.23 | 132.2 | —f | 54 |
| C14 | 0 | 8 | H (1.06 M) | 5.2 | 5.2 | 1.40 | 17.1 | 93.4, 107.2 (Tm) | 54 |
| C15 | 20 | 3 | D (0.1 M) | 4.5 | 13 | 1.49 | 26 | 105.5 (Tm) | 55 |
| C19 (Dip-H) | 100 | 3 | P (10 g) | 808 | 16 | 2.20 | nd | nd | 59 |
| C20 (Men-Ph) | 50 | 12 | P (6 g) | 140 | 21 | 1.90 | nd | −9.6 (Tg), 46.6, 63.9 (Tm) | 60 |
3.1 Cationic catalysts bearing neutral ligands
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and a Tg of −43 °C rather than the ca. −5 °C value observed in isotactic or atactic polypropylenes.3,18 The traditional Brookhart's Ni(II) catalyst C4, activated with MAO, can generate polymers with Mn of 150000 and Tg of −20 °C. The branching densities of these two polymers are 213 and 300 per 1000 carbon atoms, which are significantly lower than the expected value of 333.3,18 Generally, both 1,2- and 2,1-insertions can occur in α-diimine Ni(II) and Pd(II) catalyzed α-olefin polymerization. Higher α-olefin leads to a higher polymer Tm, which is due to the increased length of the linear segment in the polymer backbone formed from 1,ω-enchainment. For α-diimine Pd(II) catalysts, insertion occurs only into primary Pd(II)–alkyl (Pd–CH2P) bonds. In contrast, insertion can occur at secondary Ni(II)–alkyl (Ni–CH(R)P) bonds for α-diimine Ni(II) catalysts.29 In addition, the traditional Brookhart's Pd(II) catalysts give higher degrees of chain straightening than the comparable Ni(II) catalysts. For example, room temperature polymerization of propylene catalyzed by Ni(II) catalyst C4 yields atactic polypropylene with 272 branches per 1000 carbon atoms,21 while polypropylene made from Pd(II) catalyst C2 under similar conditions displays only 213 branches per 1000 carbon atoms.18
000 and a Tg of −43 °C rather than the ca. −5 °C value observed in isotactic or atactic polypropylenes.3,18 The traditional Brookhart's Ni(II) catalyst C4, activated with MAO, can generate polymers with Mn of 150000 and Tg of −20 °C. The branching densities of these two polymers are 213 and 300 per 1000 carbon atoms, which are significantly lower than the expected value of 333.3,18 Generally, both 1,2- and 2,1-insertions can occur in α-diimine Ni(II) and Pd(II) catalyzed α-olefin polymerization. Higher α-olefin leads to a higher polymer Tm, which is due to the increased length of the linear segment in the polymer backbone formed from 1,ω-enchainment. For α-diimine Pd(II) catalysts, insertion occurs only into primary Pd(II)–alkyl (Pd–CH2P) bonds. In contrast, insertion can occur at secondary Ni(II)–alkyl (Ni–CH(R)P) bonds for α-diimine Ni(II) catalysts.29 In addition, the traditional Brookhart's Pd(II) catalysts give higher degrees of chain straightening than the comparable Ni(II) catalysts. For example, room temperature polymerization of propylene catalyzed by Ni(II) catalyst C4 yields atactic polypropylene with 272 branches per 1000 carbon atoms,21 while polypropylene made from Pd(II) catalyst C2 under similar conditions displays only 213 branches per 1000 carbon atoms.18
The activities of α-diimine Ni(II) and Pd(II) catalysts in propylene polymerization are lower than in ethylene polymerization. For the α-diimine Ni(II) catalyzed propylene polymerization, the rate-limiting step is the trapping of propylene, making the chain growth process overall first order in olefin concentration.31 In contrast, the chain propagation is limited by migratory insertion rates in the α-diimine Pd(II) system.18,20 Furthermore, these catalysts can polymerize α-olefin in a living fashion and afford block copolymers with α-olefins.20,32,33
The structure of the polymers depends on the structures of the α-diimine Ni(II) and Pd(II) catalysts and the polymerization conditions. Considerable research efforts have been made in this field. Pellecchia et al. found that the regioselectivity of the α-diimine Ni(II) catalysts is temperature dependent. At below −45 °C, C3 and C4 showed preferred 1,2-propylene insertion and afforded regioregular syndiotactic polypropylene with rr values of above 74%.34,35 However, regioirregular polypropylene was obtained at 0 °C.36
Brookhart et al. reported the effects of ligand symmetry on α-diimine Ni(II)-catalyzed 1-hexene polymerization in an effort to probe the possibility of realizing stereoselective α-olefin polymerization.29 The Ni(II) catalyst C4/MAO exhibited a high propensity for 1,2-insertion and the preference decreased with temperature increasing from 0 °C to 50 °C. In contrast, 2,1-insertion was preferred when the “di-t-butyl” catalyst C5/MAO was used (Scheme 3). This is mainly due to steric effects exerted from different ligand structures.
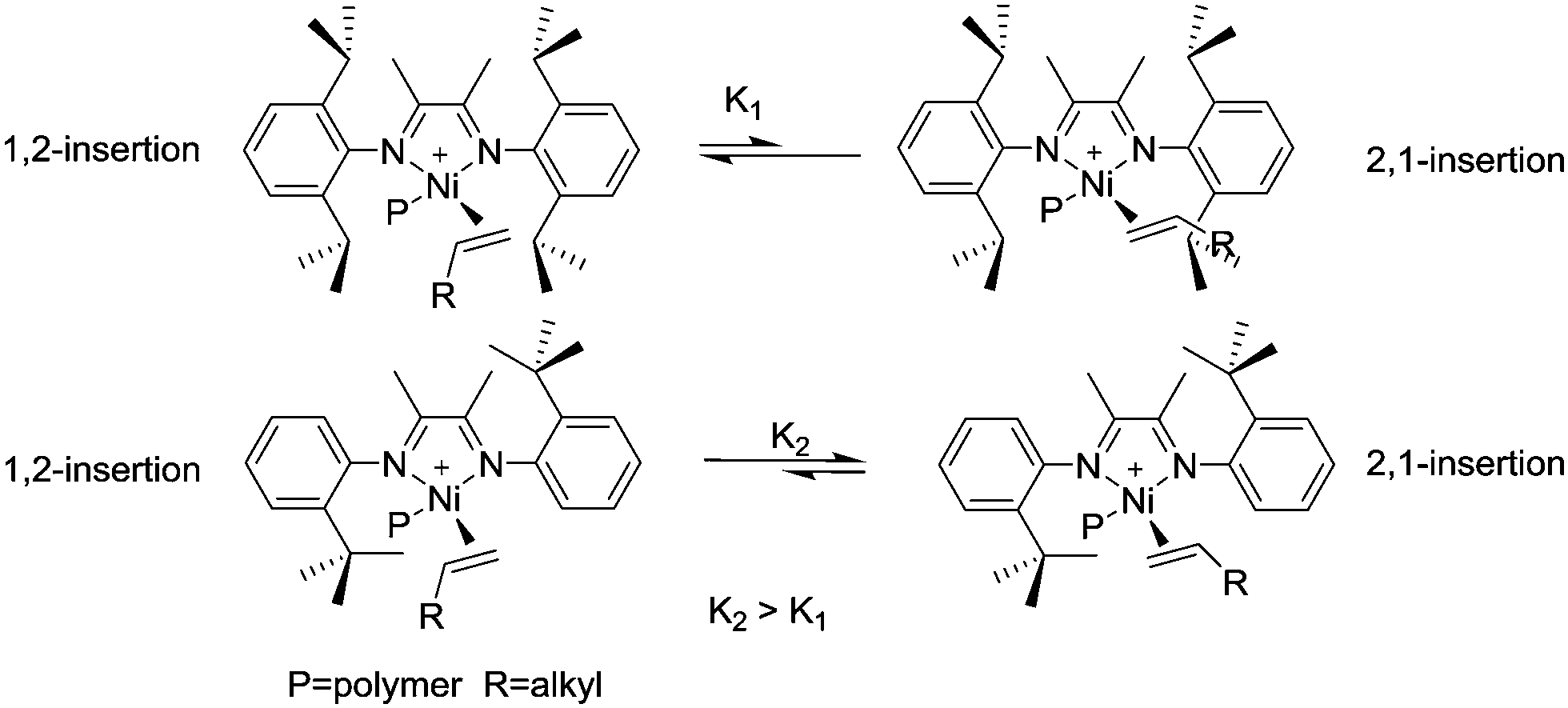 | ||
| Scheme 3 Effects of ligand symmetry on C4 and C5 catalyzed α-olefin polymerization. | ||
Coates et al. reported that C2-symmetric α-diimine Ni(II) catalyst C6 bearing a chiral sec-phenethyl moiety polymerized propylene and higher α-olefins in a living fashion and showed an interesting temperature-dependent regioselectivity (Scheme 4).37–41 The regioregular and highly isotactic polypropylene was obtained at low temperature (e.g. −78 °C) while regioirregular polypropylene with 1,2- and 3,1-enchainments was generated at higher temperature (e.g. 0 °C). An isotactic–atactic regioregular–regioirregular block copolymer can be synthesized by changing the reaction temperature during propylene polymerization. When this catalyst was used for higher α-olefin polymerization, poor regio-control was observed and an ethylene–propylene copolymer structure was obtained through 2,ω-enchainment.
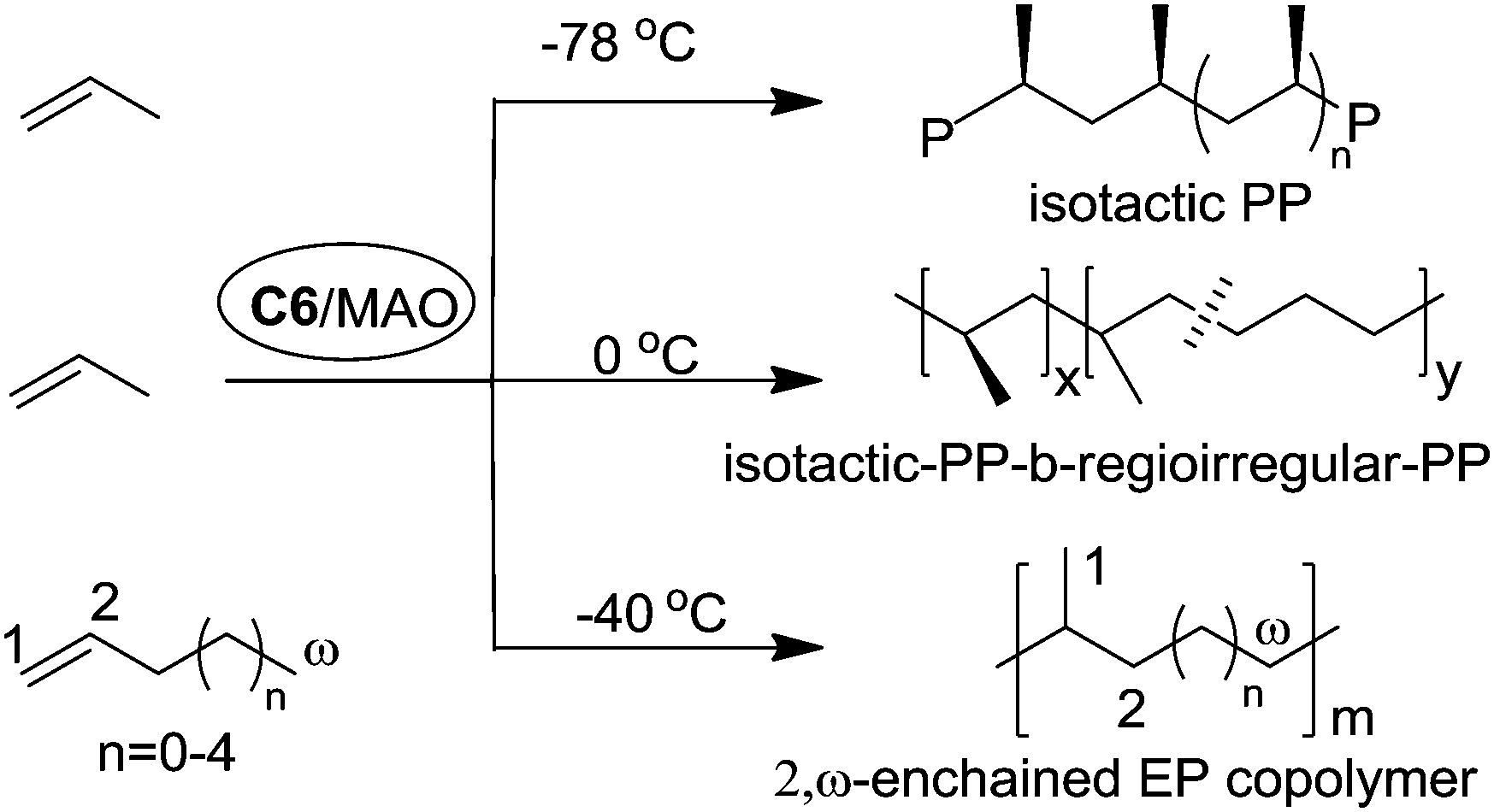 | ||
| Scheme 4 Polymerization of α-olefins with Ni(II) catalyst C6. | ||
As outlined in the mechanism shown in Scheme 2b, 1,ω-enchainment can lead to a polymer product with a highly linear microstructure. However, it is highly challenging to achieve this via precise α-olefin 2,1-insertion and a chain straightening mechanism using α-diimine Ni(II) and Pd(II) catalysts. Wu et al. reported the synthesis of polypropylene and poly(1-hexene) with an obvious Tm using camphyl based α-diimine Ni(II) catalyst C7.33,42 Coates et al. recently reported high selectivity of 1,ω-enchainment using C8 and some derivative catalysts bearing the “sandwich” type α-diimines.43 These catalysts polymerized higher α-olefins such as 1-decene to semicrystalline polyethylene-type polymers with linear units of up to 76% and melting points (Tm) of above 100 °C. Most interestingly, a similar polymer microstructure could be obtained using a mixture of α-olefins (Scheme 5). Guan et al. reported that cyclophane-based α-diimine Ni(II) catalyst C9 promoted living polymerizations of propylene and 1-hexene at elevated temperatures of up to 75 °C.44 The significantly lower level of the branching density in polypropylene and poly(1-hexene), which is only half of the value compared with traditional α-diimine Ni(II) catalysts, indicated higher selectivity for 1,ω-enchainment through 2,1-insertion of α-olefin and chain walking. Our group recently reported the tuning of polypropylene and poly(1-hexene) microstructures using a series of α-diimine Pd(II) catalysts C10 bearing both the dibenzhydryl moiety and systematically varied ligand sterics.45 These polymers are amorphous with no melting point, suggesting poor regio-control for the polymerization process.
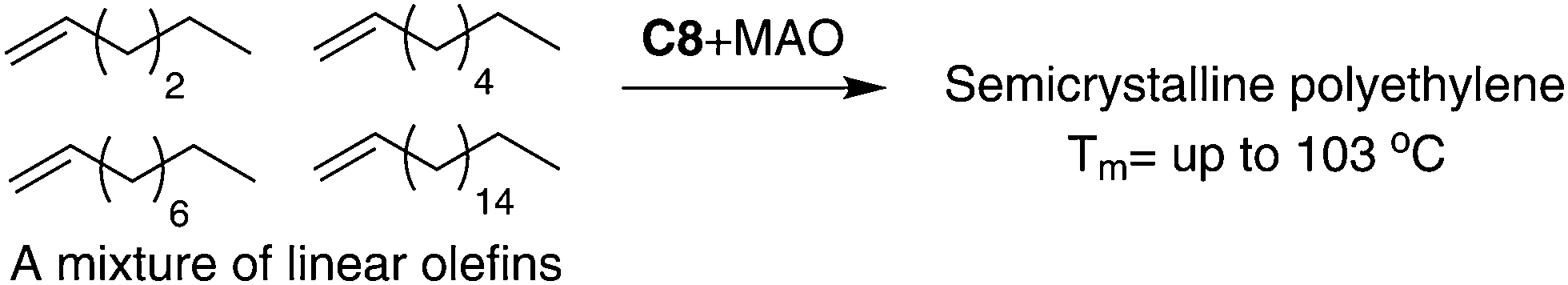 | ||
| Scheme 5 Polymerization of α-olefins with Ni(II) catalyst C8. | ||
In addition to linear α-olefin polymerization, the polymerization of branched α-olefins such as 4-methyl-1-pentene has also been investigated using conventional α-diimine Ni(II) and Pd(II) catalysts.46–48 The obtained polymers are amorphous elastomers or oils with various types of branches generated through the chain walking process.
000) could be obtained in propylene polymerization.51 Pellecchia’s group and Brookhart’s group both explored the polymerization mechanism.51,52 They found that 2,1-propylene insertion is preferred during propagation and the isotacticity originates from a chain-end control mechanism.
Bazan et al. reported that α-keto-β-diimine Ni(II) catalyst C12 can realize stereo-controlled propylene polymerization at low temperature (Scheme 6).53 For example at −60 °C, the chain walking process can be significantly suppressed and isotactic polypropylenes can be obtained. At high temperatures, atactic and regioirregular polypropylenes with 1,3-enchainment errors were obtained.
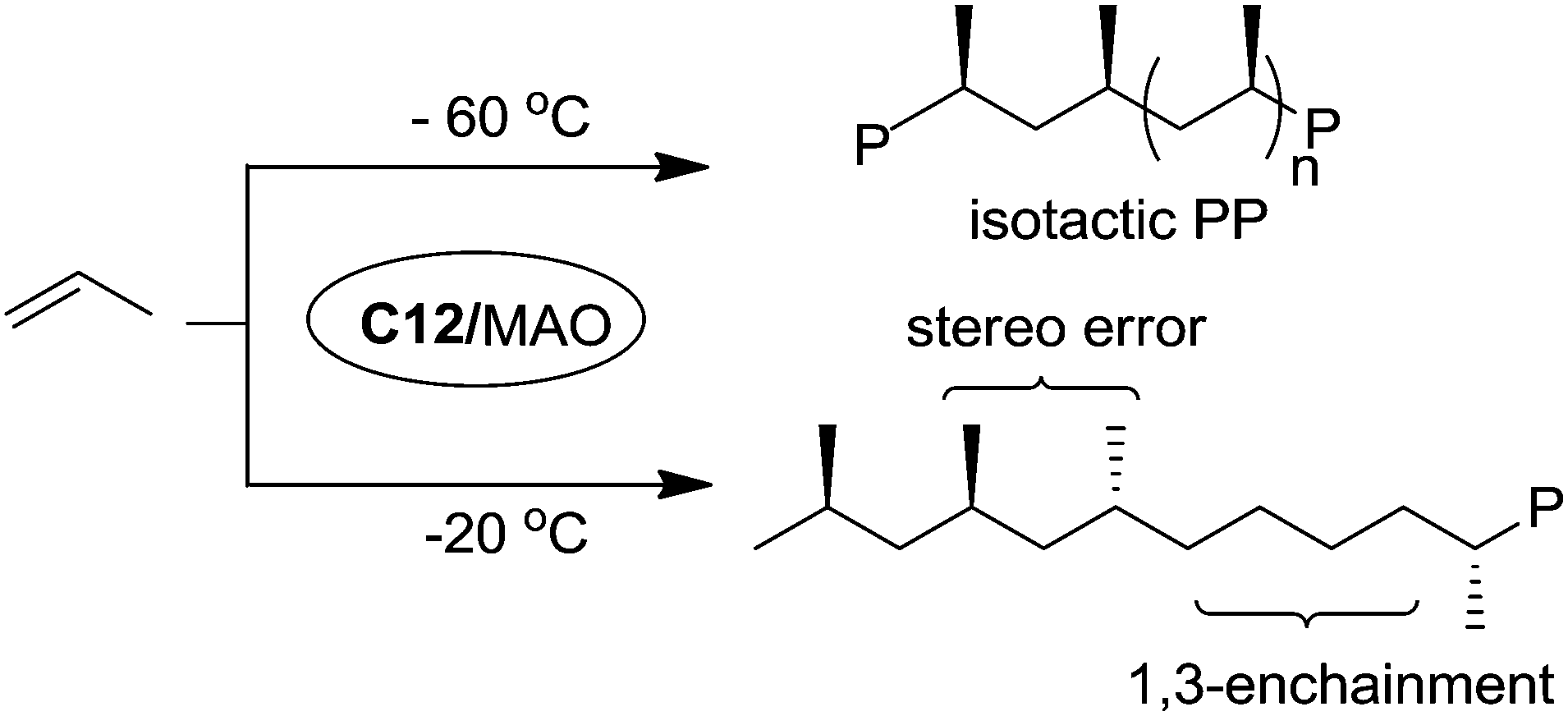 | ||
| Scheme 6 Polymerization of propylene with Ni(II) catalyst C12. | ||
The ligand structure in Ni(II) catalysts C13 and C14 can dictate the insertion fashion of 1-hexene (Scheme 7).54 Catalyst C13 with a tert-butyl substituent showed 80% selectivity of 1,2-insertion of 1-hexene, producing amorphous polymers with predominant methyl branches. In contrast, catalyst C14 with two methyl substituents showed 90% selectivity of 2,1-insertion of 1-hexene, producing semicrystalline “polyethylene” with melting points of up to 107 °C.
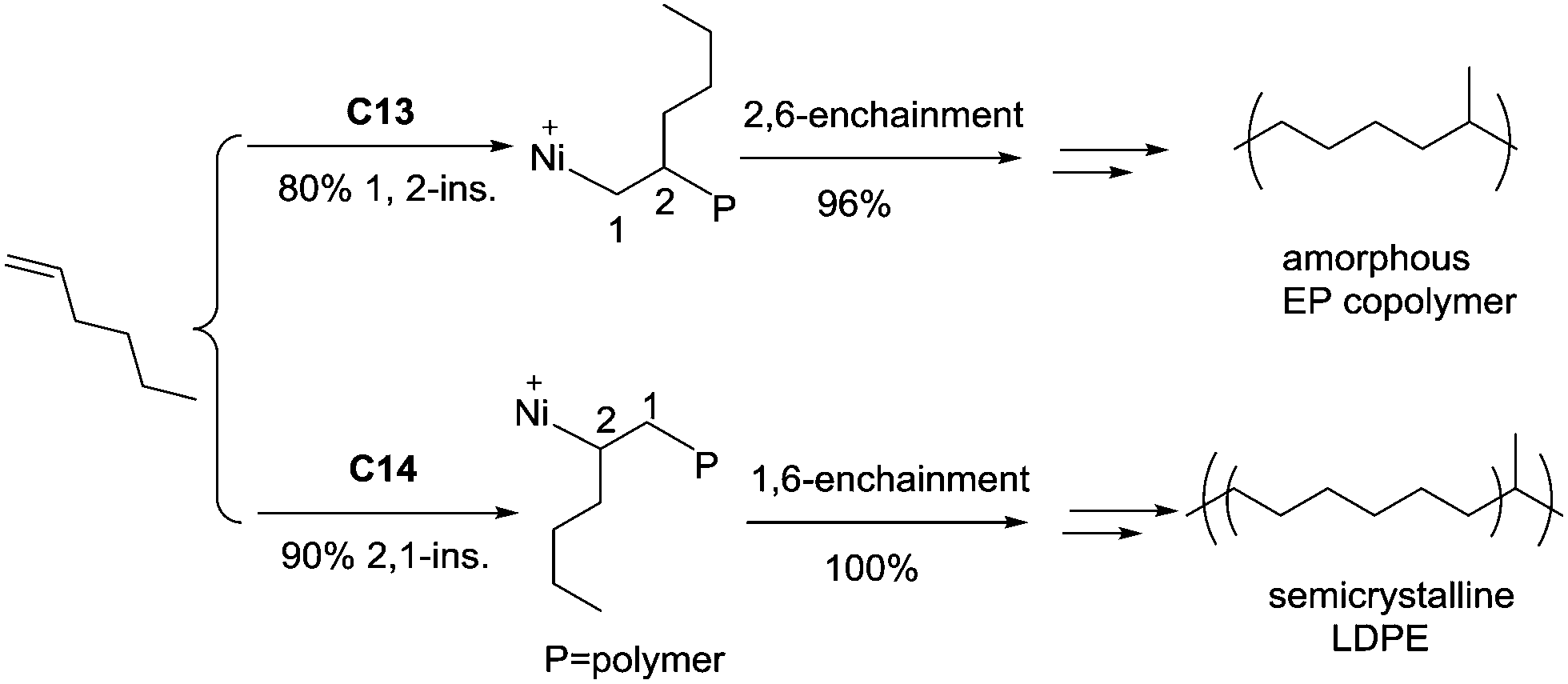 | ||
| Scheme 7 The regio-selectivity in 1-hexene polymerization mediated by Ni(II) catalysts C13 and C14. | ||
Our group recently reported that iminopyridyl Ni(II) catalyst C15 bearing both dibenzhydryl and naphthyl moieties can polymerize α-olefins with high selectivity of 1,ω-enchainment to afford polymers with melting points of up to 105 °C.55
3.2 Neutral catalysts bearing anionic ligands
Neutral catalysts bearing anionic ligands involve neutral active species in α-olefin polymerization. So far, there have been very few related reports of neutral late transition metal catalysts for α-olefin polymerization. Möhring and Fink showed that catalyst C16 with an aminobis(imino)phosphorane ligand,56 which was previously reported by Keim et al. for ethylene polymerization,57 can oligomerize propylene and higher α-olefins to give oligomers with methyl branches and well-defined repeating units. This was proposed to be due to 1,2-insertion followed by a chain walking process. The amino–imino Ni(II) catalyst C17 exhibited moderate activities for propylene polymerization and gave rubbery polypropylene at low propylene pressure (48 kPa). The salicylaldimine Ni(II) catalyst C18 was also active for propylene oligomerization and gave liquid oligomers. GC analysis indicated the presence of pentamers, hexamers and heptamers.58Nozaki et al. recently reported that C19 and related catalysts bearing imidazo-[1,5-a]quinolin-9-olate-1-ylidene ligands can suppress chain walking and exhibit high 1,2-selectivity for propylene insertion, generating polypropylenes with regio-defects of less than 1.5 mol%.59 These catalysts were also effective for 1-butene polymerization and afforded poly(1-butene)s with regio-defects of less than 0.1 mol%. However, stereo-control was not achieved in this system.
Using C20 and related phosphine-sulfonate palladium catalysts, Nozaki et al. accomplished regio- and stereo-controlled homopolymerization of propylene at 50 °C.60 A moderate triad ratio (mm = 0.59) could be achieved in this system. It is worth noting that most previously reported catalysts based on group 10 metals such as α-diimine Ni(II) complexes can only achieve stereo-control at very low temperatures (e.g. −60 °C).
4 Copolymerization of α-olefin with polar monomers
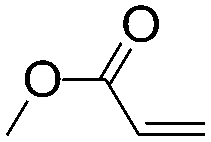
As mentioned in the Introduction section, late transition metal catalysts possess superior properties in terms of tolerance towards polar functional groups. Late transition metal catalyzed ethylene copolymerization with polar monomers has enjoyed great advances in recent years. In contrast, late transition metal catalyzed copolymerization of α-olefin with polar monomers has received much less attention. In particular, there have been very few successful examples for regio- or stereo-controlled copolymerization cases. Some of the results for copolymerization of α-olefin with various polar monomers catalyzed by different late transition metal catalysts are summarized in Table 2. It should be noted that α-diimine Ni(II) catalysts are generally very poor at incorporating polar comonomers, although they are among the most widely studied late transition metal catalysts.| Catalyst | T (°C) | Time (h) | Propylene | Comonomer | Activity (kg mol−1 h−1) | M n (×10−3) | PDIa | Incorpb (mol%) | Thermal analysisc (°C) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by GPC. b Determined by 1H or 13C NMR. c Determined by DSC. nd: not determined. | ||||||||||
| C2 | 35 | 18.5 | 6 atm |
|
2.7 | 37 | 1.8 | 0.6 | nd | 20 |
| C11 | 30 | 16 | 5.7 g |
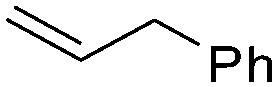
|
7.2 | 0.97 | 2.1 | 1.4 | nd | 61 |
| C11 | 30 | 16 | 7.0 g |
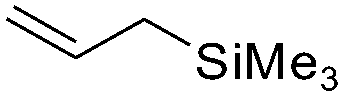
|
4.5 | 1.3 | 7.8 | 2.1 | nd | 61 |
| C11 | 30 | 16 | 7.3 g |

|
2.1 | 2.0 | 1.9 | 1.7 | nd | 61 |
| C11 | 0 | 16 | 11 g |

|
1.3 | 3.2 | 2.4 | 0.3 | nd | 61 |
| C19 | 100 | 6 | 10 g |
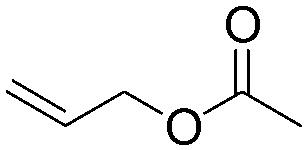
|
2.8 | 11 | 2.3 | 0.9 | nd | 59 |
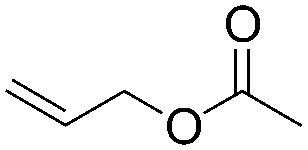
| C19 | 100 | 6 | 10 g |
|
5.4 | 13 | 2.5 | 0.5 | nd | 59 |
| C19 | 100 | 6 | 10 g |
|
0.74 | 3.0 | 3.0 | 1.1 | nd | 59 |
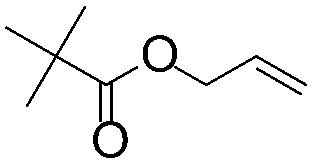
| C20 | 50 | 12 | 6.0 g |
|
0.38 | 5.8 | 2.6 | 2.9 | nd | 60 |
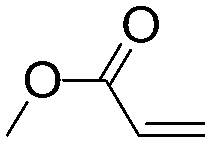
| C20 | 50 | 12 | 6.0 g |
|
0.79 | 7.5 | 1.9 | 1.5 | nd | 60 |
| C20 | 50 | 12 | 6.0 g |
|
0.58 | 11 | 2.2 | 1.7 | −10.0 (Tg), 42.9, 61.7 (Tm) | 60 |
| C20 | 50 | 12 | 6.0 g |
|
0.83 | 4.6 | 2.2 | 1.6 | −14.3 (Tg), 43.3, 61.8 (Tm) | 60 |
The typical cationic α-diimine Pd(II) catalyst C2 is effective for copolymerization of α-olefins with methyl acrylate.20 The majority of ester groups are located at the end of the branches in the resulting copolymers. The branching density of the copolymers is lower than the theoretical value from an exclusive 1,2-insertion due to the chain straightening mechanism, which is similar to the α-olefin homopolymerization scenario. The acrylate incorporation ratio is higher than that of the ethylene copolymers. For example, a 1-hexene to methyl acrylate ratio of 7:1 at equal molar concentration of the two monomers was observed in 1-hexene/methyl acrylate copolymerization, whereas the ratio was ca. 150:1 for the ethylene case. In addition, the catalytic activity and the copolymer molecular weight are markedly suppressed in the presence of polar comonomers.
Our group recently reported that sterically bulky α-diimine palladium catalysts C10 could copolymerize 1-octene with biorenewable comonomer acrylic acid, with high comonomer incorporation and high copolymer molecular weights.45a This provides an alternative strategy to access branched carboxylic acid functionalized polyolefins.
Nozaki et al. reported C11 catalyzed copolymerization of propylene with allylbenzene and various allyl sily ethers.61 The allyl comonomers were incorporated mainly into the terminal chain-end position of the copolymers.
Despite the successful synthesis of various functional copolymers, the above-mentioned late transition metal catalysts suffer from poor regio- and stereo-control in olefin (co)polymerization mainly because of the chain straightening process. If this process can be suppressed using novel catalysts, polar functionalized isotactic copolymers may be synthesized.
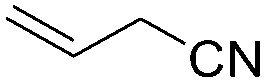
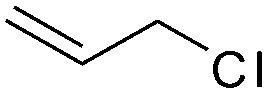
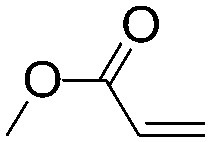
Recently, Nozaki et al. reported regio-selective copolymerization of propylene with a series of polar monomers using Pd(II) catalysts C19.59 However, the obtained functional polypropylenes exhibited an atactic stereoregularity. Using the phosphine–sulfonate palladium catalysts C20, the same group achieved moderate isotacticity in the copolymerization of propylene with various polar monomers (see Scheme 8).60 Under optimized conditions, semicrystalline isotactic polar polypropylene was obtained bearing various functional groups such as acetoxy, cyano, chloro and alkoxycarbonyl moieties. This is a very nice demonstration of the great potentials of late transition metal catalysts in achieving regio- and stereo-controlled copolymerization of α-olefin with various polar monomers.
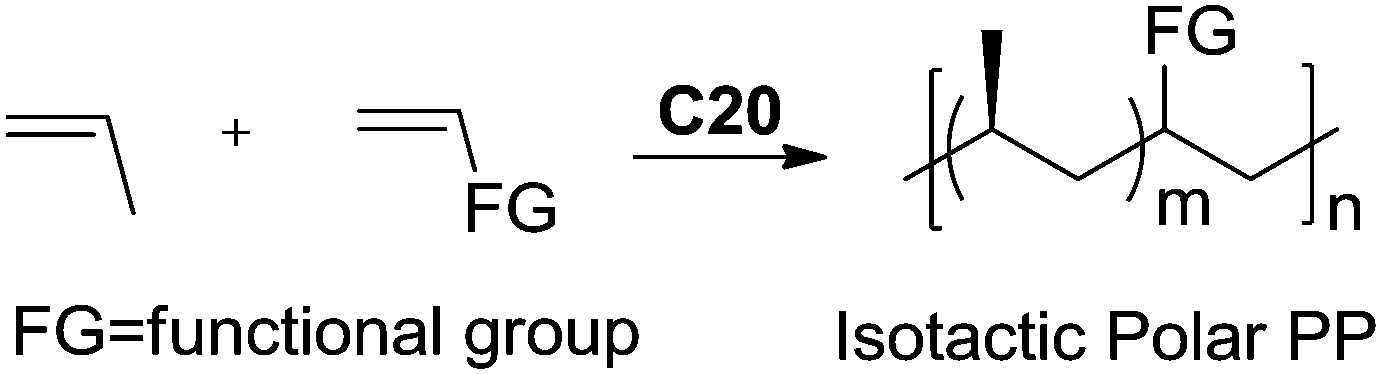 | ||
| Scheme 8 Copolymerization of propylene and polar monomers with Pd(II) catalyst C20. | ||
5 Conclusions and perspectives
Since Brookhart's seminal work on α-diimine Pd(II) and Ni(II) catalysts, a huge amount of late transition metal catalysts have been designed and studied for olefin polymerization. However, a very small fraction of these catalysts have been investigated in α-olefin polymerization. Two research directions are highly fascinating in the field of late transition metal catalyzed α-olefin polymerization: the suppression of the chain walking process to achieve regio- and stereo-controlled α-olefin polymerization; and the promotion of 1,ω-enchainment through selective 2,1-insertion and precise chain walking.It is easy to achieve regio- and stereo-specific α-olefin polymerization using early transition metal catalysts, which is currently practiced on a large scale in industry. There has been a continuous interest in preparing polar functionalized iPP (isotactic polypropylene) through direct copolymerization of propylene with polar monomers. However, these early transition metal catalysts are easily poisoned by polar monomers. In this sense, late transition-metal catalysts are promising candidates because of their low oxophilicity. Unfortunately, most late transition-metal catalysts are prone to generating amorphous and atactic polymers as a result of poor regioselectivity, poor stereoselectivity and a fast chain-walking process. It is highly challenging to solve this dilemma. Meanwhile, this also represents a major driving force for future studies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (NSFC, 21690071, 21304054, 21374108 and 51522306), Foundation of Qufu Normal University (xkJ201603), National College Students Innovation Project (201610446029) and Anhui Provincial Natural Science Foundation (1608085MB29).Notes and references
- K. Ziegler, E. Holzkamp, H. Briel and H. Martin, Angew. Chem., 1955, 67, 541 CrossRef CAS.
- G. Natta, P. Pino, P. Corradini, F. Dannusso, E. Mantica, G. Mazzanti and G. Moraglio, J. Am. Chem. Soc., 1955, 77, 1708 CrossRef CAS.
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169 CrossRef CAS PubMed.
- V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283 CrossRef CAS PubMed.
- G. W. Coates, Chem. Rev., 2000, 100, 1223 CrossRef CAS PubMed.
- A. Nakamura, S. Ito and K. Nozaki, Chem. Rev., 2009, 109, 5215 CrossRef CAS PubMed.
- E. Y. X. Chen, Chem. Rev., 2009, 109, 5157 CrossRef CAS PubMed.
- Z. Guan and C. S. Popeney, Top. Organomet. Chem., 2009, 2, 179 CrossRef.
- H. Makio, H. Terao, A. Iwashita and T. Fujita, Chem. Rev., 2011, 111, 2363 CrossRef CAS PubMed.
- D. Takeuchi, Macromol. Chem. Phys., 2011, 212, 1545 CrossRef CAS.
- D. Takeuchi, Polym. J., 2012, 44, 919 CrossRef CAS.
- Z. Dong and Z. Ye, Polym. Chem., 2012, 3, 286 RSC.
- (a) L. H. Guo and C. L. Chen, Sci. China: Chem., 2015, 58, 1663 CrossRef CAS; (b) L. H. Guo, S. Y. Dai, X. L. Sui and C. L. Chen, ACS Catal., 2016, 6, 428 CrossRef CAS.
- Y. Chen, L. Wang, H. Yu, Y. Zhao, R. Sun, G. Jing, J. Huang, H. Khalid, N. M. Abbasi and M. Akram, Prog. Polym. Sci., 2015, 45, 23 CrossRef CAS.
- (a) J. P. Hogan and R. L. Banks, U.S. Patent, Polymers and Production Thereof, 2825721, 1958 Search PubMed; (b) J. P. Hogan and R. L. Banks and U.S. Patent, Polymerization Catalysts, 2951816, 1960 Search PubMed; (c) H. R. Sailors and J. P. Hogan, J. Macromol. Sci., Chem., 1981, 15, 1377 CrossRef.
- B. Cornils and W. A. Herrmann, Applied Homogeneous Catalysis with Organometallic compounds, VCH, New York, 1996, p. 1 Search PubMed.
- (a) T. C. M. Chung, Green Sustainable Chem., 2012, 2, 29 CrossRef CAS; (b) J. Y. Dong and Y. L. Hu, Coord. Chem. Rev., 2006, 250, 47 CrossRef CAS; (c) N. M. G. Franssen, J. N. H. Reek and B. Bruin, Chem. Soc. Rev., 2013, 42, 5809 RSC; (d) X. L. Sui, C. W. Hong, W. M. Pang and C. L. Chen, Mater. Chem. Front., 2017, 1, 967 RSC; (e) Y. N. Na, D. Zhang and C. L. Chen, Polym. Chem., 2017, 8, 2405 RSC.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414 CrossRef CAS.
- L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267 CrossRef CAS.
- S. Mecking, L. K. Johnson, L. Wang and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 888 CrossRef CAS.
- C. M. Killian, D. J. Tempel, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 11664 CrossRef CAS.
- (a) Z. Guan, P. M. Cotts, E. F. McCord and S. J. McLain, Science, 1999, 283, 2059 CrossRef CAS PubMed; (b) D. H. Leung, J. W. Ziller and Z. Guan, J. Am. Chem. Soc., 2008, 130, 7538 CrossRef CAS PubMed; (c) C. Chen, S. Luo and R. F. Jordan, J. Am. Chem. Soc., 2008, 130, 12892 CrossRef CAS PubMed; (d) F. S. Liu, H. B. Hu, Y. Xu, L. H. Guo, S. B. Zai, K. M. Song, H. Y. Gao, L. Zhang, F. M. Zhu and Q. Wu, Macromolecules, 2009, 42, 7789 CrossRef CAS; (e) M. M. Wegner, A. K. Ott and B. Rieger, Macromolecules, 2010, 43, 3624 CrossRef CAS; (f) C. Chen and R. F. Jordan, J. Am. Chem. Soc., 2010, 132, 10254 CrossRef CAS PubMed; (g) C. Chen, S. Luo and R. F. Jordan, J. Am. Chem. Soc., 2010, 132, 5273 CrossRef CAS PubMed; (h) H. Liu, W. Z. Zhao, X. Hao, C. Redshaw, W. Huang and W. H. Sun, Organometallics, 2011, 30, 2418 CrossRef CAS; (i) D. Zhang, E. T. Nadres, M. Brookhart and O. Daugulis, Organometallics, 2013, 32, 5136 CrossRef CAS; (j) J. L. Rhinehart, L. A. Brown and B. K. Long, J. Am. Chem. Soc., 2013, 135, 16316 CrossRef CAS PubMed; (k) J. L. Rhinehart, N. E. Mitchell and B. K. Long, ACS Catal., 2014, 4, 2501 CrossRef CAS; (l) K. E. Allen, J. Campos, O. Daugulis and M. Brookhart, ACS Catal., 2015, 5, 456 CrossRef CAS; (m) L. H. Guo, S. Y. Dai and C. L. Chen, Polymers, 2016, 8, 37 CrossRef; (n) L. H. Guo, X. Y. Jing, S. Y. Xiong, W. J. Liu, Y. L. Liu, Z. Liu and C. L. Chen, Polymers, 2016, 8, 839 Search PubMed; (o) C. Zhou, W. J. Liu, O. Daugulis and M. Brookhart, J. Am. Chem. Soc., 2016, 138, 16120 CrossRef PubMed; (p) B. K. Long, J. M. Eagan, M. Mulzer and G. W. Coates, Angew. Chem., Int. Ed., 2016, 55, 7106 CrossRef CAS PubMed.
- (a) S. Takano, D. Takeuchi, K. Osakada, N. Akamatsu and A. Shishido, Angew. Chem., Int. Ed., 2014, 53, 9246 CrossRef CAS PubMed; (b) L. Zhu, Z. S. Fu, H. J. Pan, W. Feng, C. L. Chen and Z. Q. Fan, Dalton Trans., 2014, 43, 2900 RSC; (c) R. K. Wang, X. L. Sui, W. M. Pang and C. L. Chen, ChemCatChem, 2016, 8, 434 CrossRef CAS; (d) Y. N. Na, X. Wang, K. Lian, Y. Zhu, W. Li, Y. Luo and C. L. Chen, ChemCatChem, 2017, 9, 1062 CrossRef CAS.
- (a) W. P. Zou and C. L. Chen, Organometallics, 2016, 35, 1794 CrossRef CAS; (b) R. K. Wang, M. H. Zhao and C. L. Chen, Polym. Chem., 2016, 7, 3933 RSC; (c) S. Y. Dai, X. L. Sui and C. L. Chen, Angew. Chem., Int. Ed., 2015, 54, 9948 CrossRef CAS PubMed; (d) S. Y. Dai and C. L. Chen, Angew. Chem., Int. Ed., 2016, 55, 13281 CrossRef CAS PubMed; (e) M. Li, X. B. Wang, Y. Luo and C. L. Chen, Angew. Chem., Int. Ed., 2017 DOI:10.1002/anie.201706249; (f) K. Lian, Y. Zhu, W. Li, S. Y. Dai and C. L. Chen, Macromolecules, 2017, 50, 6074 CrossRef CAS.
- J. Boor, Ziegler–Natta Catalysts and Polymerizations, Academic Press, New York, 1979, p. 1 Search PubMed.
- W. Kaminsky, Macromol. Chem. Phys., 1996, 197, 3907 CrossRef CAS.
- K. D. Hungenberg, J. Kerth, F. Langhauser, H. J. Müeller and P. Müeller, Angew. Makromol. Chem., 1995, 227, 159 CrossRef CAS.
- H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed., 1995, 34, 1143 CrossRef CAS.
- E. F. McCord, S. J. McLain, L. T. J. Nelson, S. D. Ittel, D. Tempel, C. M. Killian, L. K. Johnson and M. Brookhart, Macromolecules, 2007, 40, 410 CrossRef CAS.
- Z. Ye, W. Feng, S. Zhu and Q. Yu, Macromol. Rapid Commun., 2006, 27, 871 CrossRef CAS.
- S. A. Svejda, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1999, 121, 10634 CrossRef CAS.
- A. C. Gottfried and M. Brookhart, Macromolecules, 2003, 36, 3085 CrossRef.
- (a) J. Liu, D. Chen, H. Wu, Z. Xiao, H. Gao, F. Zhu and Q. Wu, Macromolecules, 2014, 47, 3325 CrossRef CAS; (b) G. Leone, M. Mauri, F. Bertini, M. Canetti, D. Piovani and G. Ricci, Macromolecules, 2015, 48, 1304 CrossRef CAS; (c) K. S. O'Connor, A. Watts, T. Vaidya, A. M. LaPointe, M. A. Hillmyer and G. W. Coates, Macromolecules, 2016, 49, 6743 CrossRef.
- D. Pappalardo, M. Mazzeo, S. Antinucci and C. Pellecchia, Macromolecules, 2000, 33, 9483 CrossRef CAS.
- C. Pellecchia, A. Zambelli, L. Oliva and D. Pappalardo, Macromolecules, 1996, 29, 6990 CrossRef CAS.
- C. Pellecchia and A. Zambelli, Macromol. Rapid Commun., 1996, 17, 333 CrossRef CAS.
- A. E. Cherian, J. M. Rose, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 13770 CrossRef CAS PubMed.
- J. M. Rose, A. E. Cherian and G. W. Coates, J. Am. Chem. Soc., 2006, 128, 4186 CrossRef CAS PubMed.
- J. M. Rose, A. E. Cherian, J. H. Lee, L. A. Archer, G. W. Coates and L. J. Fetters, Macromolecules, 2007, 40, 6807 CrossRef CAS.
- J. M. Rose, F. Deplace, N. A. Lynd, Z. Wang, A. Hotta, E. B. Lobkovsky, E. J. Kramer and G. W. Coates, Macromolecules, 2008, 41, 9548 CrossRef CAS.
- C. Ruiz-Orta, J. P. Fernandez-Blazquez, A. M. Anderson-Wile, G. W. Coates and R. G. Alamo, Macromolecules, 2011, 44, 3436 CrossRef CAS.
- F. Liu, H. Gao, Z. Hu, H. Hu, F. Zhu and Q. Wu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3859 CrossRef CAS.
- (a) T. Vaidya, K. Klimovica, A. M. LaPointe, I. Keresztes, E. B. Lobkovsky, O. Daugulis and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 7213 CrossRef CAS PubMed; (b) K. S. O'Connor, A. Watts, T. Vaidya, A. M. LaPointe, M. A. Hillmyer and G. W. Coates, Macromolecules, 2016, 49, 6743 CrossRef.
- D. H. Camacho and Z. Guan, Macromolecules, 2005, 38, 2544 CrossRef CAS.
- (a) L. H. Guo, C. Zou, S. Y. Dai and C. L. Chen, Polymers, 2017, 9, 122 CrossRef; (b) S. Y. Dai, S. X. Zou, W. Zhang and C. L. Chen, Macromolecules, 2016, 49, 8855 CrossRef CAS.
- H. Gao, X. Liu, Y. Tang, J. Pan and Q. Wu, Polym. Chem., 2011, 2, 1398 RSC.
- H. Gao, J. Pan, L. Guo, D. Xiao and Q. Wu, Polymer, 2011, 52, 130 CrossRef CAS.
- L. Guo, H. Gao, L. Li and Q. Wu, Macromol. Chem. Phys., 2011, 212, 2029 CrossRef CAS.
- B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049 CrossRef CAS.
- G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 849 RSC.
- B. L. Small and M. Brookhart, Macromolecules, 1999, 32, 2120 CrossRef CAS.
- C. Pellecchia, M. Mazzeo and D. Pappalardo, Macromol. Rapid Commun., 1998, 19, 651 CrossRef CAS.
- J. D. Azoulay, H. Gao, Z. A. Koretz, G. Kehr, G. Erker, F. Shimizu, G. B. Galland and G. C. Bazan, Macromolecules, 2012, 45, 4487 CrossRef CAS.
- H. Hu, H. Gao, D. Chen, G. Li, Y. Tan, G. Liang, F. Zhu and Q. Wu, ACS Catal., 2015, 5, 122 CrossRef CAS.
- S. Y. Dai, X. L. Sui and C. L. Chen, Chem. Commun., 2016, 52, 9113 RSC.
- V. M. Möhring and G. Fink, Angew. Chem., Int. Ed., 1985, 24, 1001 CrossRef.
- W. Keim, R. Appel, A. Storeck, C. Kruger and R. Goddard, Angew. Chem., Int. Ed., 1981, 20, 116 CrossRef.
- L. K. Johnson, A. M. A. Bennett, S. D. Ittel, L. Wang, A. Parthasarathy, E. Hauptman, R. D. Simpson, J. Feldman and E. B. Coughlin, WO Patent, Polymerization of olefins, WO1998030609, 1997 Search PubMed.
- R. Nakano and K. Nozaki, J. Am. Chem. Soc., 2015, 137, 10934 CrossRef CAS PubMed.
- W. J. Tao, R. Nakano, S. Ito and K. Nozaki, Angew. Chem., Int. Ed., 2016, 55, 2835 CrossRef CAS PubMed.
- T. Kawakami, S. Ito and K. Nozaki, Dalton Trans., 2015, 44, 20745 RSC.
| This journal is © the Partner Organisations 2017 |