
Recent advances in AIEgen-based luminescent metal–organic frameworks and covalent organic frameworks
Li
Ma
,
Xiao
Feng
 *,
Shan
Wang
and
Bo
Wang
*,
Shan
Wang
and
Bo
Wang
Beijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, Key Laboratory of Cluster Science, Ministry of Education, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: fengxiao86@bit.edu.cn
First published on 14th August 2017
Abstract
AIEgens have evoked an immense amount of interest and progressed significantly since the first report of the aggregation-induced emission (AIE) concept in 2001. Metal–organic frameworks (MOFs) and covalent organic frameworks (COFs) are porous crystalline solids that allow molecular building units to be aligned based on strong interactions and bonding in a well-defined manner, offering possibilities to introduce functionalities and complexities within their backbones. Introducing AIEgens to construct such crystalline solids, in which the AIEgens are stitched together by covalent or coordinate bonds and placed in specific geometric and spatial arrangements, provides a powerful platform for the investigation of the underlying AIE mechanisms and the design of highly emissive porous materials. This mini review will give a brief introduction to AIEgen-based MOFs and COFs and focus on their photoluminescence properties and potential applications. We will discuss how the AIEgens are rigidified in these crystalline solids and review how such a concept can be utilized to remarkably improve the performance of the corresponding optoelectronics and sensors.
 Li Ma | Li Ma received her MS degree in organic chemistry from the Capital Normal University in 2014. Then she joined Prof. Bo Wang’s group as a doctoral student at the Beijing Institute of Technology. Her current research interest focuses on developing porous organic cages and covalent organic framework materials. |
 Xiao Feng | Professor Xiao Feng received his BS degree in material chemistry in 2008 and PhD degree in material science in 2013 from the Beijing Institute of Technology. He carried out a joint PhD research program at the Institute for Molecular Science, Japan (2009–2012). Now he is an associate professor at the Beijing Institute of Technology and his current research interests focus on functional porous materials, including metal–organic frameworks and covalent organic frameworks. |
 Shan Wang | Shan Wang received her MS degree in organic chemistry from the Central China Normal University in 2014. Then she joined Prof. Bo Wang’s group as a doctoral student at the Beijing Institute of Technology. Her current research interest focuses on developing new metal–organic framework and covalent organic framework materials. |
 Bo Wang | Professor Bo Wang obtained his BS and MS degrees from the Peking University in 2004 and the University of Michigan in 2006, respectively, and his PhD from the University of California Los Angeles in 2008. He has been a professor in the School of Chemistry and Chemical Engineering, Beijing Institute of Technology since 2011. His research interests focus on novel functional porous material design and synthesis, energy storage, advanced battery materials, hydrogen and methane storage, gas purification, and toxicant capture and sensing. |
1. Introduction
Developing luminescent materials is indispensable to human life in the areas of photoluminescence and electroluminescence devices, bioimaging, therapeutics, and chemosensing. Despite high emission in dilute solutions, most luminophores suffer from low luminous efficiency in the solid state, which greatly limits their applications. The partial or complete quenching of emission upon molecular aggregation, known as aggregation-caused quenching (ACQ), is attributed to close packing of luminophores with a large planar π component. In 2001, Ben Zhong Tang pioneered an intriguing area of aggregation-induced emission (AIE), in which the luminophores are non- or less-emissive in dilute solution and can emit strongly in the aggregated state.1 The development of AIE has raised a revolution in the molecular design of luminescent materials.2–11 A variety of AIE luminogens (AIEgens), featuring one conjugated stator pendant with multiple aromatic peripheral rotors, have been prepared. The representative AIEgen systems include tetraphenylethylene (TPE),9 hexaphenylsilole,12 hexaphenylbenzene,13 multiaryl-substitute pyrroles,14,15 tetraaryl-substitute 1,3-butadienes,16,17etc.Numerous efforts have been devoted to exploring the underlying mechanism of AIE.2–5 Several plausible theories have been proposed, including restriction of intramolecular rotation (RIR), formation of J-aggregates or excimers, conformational planarization, and excited-state intramolecular proton transfer. A more comprehensive AIE mechanism involves the restriction of intramolecular vibration (RIV), which is an integration of RIR and restriction of intramolecular motion (RIM). In this theory, AIEgens consume the excited energy through a nonradiative emission process via free rotation, vibration and movement in dilute solutions, where the AIEgen molecules have limited interactions with each other. In the aggregated state, these nonradiative pathways are blocked due to RIM originating from structural rigidification and permit deactivation by either fluorescence or phosphorescence. Compared with ACQ luminophores, the AIEgens mostly interact with each other or with guest molecules through intermolecular steric interactions with the absence of close π–π interactions.
Metal–organic frameworks (MOFs) and covalent organic frameworks (COFs) are two emerging classes of crystalline polymeric and porous materials. MOFs are assembled by metal ions/clusters and organic linkers, whereas COFs are constructed by pure organic building blocks. In recent years, utilizing AIEgens to construct MOFs18–30 and COFs31–36 has been of great interest to researchers. Unlike the AIEgens, which interact via non-covalent bonds in their aggregated state in an unpredictable manner, the AIE building units are fixed in predesignable positions by covalent/coordinate bonds with the aid of non-covalent interactions in COFs and MOFs. The topologies, atomic positions, and packing modes can be easily tuned in these crystalline polymeric materials, thus enabling deep understanding of the underlying mechanism of AIE. In addition, from a functional point of view, integration of AIEgens into MOFs and COFs exhibits several unique advantages: (1) the optical properties and electronic transition energies of AIEgens, including emission color, wavelength and bandgap, can be easily engineered by varying the metal nodes or organic linkers to meet the demand of practical applications; (2) the rigid skeletons and specific topologies based on reticular chemistry can efficiently restrict the intramolecular rotations, vibrations, and motions and avoid self-quenching, thus dramatically boosting the emission efficiency; (3) the highly porous nature, tunable inner chemical environment, open channels in the framework, and adjustable pore size and shape enable the guest molecules to fully and specifically interact with the AIEgen anchored to the frameworks, leading to a significant improvement in sensing performance.
These advantages provide a platform for the design of novel luminescent materials with intriguing properties. It should be noted that there are a lot of AIEgen-based amorphous porous materials and highly luminescent crystalline porous materials constructed by non-AIEgens have been prepared, and several excellent reviews have covered these exciting developments.37–43 This review is not intended to be comprehensive, but will focus on the recent advances of AIEgen-based MOFs and COFs in terms of their luminescent properties and potential applications, and we hope that this work will promote further development of luminescent porous materials.
2. AIEgen-based MOFs
MOFs are inorganic–organic hybrid materials bearing diverse structures, large surface areas, and versatile functionalities. They are the first family of highly ordered crystalline porous materials designed and synthesized using reticular chemistry. Owing to the rich choice of metal species, organic linkers and coordination patterns, thousands of MOFs have been designed and synthesized and have presented remarkable potential in gas storage and separation, sensing, catalysis, energy storage, drug delivery, etc.38–41,44–51 Plenty of research efforts have been devoted to designing luminescent MOFs due to their easy-to-functionalize surface and tunable porosity that can facilitate guest–host interactions.38–41 Particularly, AIEgen-based MOFs have emerged in recent years and attracted extensive attention thanks to their intriguing optical features.2.1 Mechanism study: tuning fluorescence by coordination in a rigid MOF matrix
Investigation of the underlying mechanism is crucial for both fundamental understanding and advanced applications. The AIE mechanism has been widely studied in small molecule systems through theoretical and experimental methods, and many theories have been proposed.2–5 As aforementioned, restriction of intramolecular motion (RIM), which could ensure the dissipation of excited state energy via a radiative pathway, has become the main mechanistic picture for the AIE effect. The AIEgen-based MOFs with atomically precise structures offer a great platform to gain a deeper understanding and further insight into the AIE mechanisms.Tetraphenylethylene (TPE) derivatives, an iconic class of AIEgens, have been widely used to construct MOFs (Fig. 1). In TPE-based small molecules, the fast rotation of the phenyl rings as well as ethylenic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond twist vibrations can quench the fluorescence in dilute solutions, and these motions are suppressed and turn-on the fluorescence in the aggregated state.
C bond twist vibrations can quench the fluorescence in dilute solutions, and these motions are suppressed and turn-on the fluorescence in the aggregated state.
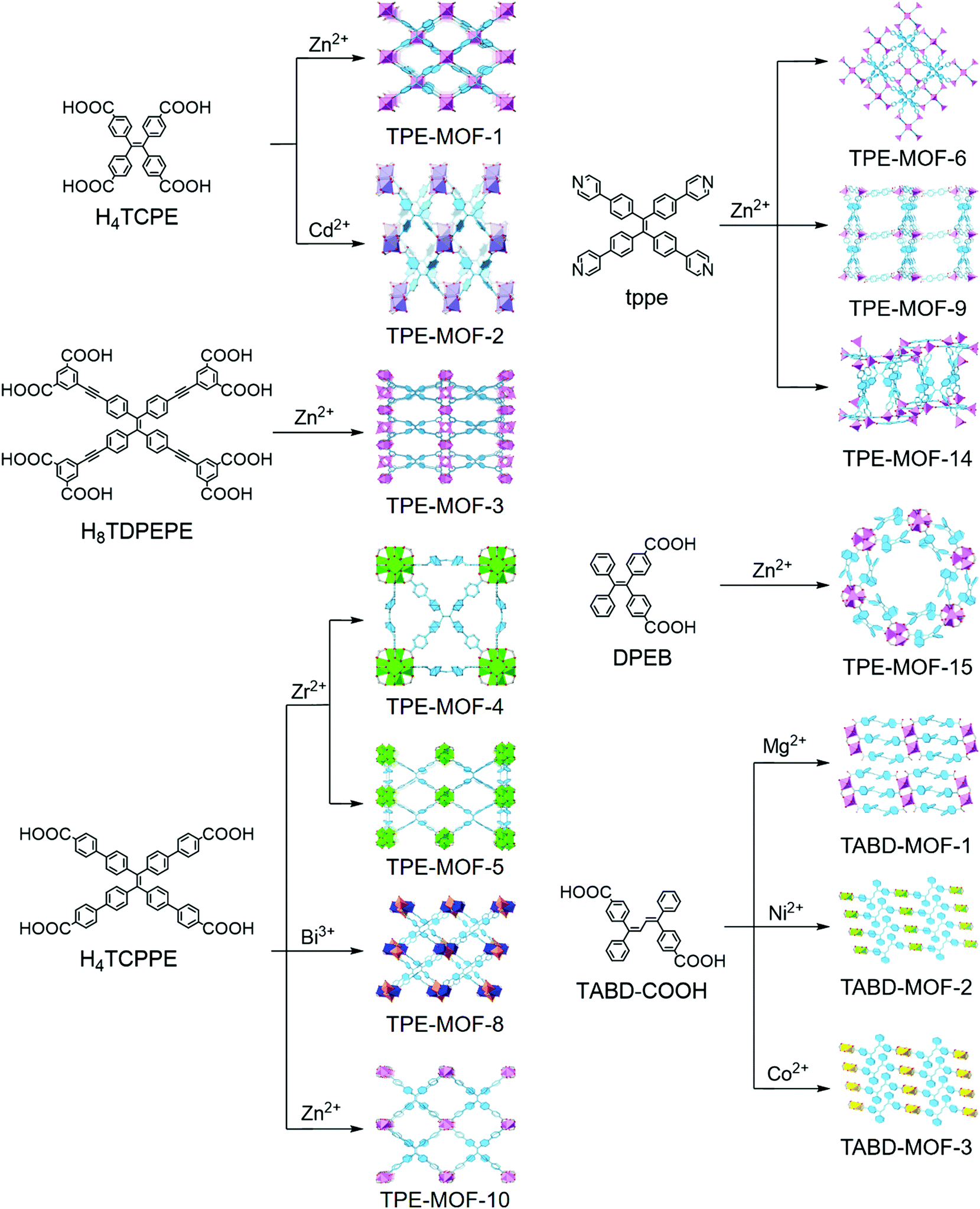 | ||
| Fig. 1 Syntheses of AIEgen-based MOFs. | ||
Dincă and co-workers were the first to report the TPE-based luminescent MOFs, which are constructed by using tetrakis(4-carboxyphenyl)ethylene (H4TCPE) as organic struts and Zn2+ or Cd2+ as metal nodes.18 Despite the spatial separation between the neighboring TPE cores imposed by coordination, both Zn2(TCPE)(H2O)2·4DEF (TPE-MOF-1) and Cd2(TCPE)(DEF)(C2H5OH)2·DEF (TPE-MOF-2) are luminescent and exhibit biexponential fluorescence decays in line with those observed for H4TCPE aggregates. They attribute the biexponential decay to either the formation of excimers or the existence of inhomogeneous phenyl ring rotation or flipping kinetics. Notably, activated TPE-MOF-1 exhibits a distinct response when exposed to ethylenediamine, cyclohexanone, and acetaldehyde. To further study the mechanism that induces fluorescence in TPE-based MOFs, they have prepared a deuterated MOF (Zn2(TCPE-d16)(DEF)2·2DEF, d16-TPE-MOF-1) that is structurally analogous to TPE-MOF-1. d16-TPE-MOF-1 is a 2D framework composed of paddlewheel Zn2(O2C–)4 secondary building units (SBU) linked by TCPE4-d16 ligands. The DEF molecules occupy both the axial sites on the Zn atoms in the SBU and the pores. They combined solid-state nuclear magnetic resonance (NMR) and density functional theory (DFT) studies with variable-temperature XRD experiments to evaluate the dynamics of the phenyl ring in the rigid TPE-based MOF structures.20 The results show that the guest DEF molecule in the pores likely prevents fast flipping of the TPE phenyl rings with an energy barrier of 43(6) kJ mol−1, and the phenyl ring flips will be activated by elevating the temperature higher than 373 K, at which temperature the guest DEF molecules will start to be liberated. They proposed that the twisting of the ethylenic CC bond may gate the torsion of the phenyl rings, and both of them are crucial for quenching the emission in TPE.
Considering the distinct demand for different applications, the fluorescence quantum yield should be either high enough to improve the energy utilization (e.g., light emitting diodes) or low enough to enable turn-on detection (e.g., chemo- or bio-sensors). Tuning the torsional barrier for phenyl ring flipping in empty porous AIEgen-based MOFs is one of the most important factors to adjust their luminous efficiency.
With an aim toward designing an AIEgen-MOF with a default dark state through decreasing the torsional barrier of phenyl ring torsion, Dincă and co-workers activated TPE-MOF-1 to afford a guest-free material. However, the intrinsic barrier is already too high to achieve a turn-off state. Therefore, to further reduce the internal torsional potential, they incorporated an ethynyl-extended TPE-based ligand (H8TDPEPE) into a zinc-based MOF to form Zn4(TDPEPE) (H2O)4(DEF)4·(H2O)3(DEF)9.5 (TPE-MOF-3).19 The p-ethynyl groups in H8TDPEPE are able to promote almost barrierless torsional motion for neighboring phenyl rings. The DFT calculations show that the activation barrier of the phenyl rings in TDPEPE8− constrained within TPE-MOF-3 (9 kcal mol−1) is 3 kcal mol−1 smaller than that of TPE-MOF-1 and 3 kcal mol−1 larger than the flipping barrier in free TPE. The relative values of the activation barrier for phenyl ring flipping agree with their fluorescence quantum yields (TPE-MOF-3, 9%; TPE-MOF-1, 35%; free TPE, non-emissive). They found that the surprising instability of TPE-MOF-3, which will collapse upon activation, originates from the inherent high strain energy. These findings may be useful for empirical prediction of the stabilities of certain MOFs.
With an eye toward increasing the luminous efficiency of an AIEgen-MOF, the AIEgen linkers are suggested to be rigidified in MOFs by structural constraint. Omary, Zhou, and co-workers have demonstrated that fixing an extended TPE-based linker (tetrakis[4-(4-carboxyphenyl)phenyl]-ethene, H4TCPPE) into a Zr-based MOF, PCN-94 (TPE-MOF-4), can efficiently control the linker conformation and enhance the quantum yield.22 TPE-MOF-4 adopts a 4,12-c 2-nodal net with ftw topology if the TCPPE is simplified as a 4-connected node. It is composed of a 12-connected Zr6 cluster and TPE-core linker. Compared with TCPPE, the average dihedral angles in TPE-MOF-4 are much larger, resulting in a breakdown of the conjugate system and a corresponding 75 nm blue shift in the emission bands (TPE-MOF-4, 470 nm, blue light; H4TCPPE, 545 nm, yellow light). The fluorescence quantum yield of TPE-MOF-4 can reach 99.9 ± 0.5% under Ar atmosphere, which is much higher than that of H4TCPPE in the solid state (30%). Unlike H4TCPPE which exhibits a biexponential decay profile attributed to monomer and excimer emission, TPE-MOF-4 displays a monoexponential and fast radiative decay. It demonstrates that neither the intramolecular nor intermolecular excited state distortion is permitted, since the pertinent aromatic units are constrained via the coordination to the Zr6 cluster centers. It is concluded that the reduction of intra- and intermolecular interactions and the restricted motion of the phenyl rings by rigidifying AIEgens in MOFs can remarkably increase the fluorescence quantum yield of MOFs, and may be beneficial for potential applications in optoelectronics and turn-off sensors.
Apart from tuning the linkers or metal ions, varying topologies can also adjust the photophysical properties of MOFs. By changing the reaction conditions, Su, Zhou, and co-workers23 prepared another Zr-MOF (PCN-128W, denoted as TPE-MOF-5W, W stands for white color under room light) starting from the same ligand (H4TCPPE) and metal salt (ZrCl4) as TPE-MOF-4. TPE-MOF-5W is composed of a TCPPE core linker and an 8-connected rather than 12-connected Zr6 cluster. It adopts a csq-a topology and emits bright blue light (470 nm). Notably, TPE-MOF-5W exhibits a unique piezofluorochromic behavior. Upon grinding the activated sample of TPE-MOF-5W, it will change to a green-light emitting (538 nm) framework (PCN-128Y, denoted as TPE-MOF-5Y, Y stands for yellow color under room light). The framework can be viewed as a microscissor lift, while TPE-MOF-5W and TPE-MOF-5Y can be considered as the ascent mode and descent mode, respectively. The coordination sites between the Zr6 clusters are occupied by trifluoroacetate (TFA) groups in the freshly prepared sample of TPE-MOF-5W, locking the TCPPE linkers in an “open” conformation. When these TFA groups are removed by treating with strong acid and replaced by OH/H2O groups, TCPPE linkers could be relaxed to generate TPE-MOF-5Y. Therefore, the optical properties of MOFs are significantly changed along with varying the conformation of the TCPPE linker. Importantly, the MOFs maintained their crystallinity throughout, and the changes of the color and the emission are fully reversible.
The motivation behind in-depth exploration of the underlying mechanisms in these AIEgen-MOFs is related to both a fundamental understanding of the dynamic behavior in crystalline solids and a desire to design and prepare high-performance materials.
2.2 Applications as phosphors for light-emitting diodes (LEDs)
White LEDs (WLEDs) have rapidly emerged in the market for illuminants owing to their low energy consumption and long lifetime. Phosphor conversion, which produces white light from the combined emissions of a phosphor coated on and excited by an LED chip, is one of the approaches to fabricate WLEDs. Among various types of phosphor conversion, a yellow phosphor coupled with a blue chip is preferred owing to the low cost and wide availability of blue LEDs. However, commercially available yellow phosphors highly rely on rare-earth elements, whose cost has rapidly grown in recent years. As aforementioned, the quantum yield and optical wavelength of the AIEgen-MOFs can be easily tuned. The AIEgen-MOFs have become promising alternatives as rare-earth free yellow phosphors. According to the definition, a phosphor is a substance that can exhibit luminescence, including both phosphorescence and fluorescence. It is noted that the AIEgen-MOFs mentioned below in this section are all fluorescent materials that decay energy from the excited singlet state to the ground state.Li and coworkers have reported a new strategy to design and synthesize a rare-earth free yellow phosphor by integrating a highly emissive AIEgen and a bandgap modulating co-ligand into a MOF.21 They used 1,1,2,2-tetrakis(4-(pyridin-4-yl)phenyl)ethane (tppe) and 1,3,5-benzentricarboxylate (btc) as organic linkers and Zn2+ as metal nodes to achieve [Zn6(btc)4(tppe)2(DMA)2]·9DMA·12H2O (TPE-MOF-6), which emits yellow light driven by a blue light source. TPE-MOF-6 exhibits exceptionally high internal quantum yields (IQY, defined as the ratio of the number of photons emitted to the number of photons absorbed) and external quantum yields (EQY, defined as the ratio of the number of photons emitted to the number of incident photons) with values of 90.7% and 96% (λex = 400 nm), respectively. They have fabricated a prototype WLED device (Fig. 2) by coating TPE-MOF-6 on the surface of a 5 mm 455–460 nm blue LED bulb. Upon applying a 3 V bias, the prototype device generates bright white light with a luminous efficacy of 47.4 lm W−1. TPE-MOF-6 also shows high moisture and thermal stability.
 | ||
| Fig. 2 (a–e) Images of dispersed sample TPE-MOF-6 coated on thin cotton strings and an LED bulb. Top row: Images of samples under daylight. Bottom row: Images of samples illuminated by a 450 nm blue LED. (a and b) TPE-MOF-6 coated on a single cotton string. (c and d) A wound-up string with the bottom half coated with TPE-MOF-6 and the top half uncoated. (e) WLED assembly. Left half: Blue LED bulb before (top) and after (bottom) being illuminated. Right half: The same blue LED coated by a dispersed sample of TPE-MOF-6 before (top) and after (bottom) being illuminated. (Reprinted with permission from ref. 21. Copyright 2014 American Chemical Society.) | ||
They further tuned the bandgap of the MOF phosphor by increasing the arm length extended from the TPE core.24 They have immobilized H4TCPPE into a Zn-based MOF (LMOF-231, denoted as TPE-MOF-7), which displays emission in the near-yellow region. The internal quantum yields of TPE-MOF-7 and desolvated TPE-MOF-7 were 82.5% and 95.1%, respectively, rivaling that of the commercially available yellow phosphor YAG:Ce3+. A PC-WLED assembly (bulb type, 455–460 nm, 5 mm chip, 20 mA, 3 V) was fabricated. It exhibits outstanding luminous efficacy (58.9 ± 1.5 lm W−1), well above that of LED directional and omnidirectional lamps (40 lm W−1 and 50 lm W−1, respectively). A similar strategy for producing a high-performance blue-excitable yellow phosphor has been reported by immobilization of an AIE-based H4TCPPE linker into porous 3D Bi-MOF (TPE-MOF-8).25 The as-made TPE-MOF-8 emits strong blue light at 459 nm. After activation, it undergoes irreversible structural change paired with optical red-shifting. The activated sample is able to emit strong yellow emission excited by 455 nm blue light with a quantum yield of 74%.
2.3 Applications as chemosensors
The chemically tailorable MOFs with porous facets are promising sensory materials, since they exhibit distinct advantages in terms of selectivity and sensitivity, two of the premier performance metrics in fluorescent chemical sensing. Chemical functionalities can be rationally implanted into the inner surface of MOFs, thereby offering high selectivity toward specific analytes. On the other hand, MOFs with high surface areas and tunable inner environment can provide a high density of analyte adsorption/binding sites for enriching and interacting with the analytes, thus enabling the improvement of sensitivity. Based on the reticular chemistry and structure–property correlations, a number of topologically and chemically diverse MOFs have been designed and applied as chemo-/bio-sensors. Many reviews have thoroughly discussed the design principles, detection mechanisms, and types of luminescent MOF sensory materials;39–42 herein we focus on MOF probes assembled with AIEgens.As aforementioned, originating from the nature of AIEgens, one can fine tune the fluorescent quantum yield of AIEgen-MOFs from nearly one unit to almost non-fluorescent. In the meanwhile, the guest molecules can strongly influence the flipping motions of phenyl rings in the AIEgen-MOFs. Therefore, AIEgen-MOFs are imparted with the ability to serve as high-performance “turn-off” or “turn-on” sensors.
Li and coworkers have designed a highly luminescent tppe-based 3D Zn-MOF, LMOF-241 (TPE-MOF-9) for turn-off detection of mycotoxins.29 TPE-MOF-9 is highly porous and shows strong blue-green emission with 92.7% internal quantum, attributed to the restriction of intramolecular vibrational and rotational motions and reduction of nonradiative relaxation pathways. Monitoring aflatoxins, one of the most dominant mycotoxins worldwide, is crucial to ensure food safety and avoid food crop contamination. TPE-MOF-9 exhibits a very fast and extremely sensitive response to aflatoxins by a fluorescence quenching mechanism. The detection limit towards aflatoxin B1, the most toxic among the major types of alfatoxins and one of the strongest known natural carcinogens, can reach 46 ppb, making it the best performing fluorescent chemo-sensor to date. The theoretical calculations and experimental data reveal that the detection mechanism is based on electron transfer rather than an energy transfer process.
Liu, Zhao, and co-workers have prepared a porous 3D TPE-MOF-10 with 1D channels.30 The fluorescent quantum yields of the as-synthesized and activated samples are 7.2% and 11.3%, respectively. The emission maximum of the activated TPE-MOF-10 is dramatically red-shifted from 470 nm to 535 nm compared with the as-synthesized sample, and it will blue shift upon exposure to benzene, m-xylene, mesitylene or aliphatic VOCs. The response towards VOCs may be attributed to the conformational change of the phenyl rings of the TCPPE unit. Furthermore, the fluorescence of the activated TPE-MOF-10 can be quenched drastically by nitroaromatics due to the electron transfer effect.
Wang and co-workers have constructed a Zr-MOF (TPE-MOF-11) decorated with TPE functions via a mixed-ligand approach by using mixed dicarboxylate struts, H2-etpdc, and H2-mtpdc.52 The mixed-ligand approach can make it possible to accommodate the TPE motif with large steric hindrance within a framework, while maintaining the porosity that allows analyte or substrate diffusion. The TPE-MOF-11 emits blue-green emission with a quantum yield of 48%. Upon exposure to nitroaromatic explosives, it exhibits selective and dramatic fluorescence quenching in response to 2,4,6-trinitrophenol and 2,4-dinitrophenol. This phenomenon results from the strong electrostatic and/or hydrogen-bonding interactions between Lewis basic N-donor sites of the imidazole in the ligand and the acidic hydroxyl group of nitrophenols that facilitate the electron and energy transfer from the electron-rich TPE units to electron-deficient nitrophenols. In addition, TPE-MOF-11 exhibits efficient catalytic activity for visible light-driven aerobic cross-dehydrogenative coupling reactions, while no catalytic activity was detected when using MOF UiO-68-mtpdc without the TPE moiety as a photocatalyst, indicating that the AIE-active TPE moiety plays a crucial role in TPE-MOF-11 as an active photocatalyst to drive the reaction. In addition, Lin and Zhao et al. have employed a tetracarboxylate functionalized benzene-cored tetraphenylethene derivative to construct two AIE-based fluorescent MOF materials, Zn2(L)(H2O)(DMA)·DMA (TPE-MOF-12) and Zn2(H2L)2(Bpy)2(H2O)3·H2O (TPE-MOF-13).53 The absolute quantum yields of these two MOFs reach 37% and 43%, respectively. Notably, TPE-MOF-13 shows superior sensitivity and selectivity toward 2,4,6-trinitrophenol in aqueous media (detection limit: 0.49 μM, approximately 110 ppb) over other nitro explosives by fluorescence quenching.
Recently, Li and co-workers have designed a bifunctional Zn-MOF (LMOF-263, TPE-MOF-14) via the mixed-ligand approach by utilizing pyridyl-decorated TPE as the luminescent source and sulfone-functionalized ligands as the heavy metal ion capture sites.54 TPE-MOF-14 possesses impressive water stability, high BET surface area, and blue emission with the highest internal quantum yield (89.2%). Notably, it is responsive to toxic heavy metal in water at a parts-per-billion level (3.3 ppb for Hg2+ and 19.7 ppb for Pb2+) via the fluorescence quenching mechanism. Furthermore, TPE-MOF-14 showed an excellent selectivity for heavy metal ions over light metals due to the introduction of thio-functionalized sites.
Dincă and co-workers have reported AIE-based MOFs used for selectively detecting ammonia at elevated temperatures.26 The TPE-based Zn2(TCPE) (TPE-MOF-1) was chosen for this study, which maintains its strong fluorescence even at 350 °C close to its decomposition temperature (400 °C). Furthermore, it does not lose its fluorescence intensity and crystallinity within three heating–cooling cycles. However, the fluoresence of TPE itself will be drastically quenched upon heating. This phenomenon is attributed to the high thermal stability of TPE-MOF-1 and the restraint of vibrational energy dissipation in the rigid-MOF. Upon exposure of activated TPE-MOF-1 to ammonia, ethylenediamine, triethylamine, N,N-diethylformamide, and water vapor at ambient temperature, the emission maximum shifts only slightly for all of these analytes. Interestingly, the selectivity is switched on at 100 °C and only ammonia exhibits remarkable red-shift of the emission wavelength maximum from 487 to 511 nm while the other analytes are silenced. This unusual selectivity is due to the strong binding affinity between the NH3 and Zn2+ ion, which is evidenced by DFT calculations.
In another case, fluorescence “turn-on” detection for VOCs was demonstrated by Tang, Liu and Zhao, et al.28 They utilized a TPE-based chromophore bearing two dangling phenyl rings without substituent groups (DPEB) as the ligand to prepare a 2D layered MOF with wide hexagonal channels named NUS-1 (TPE-MOF-15). Notably, all the dangling phenyl rings are aligned in the wide channels and remain unrestricted even after MOF formation. This structural feature is beneficial to trigger strong interactions between the dangling phenyl rings and the analytes, which restrict the motion of the phenyl rings to achieve a turn-on fluorescence response. The desolvated NUS-1a (TPE-MOF-15a) exhibits unaltered space group and structural properties compared with TPE-MOF-15, revealing the good robustness of the framework and establishing a base for further study of chemical sensing. The fluorescence quantum yield of TPE-MOF-15a is only 15%, much lower than that of the TPE-based ligand (79%). This reduction of quantum yield can be attributed to the nonradiative exciton dissipation through unhindered phenyl ring rotation/vibration. Furthermore, TPE-MOF-15a exhibits a slightly lower quantum yield and a 19 nm red shift of the emission peak in comparison with that of TPE-MOF-15 without desolvation. The results for detecting VOCs revealed that TPE-MOF-15a displays a turn-on florescence response for most of the analytes, including mesitylene, xylene, toluene, and benzene. The TPE-MOF-15a⊃benzene takes the largest bathochromic-shift (18 nm), while TPE-MOF-15a⊃mesitylene possesses the largest hypochromatic shift (28 nm). Moreover, the fluorescence quantum yield of TPE-MOF-15a⊃benzene can be as high as 49%. The above observation indicates that guest-dependent interactions with the dangling phenyl rings allow the hindering of the motion of these phenyl rings and the reducing of the nonradiative exciton dissipation.
By appending triazole groups to the TPE module, a TTPE-based 2D network {[Mn(TTPE)Cl2]·4CHCl3}n (TPE-MOF-16) has been prepared, and it can be successfully converted into another 2D framework {[Mn(TTPE)(H2O)2](ClO4)2·0.5TTPE·5.25H2O}n (TPE-MOF-17) by encapsulating large functionalized aromatic guest 1,1,2,2-tetrakis[4-(1H-1,2,4-triazol-1-yl)phenyl]ethylene (TTPE) through single-crystal-to-single-crystal transformation.55 These two luminescent materials exhibit excellent selectivity for Cd2+. In addition, for desolvated TPE-MOF-17, the emission maximum is hypsochromically shifted when exposed to a variety of analytes such cyclohexanone, cyclohexanediol, cyclohexanediamine, and ethylenediamine. Similarly, the activated [Cd2(tppe)(bpdc)2(H2O)] (TPE-MOF-18)56 and [ZnCl2tppe·4(TCE)] (TPE-MOF-19)57 show enhanced turn-on fluorescence in response to benzene and its methyl-substituted derivatives, whereas TPE-MOF-19 quenches its emission in response to nitro-phenyl substituted VOCs.
Besides decreasing the torsional barrier of the phenyl ring torsion, introducing a ligand-to-metal charge transfer (LMCT) effect into the framework is another efficient strategy to afford a default dark state of the AIEgen-MOF sensor. Our group presented a novel strategy that employs metal nodes with distinct outer-shell electron configurations and AIEgen struts to construct MOF sensors for turn-on detection of energetic heterocyclic compounds.27 Three MOFs, TABD-MOF-1, -2, and -3, were synthesized by coordination of bisubstituted TABD-COOH with Mg2+, Ni2+, and Co2+, respectively. Notably, these three TABD-MOFs show distinct fluorescence behaviors. TABD-MOF-1 exhibits strong fluorescence, whereas barely any fluorescence and complete non- fluorescence are, respectively, observed for TABD-MOF-2 and TABD-MOF-3 due to the LMCT effect. The luminescence quantum yields of these three TABD-MOFs are 38.5%, 1.12% and 0.15%, respectively. Remarkably, the cobalt-based non-fluorescent TABD-MOF-3 displays high selectivity and sensitivity towards five-membered ring energetic heterocyclic compounds (5MR-EHCs, i.e., triazoles, tetrazoles, azoxyfurazans, and their salts, derivatives and hybrids), which cannot be detected by well-established fluorometric methods on the basis of energy or electron transfer mechanisms. CN and NN bonds in these 5MR-EHCs allow the dissociation of the metal-linker coordinate bond in TABD-MOFs through a competitive coordination effect. Along with the breaking of metal–ligand bonds, the LMCT effect is interrupted and TABD ligands are released and further aggregate to yield highly emissive solids. Specifically, for the probing of 5-nitro-2,4-dihydro-3H-1,2,4-triazole-3-one (NTO), the solid and solution detection limit in the solid state can achieve as low as 6.5 ng cm−2 and 10−9 mol L−1, respectively, by using TABD-MOF-3 (Fig. 3). The signal transduction can be easily discernible by the naked eye.
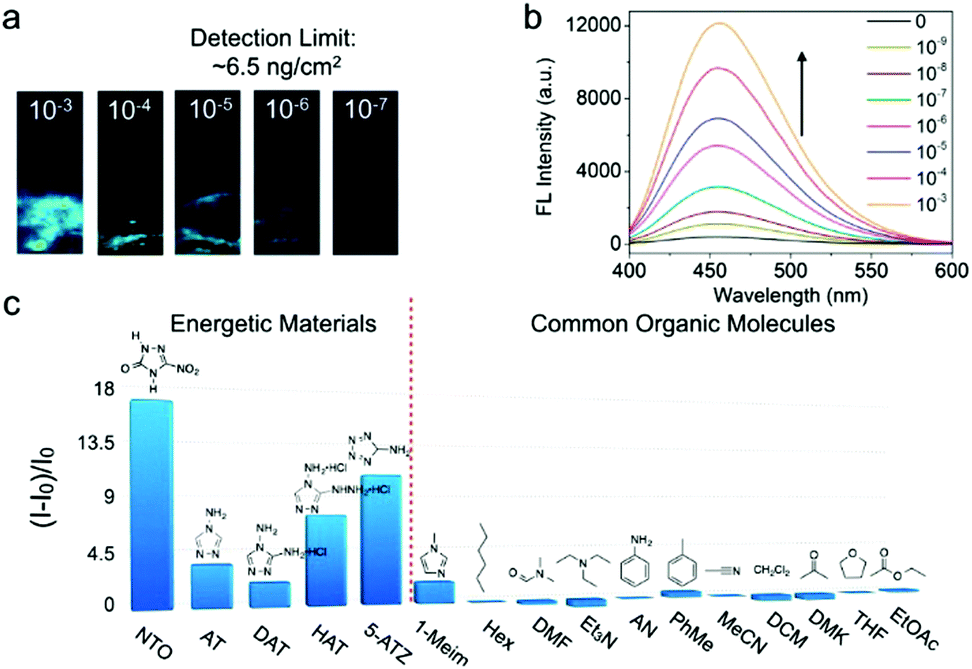 | ||
| Fig. 3 (a) Photographs of TABD-MOF-3-deposited paper strips upon addition of THF solution of NTO at different concentrations under UV light. (b) Fluorescence spectra of TABD-MOF-3 in THF upon addition of NTO solution at different concentrations followed by addition of hexane. (c) Fluorescence enhancement efficiencies ((I − I0)/I0) obtained from different analytes by TABD-MOF-3. Excitation wavelength: 360 nm. (Reprinted with permission from ref. 27. Copyright 2014 American Chemical Society.) | ||
3. AIEgen-based COFs
Compared with the formation of MOF single-crystals, the preparation of a long-range ordered network entirely held together by covalent bonds is much more challenging. Covalent organic frameworks (COFs) are composed of multidentate organic building blocks and linked through strong covalent bonds, extending organic chemistry beyond simple molecules and polymers to two- and three-dimensional frameworks. Selecting themodynamic reversible reactions that enable “error-checking” and “proof-reading” during COF growth is crucial for the formation of highly crystalline solids. The organic linkages involved in the formation of COFs include B–O (boroxine, boronate ester, spiroborate and borosilicate), CN (imine, hydrazine and squaraine), C–N (β-ketoenamine, imide and amide), etc. The chemical stability of these linkages determines the chemical robustness of the corresponding frameworks. Unlike MOFs, it is difficult to afford single crystals of COFs, therefore, their structure information is usually deduced from the powder X-ray diffraction (PXRD) profiles.
The COFs feature low skeletal density, large porosity, and high chemical and thermal stabilities. The diversities of organic building units and synthetic organic reactions and the simplicity of post-synthetic modification lead to the ease of construction of COFs with specific structures and functions. In addition to these intriguing features, the ability to control COFs on the molecular level together with high architectural and chemical robustness results in expansion of their applications in gas storage and separation, optoelectronics, catalysis, energy storage, etc.43,58–68
3.1 Construction of AIEgen-COFs
In 2D COFs, the multidentate organic building units are covalently stitched together into 2D polymeric sheets that further stack via non-covalent forces to form layered structures, affording segregated arrays of π columns that enable charge-carrier transportation and 1D channels that can accommodate guest molecules. Unlike traditional planar π-arene building blocks with high symmetry used for the construction of 2D COFs, AIEgens usually adopt non-planar and propeller-shaped configurations and relatively lower symmetries. Therefore, utilizing AIEgens to construct COFs often leads to new topologies and structural findings (Fig. 4).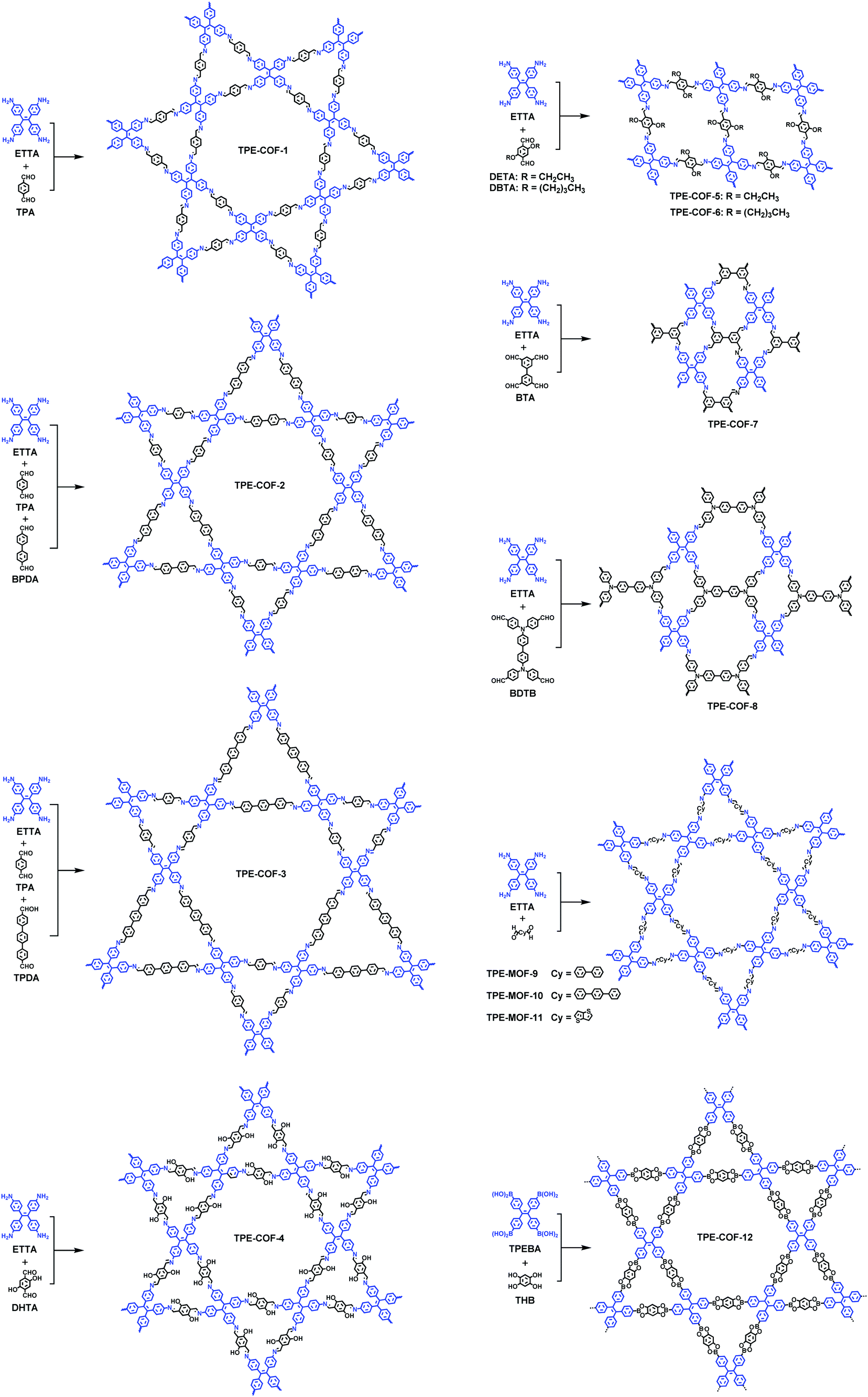 | ||
| Fig. 4 Syntheses of AIEgen-COFs. | ||
Zhao and co-workers have reported the first 2D dual-pore COF (TPE-COF-1) bearing trigonal micropores (7.1 Å) and hexagonal mesopores (26.9 Å) via co-condensation of 4,4′,4′′,4′′′-(ethene-1,1,2,2-tetrayl)-tetraaniline (ETTA, D2h symmetry) and terephthalaldehyde (C2).31 From a topology point of view, two crystallographic systems, orthorhombic and hexagonal systems, are expected to be formed by the combination of a D2h symmetric monomer and a C2 symmetric monomer. The thermodynamic-controlled COF-formation reaction allows self-repairing to form the most energy favorable structure with long-range order. According to theoretical calculations, the relative total energies of hexagonal structures with AA stacking are more stable than hexagonal structures with AB stacking and orthorhombic structures. The structural feature deduced from the PXRD profiles and nitrogen sorption isotherms are in agreement with theoretical predictions, confirming that TPE-COF-1 bears hexagonal and triangle pores arranged in a hexagonal crystallographic system alternately and periodically. They further constructed TPE-based triple-pore COFs (TPE-COF-2 and TPE-COF-3) by three-component condensation reactions of ETTA and two different dialdehydes (BPDA and TPA/TPDA) via the heterostructural mixed linker strategy.34 The successful construction of these COFs with a new topology greatly improves the hierarchy and structural complexity of COF chemistry. They further synthesized three TPE-based COFs, COF-DHTA (TPE-COF-4), COF-DETA (TPE-COF-5) and COF-DHTA (TPE-COF-6), through condensation reactions between D2h symmetric ETTA and C2 symmetric terephthalaldehydes pendant with hydroxyl, ethoxy, and butoxy groups.36 The results show that TPE-COF-4 is a dual-pore COF, while TPE-COF-5 and TPE-COF-6 adopt a single-pore topology, indicating that the substitution groups in the parent building blocks have a significant influence on the topologies of the resulting COFs. In addition, Jiang and Zhao, et al. have reported another two dual-pore (quadrangular and inequilateral hexagonal micropores) COFs, SIOC-COF-5 (TPE-COF-7) and SIOC-COF-6 (TPE-COF-8), based on co-condensation of two D2h symmetric monomers (ETTA and BTA/BDTB).35
According to theoretical studies, the adjacent layered polygon sheets favor stacking in an inclined or serrated manner with a lateral offset. Auras and Bein and co-workers considered that the lateral offset may possibly cause lattice strain and defects and thus weaken the crystallinity of the relative COFs. They proposed that employing building blocks with a lock-and-key-like molecular conformation is helpful for the minimization of the appearance of stacking faults and dislocations.32 The TPE derivatives adopting the propeller-like configuration can fulfill this requirement. Therefore, they have co-condensed a four-armed building block (TPE) with linear dialdehydes, terephthalaldehyde, biphenyl-4,4′-dicarboxaldehyde, p-terphenyl-4,4′′-dicarboxaldehyde, and thieno[3,2-b]thiophene-2,5-dicarboxaldehyde to yield imine-based COFs (TPE-COF-1, TPE-COF-9, TPE-COF-10 and TPE-COF-11). The resulting COFs are highly crystalline and exhibit robust chemical stability. Experimental and theoretical studies indicate that the four core phenylene groups undertake a screw-like arrangement (Fig. 5a) and the COF layers thermodynamically favor an eclipsed structure without any lateral offset. The highly ordered hexagonal arrangement of the mesopores can be clearly observed in domains ranging from 50 to 100 nm (Fig. 5b).
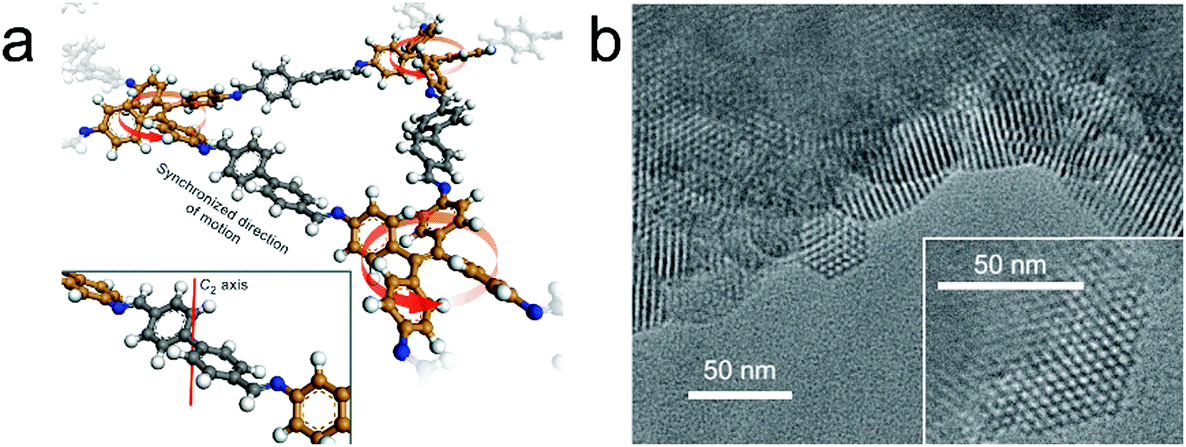 | ||
| Fig. 5 (a) Fragment of the TPE-COF-9 structure. The direction of rotation (red arrows) of TPE entities is synchronized via the molecular conformation of the linear bridging unit. Inset, a cut-out showing the C2-symmetric bridging unit that is crucial for transmitting information on the molecular conformation of the TPE entities. (b) TEM images of TPE-COF-10 that show the faceted COF crystallites with a hexagonal arrangement of the mesopores. Inset, magnified view onto a single COF crystallite. (Reprinted with permission from ref. 32. Copyright 2016 Nature Publishing Group.) | ||
These results suggested that propeller-shaped and D2h symmetric building units are useful for affording well-defined periodic docking sites and promoting formation of long-range order with unique heteropore structural features during COF growth.
3.2 Emissive COF-based AIEgens
Despite the fact that highly emissive COFs with confined porous structures are highly desired for sensing applications, examples of 2D highly luminescent COFs are rather limited. This is because, in 2D COFs, flat polymeric sheets usually stack in a face-to-face fashion with maximal π-orbital overlap, thus leading to the ACQ phenomenon. It should be mentioned that serval pyrene-based COFs connected by boronate-ester or azine linkages possess close π–π stacking yet still are highly luminescent, such as TP-COF,69 PPy-COF,70 Py-DBA COFs,71 Py-MV-DBA-COF,71 and 2D Py-Azine COF.72 This is because pyrene excimers are formed in these COFs, which only result in partial loss of the excitation energy and still permit deactivation via a radiative path.Integration of AIEgens into 2D COFs is one of the most straightforward strategies to solve the ACQ problems and make COFs highly emissive, since AIEgens usually adopt a propeller-like shape and can sufficiently avoid close π–π stacking. Jiang and co-workers co-condensed TPE-cored boronic acids (TPEBA) and 1,2,4,5-tetrahydroxybenzene (THB) to generate boronate-ester linked TPE-Ph COF (TPE-COF-12) with a layer distance of 4.5 Å, which is significantly larger than the conventional arene-based COFs (typically 3.4 Å).33 The TPE-COF-12 exhibited homogeneous brilliant blue luminescence emission at 500 nm with an absolute luminescence quantum yield of 32%, about twice that of the model compound in the solid state (15%). As evidenced by DFTB calculations, the phenyl rings of the monolayer of TPE-COF-12 require higher rotation energy (9.9 kcal mol−1, at 70° dihedral angle) in comparison with that of the TPE vertex analogue (7.1 kcal mol−1, at 70° dihedral angle). Importantly, for the AA-stacked layered TPE-COF-12, the rotation barrier energy is drastically increased to 216.7 kcal mol−1 (at 70° dihedral angle) and will increase continuously if the dihedral angle is enhanced further. Hence, integrating AIE-based units into the rigidified frameworks can observably reduce the rotation-induced excitation-energy dissipation that can be attributed to the synergistic structural locking effect. In addition, based on the Lewis acid–base interaction, boronate-ester linked TPE-COF-12 can serve as a sensitive sensor toward ammonia (Fig. 6) down to the sub ppm level. As mentioned earlier, several TPE-based COFs bearing an imine linkage have been reported, however, their luminescent properties have not been discussed. This is possibly due to the occurrence of the charge transfer from the electron-deficient AIEgen units to the electron-rich imine linkages in these imine-linked AIEgen-COFs, and their emissions are quenched. Reduction of the imine linkages or change of the linkage types may provide a chance to trigger their emissions.
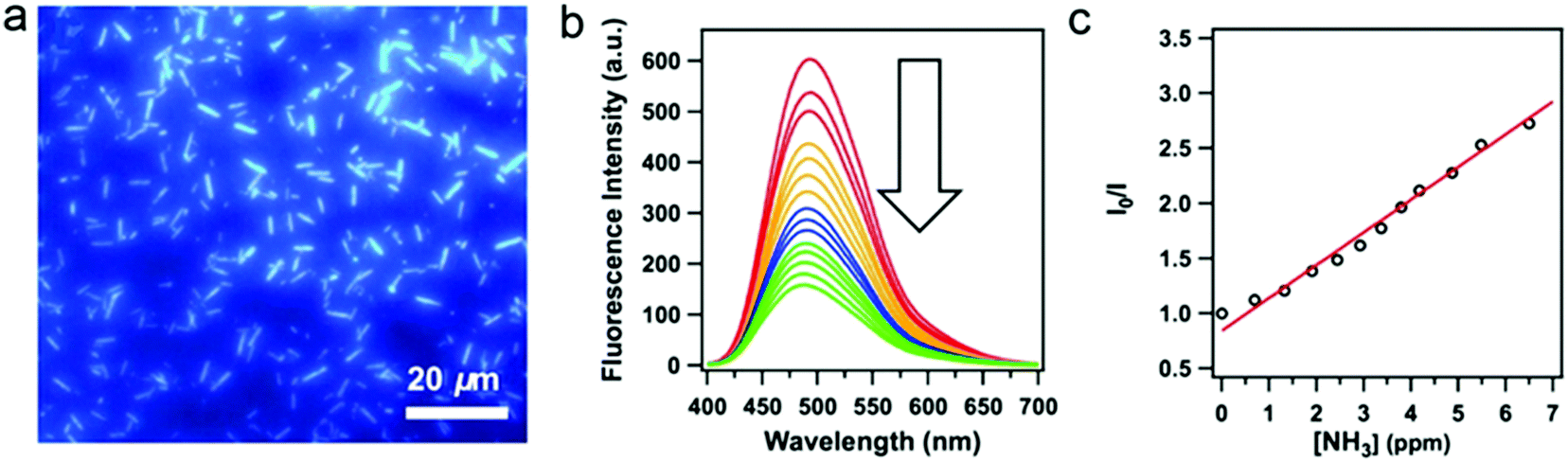 | ||
| Fig. 6 (a) Fluorescence microscopy images of TPE-COF-12. (b) Fluorescence spectral change of the TPE-COF-12 upon addition of ammonia. (c) Stern–Volmer plot of the fluorescence quenching by ammonia. (Reprinted with permission from ref. 33. Copyright 2016 American Chemical Society.) | ||
Other strategies to tackle the ACQ problems include preparation of 2D few-layered COFs and design of 3D COFs. For the former, Banerjee and co-workers have reported that covalent organic nanosheets (TfpBDH-CONs) have been prepared by an exfoliation method.73 TfpBDH-CONs with few-layer structures exhibit increased luminescence intensity in comparison with corresponding bulk COFs, and this phenomenon can be attributed to the weakening of the interlayer π–π interactions of 2D COFs. For the latter, Wang and co-workers have reported a highly luminescent pyrene-based 3D COF (3D-PyCOF) adopting a two-fold interpenetrated pts topology.74 However, up to now, no AIEgen-based 3D COF have been reported yet due to synthetic difficulties. Similar to 3D MOFs, introduction of AIEgens into 3D COFs provides a chance to fine-tune the torsional barrier for phenyl ring flipping and may show promising properties in sensing and optoelectronic applications.
4. Conclusion and outlook
In summary, reticular chemistry allows precise control of the geometries and atomic positions of 2D and 3D frameworks, offering a strong thread linking basic science and practical applications. Connecting AIEgens based on reticular chemistry affords COFs and MOFs with high internal surface area, accessible space, and outstanding optical properties. Through balancing the thermodynamics and the kinetics of the reaction via altering the reaction conditions, crystalline AIEgen-MOFs or -COFs with distinct topologies can be made even using the same organic building units and metal sources. The AIEgens are locked and reinforced in precise locations throughout the framework. Thus, the rotation, vibrations, and motions of aromatic rings in the AIEgen building units can be regulated by the steric hindrance or strains within the frameworks or guest molecules accommodated in the pores, providing a great chance to fine-tune the fluorescent quantum yield and energy band of these crystalline frameworks and achieve high performance in applications such as optical electronics and chemo-/bio-sensors. Despite great advances in this area, there still remain various challenging topics in this fields, such as processability, molecular complexity, and hierarchical structures.Both MOFs and COFs are brittle and difficult to process due to their inmeltable and insoluble nature. Although AIEgen-based crystalline materials hold great promise in many applications, fabricating them into real devices is highly demanded. This is because advanced device production technology is helpful for achieving high signal consistency, strong anti-interference ability, and better stability, as well as have benefits for cutting costs and integrating multifunction. Various cutting-edge fabrication techniques and miniaturization processes have been developed for controlling the location of individual crystalline porous solids.75–78 For example, MOF and COF thin films have been fabricated through several methods, such as Langmuir–Blodgett deposition (LB),79,80 surface functionalization,81 layer-by-layer (LBL),82 physical mixing with polymers,83,84 post-synthetic polymerization (PSP),85 and the hot-pressing (HoP) method (Fig. 7);86 the protocols to prepare MOF patterns are classified into bottom-up (e.g. surface functionalization, electrochemical deposition, microfluidics, and ink-jet) and top-down (e.g. photolithography and imprinting) patterning technologies. Nevertheless, joint efforts from different fields, including chemistry, materials, engineering and electronics, need to be devoted to this emerging area.
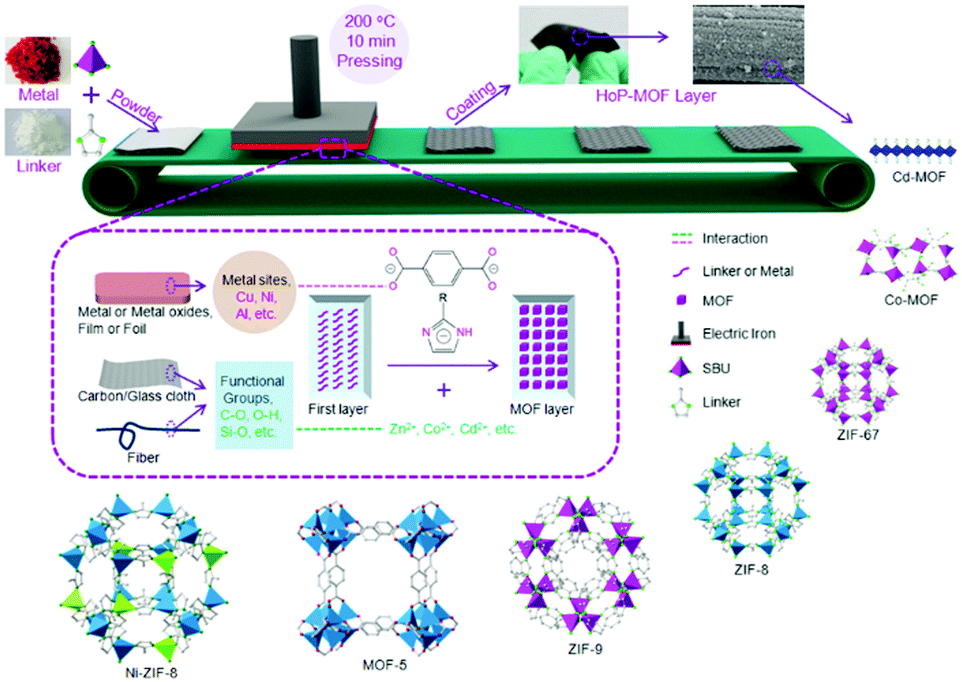 | ||
| Fig. 7 Schematic presentation of the hot-pressing (HoP) method for MOF coating. (Reprinted with permission from ref. 86. Copyright 2016 Wiley.) | ||
AIEgens can provide sensory function in a crystalline framework but usually lack enough selectivity and catalytic ability. Increasing molecular complexity (Fig. 8a)87 and preparation of hierarchical structures (Fig. 8b)88 are two ways to simultaneously achieve highly selective and highly sensitive multifunctional materials, which are long pursued yet largely unmet. For the former, the molecular complexity can be introduced through post-synthetic modification or a mixed-linker strategy, both of which are able to maintain the order of the underlying structure without losing synthetic control. Although there is still a long way to go for the realization of well-defined ‘sequence dependent materials,’ it still provides a chance to mimic nature and design smart and intelligent materials. For the latter, sequential growth or functionalization of these crystalline solids to prepare hierarchical or heterogeneous structures allows us to combine individual spatial functions and perform these functions in a logical sequence. For example, the outer shell with a particular shape and size can selectively extract and accommodate target molecules from complicated components, the intermediate layer furnished with AIEgens can produce an apparent signal output to disclose the environmental changes, and the inner part can serve as a nanoreactor to decompose hazardous substance to harmless compounds.
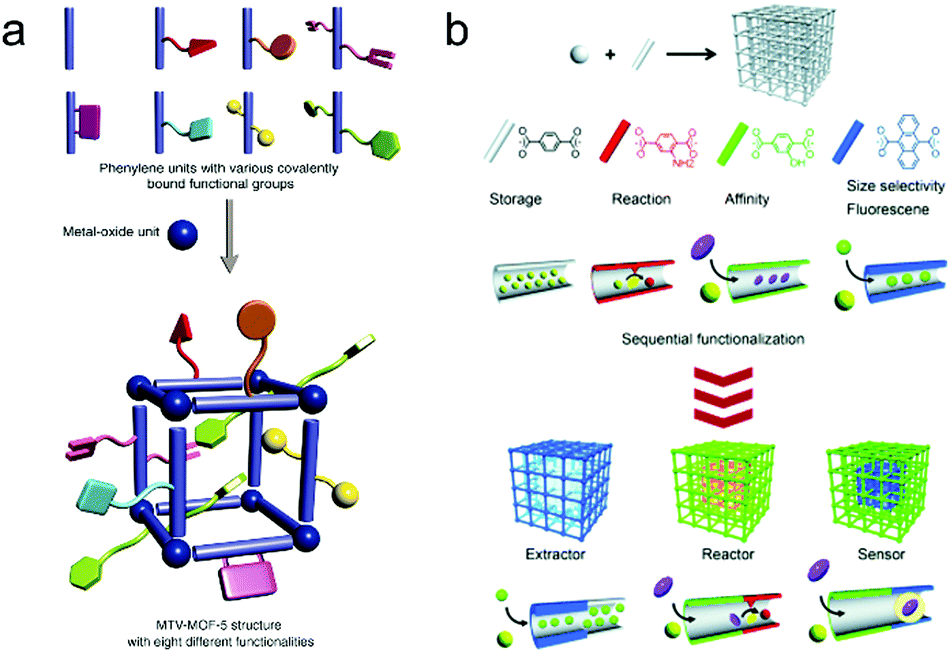 | ||
| Fig. 8 (a) Multivariate (MTV) MOF-5 type structures. (Reprinted with permission from ref. 87. Copyright 2010 The American Association for the Advancement of Science.) (b) Porous coordination polymer crystal hybridization for sequential functionalization systems. (Reprinted with permission from ref. 88. Copyright 2011 Wiley.) | ||
Last but not the least, further deciphering the exact AIE mechanism with the help of the construction of AIEgen-based MOFs and COFs with precisely controlled and well-defined molecule locations and configurations continues to be an important subject, since it would be no doubt beneficial for strengthening the scientific foundations and ab initio development of novel optical functional materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the 973 Program 2013CB834702; the National Natural Science Foundation of China (Grant No. 21625102, 21471018, 21404010, 21490570 and 21674012); and the 1000 Plan (Youth).Notes and references
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, B. Z. Tang, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed.
- M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858–1867 RSC.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726–23740 RSC.
- R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC.
- R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228–4238 RSC.
- Z. Zhao, B. He and B. Z. Tang, Chem. Sci., 2015, 6, 5347–5365 RSC.
- V. Vij, V. Bhalla and M. Kumar, Chem. Rev., 2016, 116, 9565–9627 CrossRef CAS PubMed.
- X. Feng, B. Tong, J. Shen, J. Shi, T. Han, L. Chen, J. Zhi, P. Lu, Y. Ma and Y. Dong, J. Phys. Chem. B, 2010, 114, 16731–16736 CrossRef CAS PubMed.
- T. Han, X. Feng, J. Shi, B. Tong, Y. Dong, J. W. Y. Lam, Y. Dong and B. Z. Tang, J. Mater. Chem. C, 2013, 1, 7534–7539 RSC.
- T. Han, Y. Zhang, X. Feng, Z. Lin, B. Tong, J. Shi, J. Zhi and Y. Dong, Chem. Commun., 2013, 49, 7049–7051 RSC.
- Y. Zhang, T. Han, S. Gu, T. Zhou, C. Zhao, Y. Guo, X. Feng, B. Tong, J. Bing, J. Shi, J. Zhi and Y. Dong, Chem. – Eur. J., 2014, 20, 8856–8861 CAS.
- N. B. Shustova, B. D. McCarthy and M. Dinč, J. Am. Chem. Soc., 2011, 133, 20126–20129 CrossRef CAS PubMed.
- N. B. Shustova, A. F. Cozzolino and M. Dincǎ, J. Am. Chem. Soc., 2012, 134, 19596–19599 CrossRef CAS PubMed.
- N. B. Shustova, T. C. Ong, A. F. Cozzolino, V. K. Michaelis, R. G. Griffin and M. Dinč, J. Am. Chem. Soc., 2012, 134, 15061–15070 CrossRef CAS PubMed.
- Q. Gong, Z. Hu, B. J. Deibert, T. J. Emge, S. J. Teat, D. Banerjee, B. Mussman, N. D. Rudd and J. Li, J. Am. Chem. Soc., 2014, 136, 16724–16727 CrossRef CAS PubMed.
- Z. W. Wei, Z. Y. Gu, R. K. Arvapally, Y. P. Chen, R. N. McDougald, J. F. Ivy, A. A. Yakovenko, D. W. Feng, M. A. Omary and H. C. Zhou, J. Am. Chem. Soc., 2014, 136, 8269–8276 CrossRef CAS PubMed.
- Q. Zhang, J. Su, D. W. Feng, Z. W. Wei, X. D. Zou and H. C. Zhou, J. Am. Chem. Soc., 2015, 137, 10064–10067 CrossRef CAS PubMed.
- Z. Hu, G. Huang, W. P. Lustig, F. Wang, H. Wang, S. J. Teat, D. Banerjee, D. Zhang and J. Li, Chem. Commun., 2015, 51, 3045–3048 RSC.
- B. J. Deibert, E. Velasco, W. Liu, S. J. Teat, W. P. Lustig and J. Li, Cryst. Growth Des., 2016, 16, 4178–4182 CAS.
- N. B. Shustova, A. F. Cozzolino, S. Reineke, M. Baldo and M. Dincǎ, J. Am. Chem. Soc., 2013, 135, 13326–13329 CrossRef CAS PubMed.
- Y. X. Guo, X. Feng, T. Y. Han, S. Wang, Z. G. Lin, Y. P. Dong and B. Wang, J. Am. Chem. Soc., 2014, 136, 15485–15488 CrossRef CAS PubMed.
- M. Zhang, G. X. Feng, Z. G. Song, Y. P. Zhou, H. Y. Chao, D. Q. Yuan, T. T. Y. Tan, Z. G. Guo, Z. G. Hu, B. Z. Tang, B. Liu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 7241–7244 CrossRef CAS PubMed.
- Z. Hu, W. P. Lustig, J. Zhang, C. Zheng, H. Wang, S. J. Teat, Q. Gong, N. D. Rudd and J. Li, J. Am. Chem. Soc., 2015, 137, 16209–16215 CrossRef CAS PubMed.
- X. G. Liu, H. Wang, B. Chen, Y. Zou, Z. G. Gu, Z. J. Zhao and L. Shen, Chem. Commun., 2015, 51, 1677–1680 RSC.
- T. Y. Zhou, S. Q. Xu, Q. Wen, Z. F. Pang and X. Zhao, J. Am. Chem. Soc., 2014, 136, 15885–15888 CrossRef CAS PubMed.
- L. Ascherl, T. Sick, J. T. Margraf, S. H. Lapidus, M. Calik, C. Hettstedt, K. Karaghiosoff, M. Döblinger, T. Clark, K. W. Chapman, F. Auras and T. Bein, Nat. Chem., 2016, 8, 310–316 CrossRef CAS.
- S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, J. Am. Chem. Soc., 2016, 138, 5797–5800 CrossRef CAS PubMed.
- Z. F. Pang, S. Q. Xu, T. Y. Zhou, R. R. Liang, T. G. Zhan and X. Zhao, J. Am. Chem. Soc., 2016, 138, 4710–4713 CrossRef CAS PubMed.
- Y. Tian, S. Q. Xu, C. Qian, Z. F. Pang, G. F. Jiang and X. Zhao, Chem. Commun., 2016, 52, 11704–11707 RSC.
- Z. Pang, T. Zhou, R. Liang, Q. Qi and X. Zhao, Chem. Sci., 2017, 8, 3866–3870 RSC.
- S. Dalapati, C. Gu and D. Jiang, Small, 2016, 12, 6513–6527 CrossRef CAS PubMed.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- U. Díaz and A. Corma, Coord. Chem. Rev., 2016, 311, 85–124 CrossRef.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- S. Qiu, M. Xue and G. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- Y. Bai, Y. Dou, L. H. Xie, W. Rutledge, J. R. Li and H. C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- L. Wang, Y. Han, X. Feng, J. Zhou, P. Qi and B. Wang, Coord. Chem. Rev., 2016, 307, 361–381 CrossRef CAS.
- A. Schoedel, M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2016, 116, 12466–12535 CrossRef CAS PubMed.
- Q. Li, Z. Ma, W. Zhang, J. Xu, W. Wei, H. Lu, X. Zhao and X. Wang, Chem. Commun., 2016, 52, 11284–11287 RSC.
- Y. Deng, N. Chen, Q. Li, X. Wu, X. Huang, Z. Lin and Y. Zhao, Cryst. Growth Des., 2017, 17, 3170–3177 CAS.
- N. D. Rudd, H. Wang, E. M. A. Fuentes-Fernandez, S. J. Teat, F. Chen, G. Hall, Y. J. Chabal and J. Li, ACS Appl. Mater. Interfaces, 2016, 8, 30294–30303 CAS.
- Y. Wang, B. Yuan, Y. Y. Xu, X. G. Wang, B. Ding and X. J. Zhao, Chem. – Eur. J., 2015, 21, 2107–2116 CrossRef CAS PubMed.
- X. Zhao, Y. Li, Z. Chang, L. Chen and X. H. Bu, Dalton Trans., 2016, 45, 14888–14892 RSC.
- S. L. Jackson, A. Rananaware, C. Rix, S. V. Bhosale and K. Latham, Cryst. Growth Des., 2016, 16, 3067–3071 CAS.
- X. Feng, X. S. Ding and D. L. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453–465 CrossRef CAS PubMed.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- M. Dogru and T. Bein, Chem. Commun., 2014, 50, 5531–5546 RSC.
- X. H. Liu, C. Z. Guan, D. Wang and L. J. Wan, Adv. Mater., 2014, 26, 6912–6920 CrossRef CAS PubMed.
- P. J. Waller, F. Gandara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- J. L. Segura, M. J. Mancheño and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. L. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. L. Jiang, Angew. Chem., Int. Ed., 2009, 48, 5439–5442 CrossRef CAS PubMed.
- J. W. Crowe, L. A. Baldwin and P. L. McGrier, J. Am. Chem. Soc., 2016, 138, 10120–10123 CrossRef CAS PubMed.
- S. Dalapati, S. B. Jin, J. Gao, Y. H. Xu, A. Nagai and D. L. Jiang, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed.
- G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931–3939 RSC.
- G. Lin, H. Ding, D. Yuan, B. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302–3305 CrossRef CAS PubMed.
- P. Falcaro, R. Ricco, C. M. Doherty, K. Liang, A. J. Hill and M. J. Styles, Chem. Soc. Rev., 2014, 43, 5513–5560 RSC.
- P. Falcaro, D. Buso, A. J. Hill and C. M. Doherty, Adv. Mater., 2012, 24, 3153–3168 CrossRef CAS PubMed.
- A. Carné, C. Carbonell, I. Imaz and D. Maspoch, Chem. Soc. Rev., 2011, 40, 291–305 RSC.
- M. D. Allendorf, A. Schwartzberg, V. Stavila and A. A. Talin, Chem. – Eur. J., 2011, 17, 11372–11388 CrossRef CAS PubMed.
- R. Makiura, S. Motoyama, Y. Umemura, H. Yamanaka, O. Sakata and H. Kitagawa, Nat. Mater., 2010, 9, 565–571 CrossRef CAS PubMed.
- W. Dai, F. Shao, J. Szczerbiński, R. McCaffrey, R. Zenobi, Y. Jin, A. D. Schlüter and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 213–217 CrossRef CAS PubMed.
- D. Zacher, R. Schmid, C. Wöll and R. A. Fischer, Angew. Chem., Int. Ed., 2011, 50, 176–199 CrossRef CAS PubMed.
- O. Shekhah, H. Wang, S. Kowarik, F. Schreiber, M. Paulus, M. Tolan, C. Sternemann, F. Evers, D. Zacher, R. A. Fischer and C. Wöll, J. Am. Chem. Soc., 2007, 129, 15118–15119 CrossRef CAS PubMed.
- Y. Zhang, X. Feng, S. Yuan, J. Zhou and B. Wang, Inorg. Chem. Front., 2016, 3, 896–909 RSC.
- B. P. Biswal, H. D. Chaudhari, R. Banerjee and U. K. Kharul, Chem. – Eur. J., 2016, 22, 4695–4699 CrossRef CAS PubMed.
- Y. Zhang, X. Feng, H. Li, Y. Chen, J. Zhao, S. Wang, L. Wang and B. Wang, Angew. Chem., Int. Ed., 2015, 54, 4259–4263 CrossRef CAS PubMed.
- Y. Chen, S. Li, X. Pei, J. Zhou, X. Feng, S. Zhang, Y. Cheng, H. Li, R. Han and B. Wang, Angew. Chem., Int. Ed., 2016, 55, 3419–3423 CrossRef CAS PubMed.
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS PubMed.
- K. Hirai, S. Furukawa, M. Kondo, H. Uehara, O. Sakata and S. Kitagawa, Angew. Chem., Int. Ed., 2011, 50, 8057–8061 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2017 |