
Conducting polymer composites: material synthesis and applications in electrochemical capacitive energy storage
Jing
Yang
,
Ying
Liu
,
Siliang
Liu
,
Le
Li
,
Chao
Zhang
 * and
Tianxi
Liu
*
* and
Tianxi
Liu
*
State Key Laboratory for Modification of Chemical Fibers and Polymer Materials & College of Materials Science and Engineering, Donghua University, Shanghai 201620, P. R. China. E-mail: czhang@dhu.edu.cn; txliu@fudan.edu.cn; txliu@dhu.edu.cn
First published on 6th October 2016
Abstract
In recent years, high efficiency, low cost and environmental friendly energy storage has drawn attention to meet the constantly escalating energy crisis. Conducting polymers in their pristine form have difficulty in achieving satisfying characteristics required for practical applications in electrochemical capacitive energy storage. Considering that conducting polymer composites have emerged as pertinent and beneficial resources for electrochemical capacitive energy storage, this review investigates the relevant topics by presenting the approaches in the design and fabrication of conducting polymer composites as electrode materials for electrochemical capacitive energy storage. The key issues for achieving optimized supercapacitive performances, such as fabricating nanostructured electrodes and tailoring microstructures of conducting polymer composites, are described and concisely discussed in this review. Finally, an outlook of the prospects and challenges in terms of synthesis and applications of conducting polymer composites for supercapacitors is presented.
 Jing Yang | Jing Yang received her BS degree in Polymer Science from Wuhan Textile University in 2015. She is now pursuing her MS degree under the supervision of Prof. Tianxi Liu at Donghua University. Her research interests mainly focus on the design and synthesis of layered transition metal dichalcogenides and their composites for energy conversion and storage. |
 Ying Liu | Ying Liu received her BS degree in Materials Science and Engineering in 2016 from Donghua University. Currently, she is a graduate student in the group of Prof. Tianxi Liu at Donghua University. Her research focuses on the development of organic/inorganic hybrids for energy storage. |
 Siliang Liu | Siliang Liu received his BS degree in Materials Science and Engineering from Qiqihar University in 2014. He is now a Master candidate under the supervision of Prof. Tianxi Liu at Donghua University. Currently, his research interests focus on the synthesis of carbon-based materials for electrochemical energy storage. |
 Le Li | Le Li received his BE degree from the North University of China in 2012. After he received his MS degree from Sichuan University, he became a PhD candidate under the supervision of Prof. Tianxi Liu at Donghua University. His research interests focus on the synthesis, assembly and applications of conducting polymers and their composite materials. |
 Chao Zhang | Chao Zhang received his PhD degree from Fudan University under the supervision of Prof. Tianxi Liu in 2013. From 2013 to 2015, he worked with Dr Tim-Patrick Fellinger and Prof. Markus Antonietti at the Max Planck Institute of Colloids and Interfaces as a Postdoctoral Research Fellow. In 2015, he joined the College of Materials Science and Engineering, Donghua University, as a professor. He has published more than 40 articles in peer-reviewed journals (cited more than 1300 times with an H-index of 20). His current research interests focus on synthesis of low-dimensional nanostructured functional materials as well as their applications in polymer nanocomposites, energy storage and conversion (supercapacitors, fuel cell, lithium ion battery, etc.). |
 Tianxi Liu | Tianxi Liu received his PhD degree in Polymer Chemistry and Physics from the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences in 1998. He was an Alexander von Humboldt Research Fellow from 1998 to 2000 at the University of Dortmund, Germany. He was a Research Associate from 2000 to 2001 and a Research Scientist from 2002 to 2004 at the Institute of Materials Research and Engineering (IMRE), Singapore. He joined as a full professor (since 2004) at the Department of Macromolecular Science, Fudan University, and then moved to the College of Materials Science and Engineering, Donghua University in 2016. He has published more than 230 articles in peer-reviewed journals (cited more than 7000 times with an H-index of 47). His current research interests include polymer nanocomposites, nanofibers and their composites, organic/inorganic hybrid materials, and new energy materials and devices. |
1. Introduction
The continuous depletion of fossil fuels such as gasoline, coal and natural gas, global warming issues and environmental pollution necessitate the development of high-performance energy storage devices. In particular, the widespread applications of energy storage issues require finding alternative energy sources with high power/energy density, long cycle life and enhanced operation safety. In this regard, supercapacitors, also called ultracapacitors or electrochemical capacitors, have been considered as leading candidates for advanced energy storage devices and are widely used for automotive systems, portable electronics, and so on.1 Supercapacitors have been involved in a wide and growing range of research interests because of their superior features compared to lithium ion batteries.2–7 When two electrodes are polarized by a bias voltage, two layers of opposite charges form at the electrode/electrolyte interface. The distance between the two layers with opposite charges is low and equal to an atomic distance and leads to significantly more energy compared to conventional capacitors. Therefore, supercapacitors are expected to fill the gap between lithium ion batteries and conventional capacitors in terms of both high power and energy density.8 Consequently, supercapacitors with a long cycling life and high power density are more attractive and versatile for applications in energy back-up systems, portable devices and electric vehicles.9Supercapacitors typically consist of two electrodes. To prevent the two electrodes from electrical contact, a separator that is non-electrically conductive is interposed. In addition, an electrolyte is employed to ionically connect both electrodes.2 The most widely used electrolytes for supercapacitors include aqueous electrolytes, organic electrolytes and solid-state electrolytes. Aqueous electrolytes, such as aqueous solution of H2SO4 and KOH, can offer higher ionic concentrations and lower resistances, thus leading to a higher capacitance and higher power density. However, the small voltage window for aqueous electrolytes (typically lower than 1.2 V) may restrict the achievement of high energy density. Compared with aqueous electrolytes, organic electrolytes such as acetonitrile and propylene carbonate possess a higher voltage window, up to 3.5 V, but they can easily generate environmental and toxic problems. With respect to safety issues, the most widely used solid-state electrolytes in supercapacitors are gel polymers, which can avoid leakage problems and reduce the packaging cost.
According to the fundamentally different mechanisms of energy storage, supercapacitors can be classified into two types, electric double layer capacitors (EDLCs) and pseudocapacitors.10,11 EDLCs store the energy electrostatically by accumulating ions on a conductive surface of the electrode material. The other type is a pseudocapacitor, in which the capacitance originates from a fast and reversible faradic reaction of the electrochemically active electrode material with the electrolyte.5,12 The most commonly known active species investigated for pseudocapacitors include conducting polymers and transition metal oxides.13–16 Conducting polymers are attractive as they have a high charge density, low cost, considerable structural diversity and mechanical flexibility, which make them appropriate as electrode materials for supercapacitors. Compared to EDLCs with high-surface-area carbon as the electrode material, conducting polymer-based pseudocapacitors can store a greater amount of charge in the electrical double layer and through a rapid faradic charge transfer since the electrochemical process occurs both on the surface and in the bulk of the electrode material. However, conducting polymers used as electrode materials often suffer remarkable degradation because of the mechanical instability caused by swelling and shrinking during the charge and discharge processes. The poor cycle life of the conducting polymer electrodes greatly restricts their practical applications because conducting polymers tend to irreversibly agglomerate during electrode preparation and peel off from the current collector. Hence, it is of great importance to combine conducting polymers with other materials to form a conducting polymer composite, which is beneficial for the construction of electrode materials with a high specific capacitance and excellent cycling stability. The discussions on the construction of electrode materials for supercapacitors in this review will be limited to conducting polymer composites.
2. Properties of conducting polymers and their composites
Conventional polymers such as plastics, rubbers and resins exhibit significant resistance and are either insulators or dielectrics. Since Heeger, MacDiarmid and Shirakawa were jointly rewarded with the Nobel Prize in Chemistry in 2000 for their pioneering work on conducting polymers, conducting polymers have received more and more attention from both the academic and industrial community because of their wide applications in electrochromic devices17 and sensors.18,19 Conductive polymers are organic polymers with conjugated double bonds. They can combine the electrically conductive properties of metals or semiconductors with the advantages of traditional polymers such as low cost, considerable structural diversity, high flexibility and durability, which make them ideal for electrode materials for supercapacitors.20The conducting polymers that are most commonly studied in both fundamental research and various application fields are polyaniline (PANI), polypyrrole (PPy), and polythiophene (PT).21 Compared to bulk conducting polymers, their composites show outstanding energy storage performances, which arise from their superior conductivity, high surface area, improved electrochemical activity and excellent mechanical properties. In addition, conducting polymer composites also exhibit advantages because of their synergistic performance derived from each component. For instance, inorganic materials normally exhibit a high capacitance but lack the conductivity and cycling stability required for commercial capacitive applications. To alleviate these problems, the efficient combination of inorganic materials with conducting polymers plays a key role in improving the performance of composites because the conducting polymers can interact synergistically with inorganic materials to provide a conducting backbone, electrical conductivity and plastic property.22–25
Conducting polymers are attractive because they have a very high current density and low cost when compared to carbonaceous electrode materials. Taking PANI as an example, a supercapacitor device based on PANI can exhibit a specific energy of 10 W h kg−1 with a slightly lower specific power of 2 kW kg−1,26,27 while a carbon-based supercapacitor device can only reach a specific power of 3–4 kW kg−1 and a specific energy of 3–5 W h kg−1.26,28 Supercapacitors in term of EDLCs are generally highly cycleable, namely, more than 0.5 million cycles, whereas conducting polymer pseudocapacitors often begin to degrade in less than a thousand cycles because of mechanical failure of the electrode in their physical structure.27,29 Therefore, the cyclic lift of the conducting polymer electrodes and corresponding devices will be discussed later.
Furthermore, a test fixture configuration, such as a two-electrode system and three-electrode system, is closely related to the performance of a packaged cell. In general, a test system based on a three-electrode configuration that includes a working electrode, a counter electrode and a reference electrode is widely applied in electrochemical research. However, the working electrode in a three-electrode configuration has twice the potential range when the same potential range is applied to the electrodes in a two-electrode configuration, which lead to a doubling of the calculated capacitances. Therefore, supercapacitor cells with a two-electrode configuration can provide a more accurate measurement of the capacitive performances because the three-electrode configuration may result in larger errors when calculating the energy storage capacitance. Besides, the test system based on a two-electrode configuration is easily fabricated and commercially available.
3. Strategies to fabricate nanostructured electrodes with conducting polymer composites
The conducting polymer composite electrodes have a positive impact on the final performance compared to electrodes of individual materials. The composite electrodes, including two main types of composite conducting polymers with electrochemical double-layer capacitive and pseudocapacitive materials, respectively, have been receiving more and more attention. Conducting polymer composites with a proper choice of compositing components are among the supercapacitive materials with the most potential for motivating existing supercapacitor devices toward future advanced energy storage applications because of their high redox active-specific capacitance and inherent elastic polymeric nature. Moreover, microstructural control for conducting polymer composites has been considered as an effective approach to improve the cycling stability of the conducting polymers.3.1 Compositing conducting polymers with carbon nanomaterials
Electrode materials for EDLCs usually refer to carbon materials, which possess many advantages, such as large surface area and high conductivity. In addition, the easy availability and environmental friendliness are other reasons why carbon materials have been widely used as electrode materials for EDLCs. Superior capacitive properties can be achieved when carbon materials are composited with conducting polymers with tailored nanostructures.As commonly used electrode materials for EDLCs, carbon materials not only provide efficient supporting substrates but also provide highly conductive pathways. Typically, nanostructured carbon materials that possess a high surface area and large porosity can provide effective decoration sites to promote the formation of nanostructured conducting polymers. Nanostructured carbon materials are better alternatives for boosting performances when composited with conducting polymers, and therefore, the introduction of nanostructured carbon materials with one-dimensional (1D), two-dimensional (2D) and three-dimensional (3D) architectures into conducting polymers has a significant, positive impact on the electrochemical capacitive performance.
CNT-incorporated conducting polymer composites can be fabricated by a solution-blending method37–39 and in situ polymerization method.40–48 Roberts and co-workers have prepared robust free-standing CNT/conducting polymer composite electrodes by a filtration method, which can be easily scaled up. The composite electrodes show an enhanced capacitance. This process gives a simple, low-cost and high-throughput strategy for large-scale manufacturing of CNT/conducting polymer composite electrodes. CNTs have displayed favorable flexibility and are promising as free-standing and flexible electrodes. Chen and co-workers provided an in situ electrochemical polymerization method to fabricate free-standing and flexible single walled carbon nanotube (SWNT)/PANI hybrid films. In this case, the SWNT film with a continuous reticulate architecture acts as the skeleton and the PANI layers act as the skin. This unique “skeleton/skin” structure ensures composite films with high energy and power densities of 131 W h kg−1 and 62.5 kW kg−1, respectively.49
Several studies have shown that CNTs with a random arrangement within the conducting polymer matrix have a synergistic effect on the final capacitive performance. Moreover, CNTs with ordered structures are able to significantly improve the electrochemical performance. Zhang and co-workers utilized a mechanical method to align CNT (A-CNT) electrodes with a growth of conducting polymers. Because of the unique properties of A-CNTs, the supercapacitor devices exhibit a wide operation voltage of 4 V with a maximum energy and power density of 82.8 W h L−1 and 130.6 kW L−1 in a volumetric performance, respectively.50
In addition to the widespread applications of CNTs for the construction of 1D carbon-incorporated conducting polymer composites, CNFs and GNRs have also been used to construct conducting polymer composite electrodes for the enhancement of their electrochemical capacitive performance.51,52 Compared with CNTs, CNFs have inherent advantages such as low production cost and easy mass production.52,53 Yoon and co-workers have used a one-step vapor deposition polymerization technique to fabricate PANI-coated CNFs.54 In this case, CNFs with an average diameter of 50 nm were synthesized in a quartz flow reactor with an iron catalyst powder, and a subsequent immobilization of PANI makes the CNF/PANI composites exhibit a maximum specific capacitance value of 264 F g−1.
Thanks to their high surface area, high electrical conductivity, and scalability, GNRs are considered ideal carbon substrates for the immobilization of conducting polymers.55–57 Tour and co-workers used a low-cost in situ polymerization method to fabricate GNR/PANI composites with ordered and vertically-aligned PANI nanorods grown on the GNRs. The resulting composites have a high specific capacitance of 340 F g−1 and a stable cycling performance with a 90% capacitance retention over 4200 cycles because of the synergistic combination of electrically conductive GNRs and high-capacitance PANI.58
GO, which contains oxygenated groups such as epoxides, hydroxyl groups, and carboxyl functional groups on its basal planes and edges, has good compatibility with polymers and has been widely used to fabricate conducting polymer-based composites because of its extraordinary morphology and strong hydrophilicity.63–65 Besides, during the oxidative polymerization, monomers like aniline are oxidized to PANI, and simultaneously, the removal of oxygen occurs and GO is reduced to graphene, creating good conducting networks.66,67 Xu and co-workers constructed hierarchical composites by combining PANI nanowires with 2D GO sheets by simply adjusting the ratios of aniline to GO in different nucleation processes.68 The as-obtained PANI/GO composites possess a high capacitance of 555 F g−1 and retained 92% of the initial capacitance after 2000 charge and discharge cycles.
Graphene in the form of rGO is extremely attractive for application in supercapacitors because of its unprecedented properties including excellent electrical and electrochemical performances.62,69–71 Wang and co-workers have fabricated rGO-wrapped PANI nanofiber composites via assembly of negatively charged GO with positively charged PANI nanofibers in aqueous dispersions followed by the reduction of GO to rGO. The capacitance of the composites reached 250 F g−1 at 0.5 A g−1 in a 1 M Et4N+·BF4−/propylene carbonate organic electrolyte and only 26.3% of the capacitance decreased over 1000 cycles.70 Baek and coworkers prepared conducting PANI-grafted rGO composites (PANi-g-rGO). In this case, as shown in Fig. 1, the amine-protected 4-aminophenol is first grafted onto GO, and amine terminated GO is subsequently polymerized in the presence of an aniline monomer. Because of the participation of thionyl chloride vapors, the GO is gradually reduced into rGO. Such composite materials with an electrical conductivity of 8.66 S cm−1 exhibit a specific capacitance of 250 F g−1 and good cycling stability.72
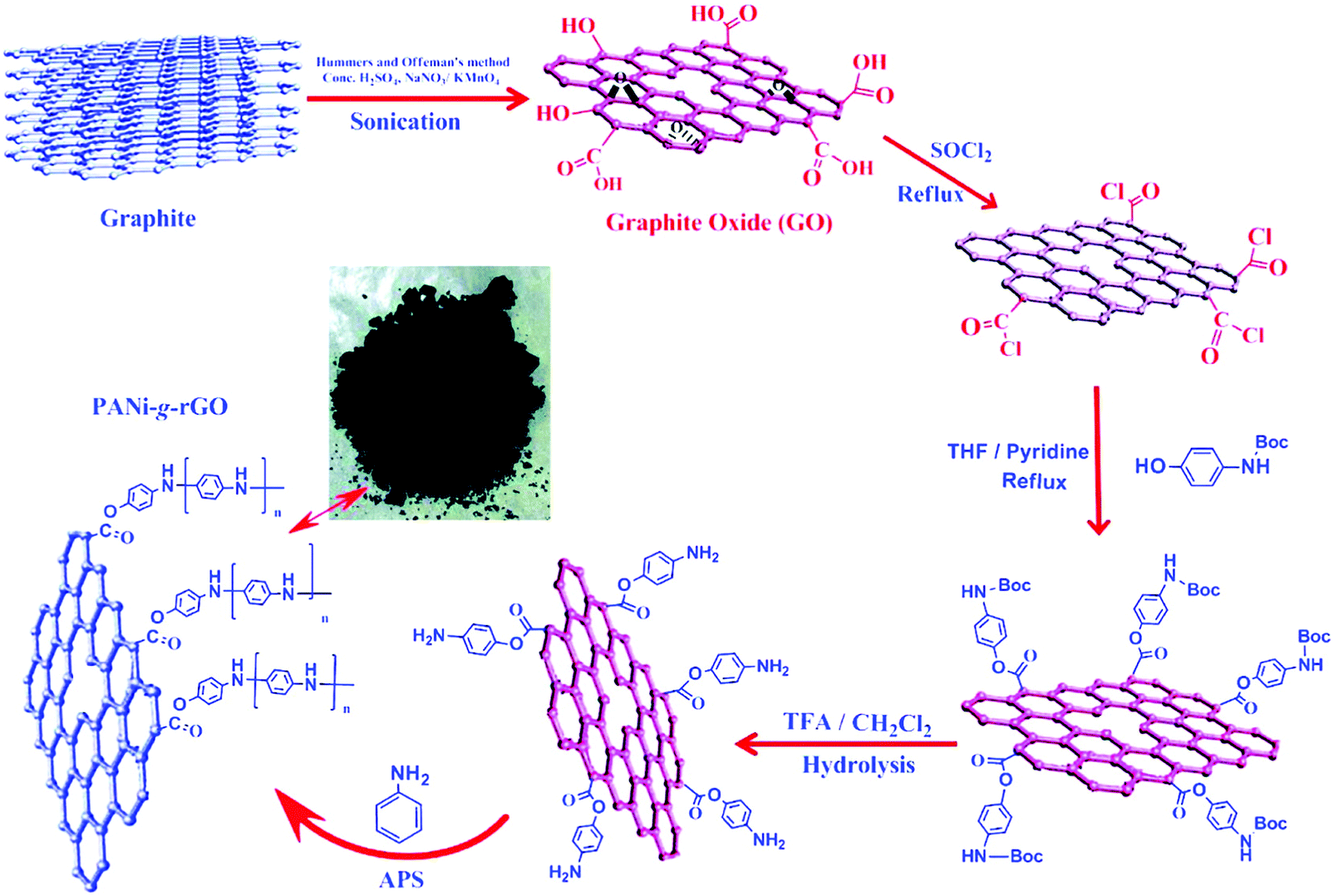 | ||
| Fig. 1 Schematic illustration of the preparation of PANi-g-rGO with a digital picture of the sample in the middle. Reprinted with permission from ref. 72 Copyright 2012 American Chemical Society. | ||
Graphite nanoflakes (GFs) have also been applied for the construction of 2D carbon-incorporated conducting polymer composites. Liu and coworkers have used a partially exfoliated graphene (Ex-GF) electrode with graphene sheets standing on a graphite foil matrix as a template for the electropolymerization of pyrrole with 1,5-naphthalene disulfonate (NDS) and 2-naphthalene sulfonate (NMS) as the “permanent” doping anions to prepare Ex-GF/PPy-NDS and Ex-GF/PPy-NMS composites, respectively. Both of the two composite electrodes displayed high energy densities. The cyclic stability of the electrodes was also improved because of the synergistic effect with partially exfoliated graphene.73 Yu and co-workers have fabricated GFs with a few-nanometer thickness by scraping a graphite rod over a commercially available polymer lapping film with grits. Then, a thin layer of p-toluene sulfonate (pTS)-doped PPy was electrochemically polymerized on the surface of the GF-coated polymer lapping film to form a hybrid flexible electrode, as shown in Fig. 2. Such micro-supercapacitor devices show a maximum capacitance of 37 mF cm−2 in a half cell using a 1 M H2SO4 electrolyte, 23 mF cm−2 in full cell, and 6 mF cm−2 in a planar cell configuration using poly(vinyl alcohol) (PVA)/H3PO4 as a solid-state electrolyte.74
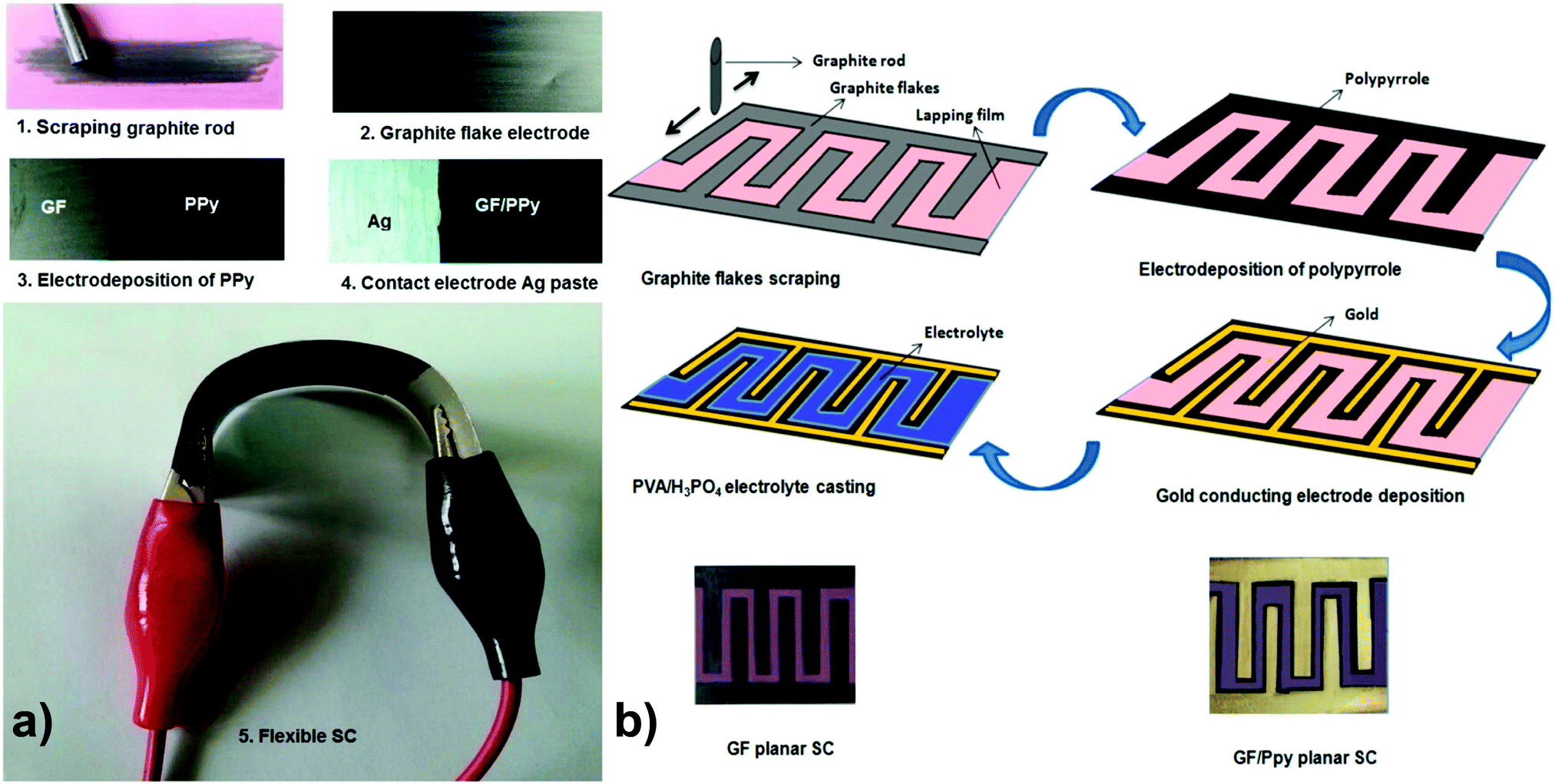 | ||
| Fig. 2 (a) Photographic demonstration of electrodes and supercapacitor fabrication steps. (b) Fabrication steps of planar supercapacitors and the photograph of supercapacitors with and without gold electrode. Reprinted with permission from ref. 74 Copyright 2015 American Chemical Society. | ||
In general, carbon fibers with diameters normally about several micrometers can be synthesized through a spinning technique with controlled pyrolysis and activation.86–88 Wu and co-workers have fabricated high-performance electrodes made of graphene-beaded carbon fibers (G/CFs) coated with conducting polymers.89In situ polymerization is utilized to coat the G/CFs with an ultrathin layer of thorn-like PANI nanorods. The highly porous PANI-G/CNF composites exhibit a large surface area, low internal resistance, and fast redox rate. Kurungot and co-workers have designed a kind of carbon fiber electrode with electrodeposited polyethylenedioxythiophene (PEDOT) for all-solid-state supercapacitors.90 The electrodeposited, flower-like PEDOT coatings on each carbon fiber led to an enhanced surface area and electrical conductivity, and the pores in this system allowed an effective immersion of the polymer-based gel electrolyte.
Carbon cloths, broadly applied in the field of free-standing and conducting substrate electrodes, are a network of carbon fibers with diameters of about several micrometers.91 They have been extensively studied as electrode materials and supporting substrates. Liu and coworkers reported well-ordered PPy nanowire arrays on the surfaces of carbon fibers in untreated carbon cloths that were used to construct hierarchical structures, which contain the 3D conductive carbon fiber skeleton and the immobilized, well-ordered electrochemically active conducting polymer nanowires.92 The well-ordered PPy nanowire array electrode exhibited a high specific capacitance of 699 F g−1 at 1 A g−1 with an excellent rate capability, and 81.5% of its capacitance was retained at 20 A g−1.
Carbon aerogels with 3D interpenetrating structures can improve the efficiency of ionic and electronic transport.93–95 The carbon aerogel-based composites are designed for flexible and free-standing energy storage devices without any binders. Chen and his coworkers reported a novel 3D interconnected nanotubular graphene/PPy composite by incorporating PPy into highly conductive and stable nanotubular graphene. As shown in Fig. 3, the 3D nanotubular graphene is synthesized by chemical vapor deposition using nanoporous nickel (np-Ni) as a template. Then, ultrathin PPy layers are loaded onto the 3D core–shell graphene@np-Ni by cyclic electrochemical deposition. Finally, the np-Ni template is etched by acid. The composite electrode reaches a maximum energy density of 21.6 W h kg−1 at a power density of 32.7 kW kg−1.96
 | ||
| Fig. 3 Schematic of preparation steps of nanotubular graphene/PPy composite. (a) Nanoporous graphene@np-Ni prepared by chemical vapor deposition. (b) Nanoporous nickel/graphene/PPy prepared by electrodeposition of PPy on the surface of graphene@np-Ni ligaments. (c) Nanotubular graphene/PPy composite after removing np-Ni template by chemical etching. Reprinted with permission from ref. 96 Copyright 2016 Elsevier. | ||
The high electric double layer capacitances of porous carbon are related to their high surface area and precise pore size distributions according to the electrolyte used. The microstructure, pore size, pore shape, and surface termination of porous carbon can be precisely controlled by changing the synthesis parameters and the precursor compositions.97,98 Zhao and coworkers prepared a conducting polymer–porous carbon composite electrode by deposition of a thin layer of PANI on the surface of 3D highly-ordered macroporous carbon (3DOM carbon). As exhibited in Fig. 4, the honeycomb-like 3DOM carbon not only favors the diffusion of electrolyte ions but also prevents the agglomeration of PANI.99
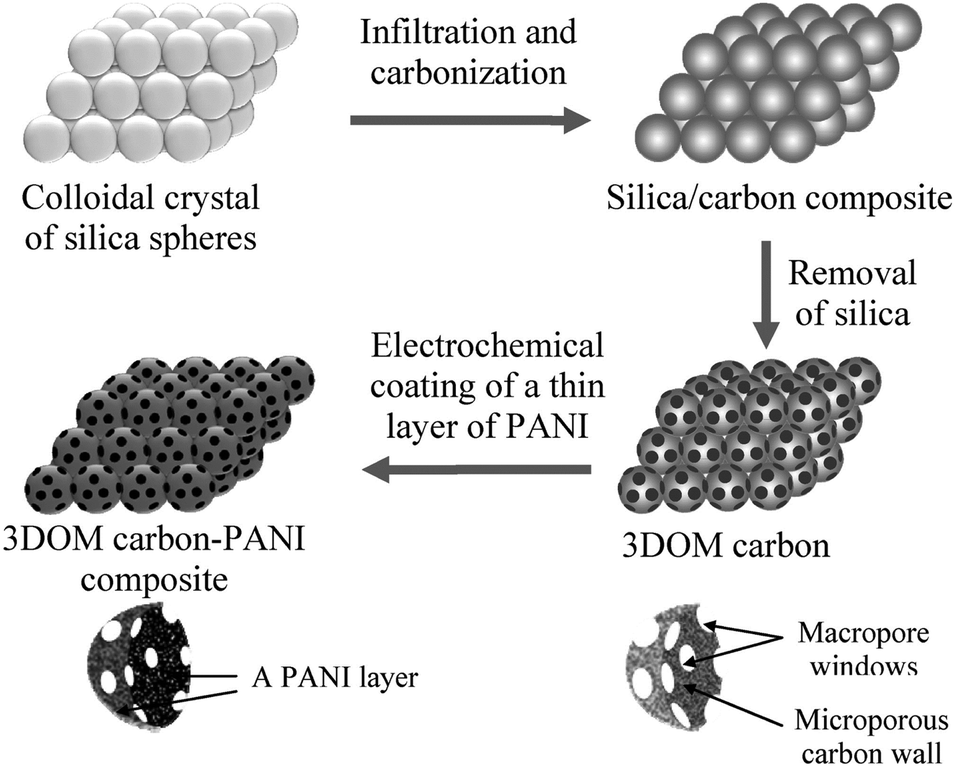 | ||
| Fig. 4 Preparation process of 3DOM carbon and 3DOM carbon–PANI composite. Reprinted with permission from ref. 99 Copyright 2010 American Chemical Society. | ||
There have been some attempts to combine 1D CNTs and 2D graphene to prepare 3D carbon materials for supercapacitors. Significant performance enhancements are observed in these hybrid materials, and the CNTs are expected to bridge the defects for efficient electron transfer and increase the basal spacing between graphene sheets with a larger surface area.100–104 For instance, Cheng and co-workers fabricated a graphene/CNT/PANI composite electrode by coating PANI nanocones onto the graphene/CNT composites. An asymmetric supercapacitor is assembled with the ternary graphene/CNT/PANI and the graphene/CNT electrode to achieve a high specific energy density. The excellent capacitive performance is attributed to the larger surface area of the 3D graphene/CNT network structure and the increased electrical conductivity from the presence of the CNTs.104 Liu and co-workers prepared a 3D GNR–CNT–PANI composite via in situ polymerization of aniline monomers on the surface of GNR–CNT hybrids, which are conveniently obtained by partially unzipping CNTs. A two-electrode cell system, which can provide the most accurate measurement of the performance, was fabricated using a PVA/H3PO4 gel as the solid-state electrolyte. The hierarchical GNR–CNT–PANI composites possess a much higher specific capacitance (890 F g−1) than the GNR–CNT hybrid (195 F g−1) and neat PANI (283 F g−1) at 0.5 A g−1. At the same time, the GNR–CNT–PANI composites display a good cyclic stability with an 89% capacitance retention after 1000 cycles.105
A proper substrate consisting of 3D carbon materials with a high porosity, excellent electrical conductivity and good flexibility to combine with conducting polymers is highly desirable for the efficient utilization of pseudocapacitance originating from conducting polymers. Liu and co-workers immobilized PANI nanoparticles onto a conducting paper-like substrate, which utilized graphene and CNTs as building blocks, to obtain flexible graphene/CNT/PANI composite films with a hierarchical nanostructure. The ternary composite film exhibited a specific capacitance of 432 F g−1 at 0.5 A g−1 and an enhanced cyclic stability with ∼96% of its original capacitance after 600 charge and discharge cycles. The superior electrical conductivity of the graphene/CNT composite films provided improved conductive pathways for charge transfer, and at the same time, the dimensional confinement of the PANI particles on the planar carbon substrate prohibits volume expansion and shrinkage upon electrolyte soakage.103
3.2 Compositing conducting polymers with pseudocapacitive materials
Conducting polymers have been a research hotspot as electrode materials for supercapacitors because of their high conductivity in a doped state, high charge storage capacity, low cost and environmental friendliness.16,106–110 Conducting polymer-based electrodes can undergo a rapid and reversible redox reaction, which allows the devices to store high-density charges and generate high faradic capacitances.111–114 However, the swelling and shrinkage of conducting polymers may inevitably occur during the electrochemical redox reaction, which leads to low cycle life and deterioration of capacitance after long-term charge and discharge cycles.115–117 For example, the PPy-based electrodes can achieve an initial specific capacitance of 120 F g−1 in a current density of 2 mA cm−2, but its specific capacitance drops by about 50% after 1000 cycles because of the volume change during the electrochemical reaction.118 Therefore, the improvement in the cyclic stability of conducting polymer electrodes has become an urgent problem to be addressed. In general, the preparation of conductive polymer composites with a combination of metal oxides, metal hydroxides or metal sulfides can buffer the volume change during the repeated redox cycles as well as improve the mechanical stability and specific capacitance of the conducting polymers.119–1223.2.1.1 Conducting polymer/ruthenium oxide composites. Among the metal oxides/hydroxides, ruthenium oxide (RuO2) has been extensively studied because of its highly reversible redox reactions and remarkably high theoretical specific capacitance (1360 F g−1). However, the expensive cost of RuO2 restricts its mass practical application in supercapacitors.125,129 Therefore, the hybridization of metal oxides/hydroxides with inexpensive conducting polymers can greatly reduce the cost and simultaneously maintain the excellent performance.130–134 For example, Lee and co-workers reported RuO2/PEDOT nanotubes with a high specific capacitance of 1217 F g−1 and a high power density of 20 kW kg−1 from the high specific surface area and fast charge/discharge of the tubular structures.135 Lokhande and co-workers prepared the PANI–RuO2 composite films using a chemical bath deposition method, which achieve a specific capacitance of 830 F g−1.136 Lian and co-workers successfully constructed a 3D arrayed nanotubular architecture with an ultrathin RuO2 layer coated on the well-aligned cone-shaped nanostructure of PPy (WACNP). The unique WACNP/RuO2 composite electrodes on an Au substrate exhibit a higher specific capacitance of 15.1 mF cm−2 (302 F g−1) at a current density of 0.5 mA cm−2, which is more than that of WACNP. Moreover, the specific capacitance of the WACNP/RuO2 electrode decays slightly during the first 30 cycles and remained at 90% after 300 charge and discharge cycles. The high capacitance and excellent stability of the WACNP/RuO2 composite electrode may be attributed to the enhanced electrochemical activity as well as the reduced diffusion resistance of electrolytes in the electrode materials.137
3.2.1.2 Conducting polymer/manganese dioxide composites. Despite the high specific capacitance of RuO2, its high cost and toxic nature have restricted its mass production and wide application as an electrode material for supercapacitors. With the advantage of low-cost and relatively high specific capacitance (1100 F g−1 of theoretical capacitance), manganese dioxide (MnO2) is expected to be an outstanding alternative to RuO2.138–141 Therefore, creating composites of MnO2 with conducting polymers as electrode materials for supercapacitors has attracted intense attention. Desu and co-workers synthesized MnO2-embedded PPy composite (MnO2/PPy) thin film electrodes on polished graphite substrates, which exhibit a remarkable specific capacitance of 620 F g−1 compared to MnO2 (225 F g−1) and PPy (250 F g−1).118 Yang and co-workers developed an ion-exchange reaction for PANI with n-octadecyltrimethylammonium-intercalated MnO2 precursors in proper solvents, such as N-methyl-2-pyrrolidone (NMP), to prepare PANI-intercalated layered MnO2 composites, which can maintain 94% of its initial specific capacitance (330 F g−1) after 1000 cycles, indicating an improved electrochemical cyclic stability compared to pristine PANI and MnO2.142 Lee and co-workers introduced a one-pot co-electrodeposition method to fabricate core–shell MnO2/PEDOT coaxial nanowires with a controlled thickness of PEDOT shells by varying applied potentials, as shown in Fig. 5. The resultant coaxial nanowires exhibit high specific capacitances and excellent mechanical properties with 85% of the capacitance (from 210 to 185 F g−1) preserved as the current density increases from 5 to 25 mA cm−2, which may be attributed to the fact that the solid core MnO2 and the flexible PEDOT shell can synergistically prevent the collapse and breaking of the coaxial nanowires.143
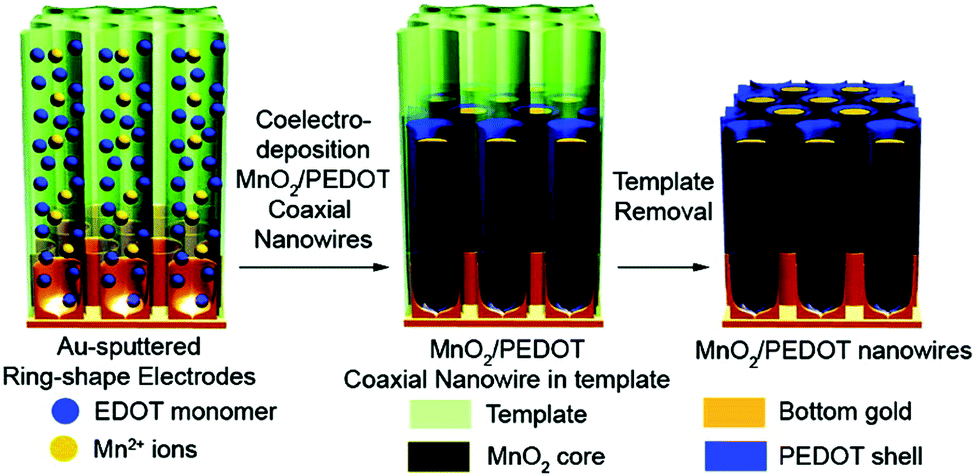 | ||
| Fig. 5 One-step synthesis of MnO2/PEDOT coaxial nanowires. Reprinted with permission from ref. 143 Copyright 2008 American Chemical Society. | ||
3.2.1.3 Conducting polymer/nickel oxide–hydroxide composites. In addition to the composites of conducting polymers with RuO2 and MnO2, nickel oxide (NiO) and nickel hydroxide (Ni(OH)2) have also been considered as alternative materials and have been used to create composites with conducting polymers as electrode materials for supercapacitors because of their low cost, easy synthesis, distinct redox reaction, and most importantly, their relatively high theoretical specific capacitance (2584 F g−1 for NiO and 2081 F g−1 for Ni(OH)2).144–148 Zhao and co-workers adopted an in situ method to synthesize flower-like PANI–NiO nanostructures on nickel foams as binder-free electrodes, which can achieve a specific capacitance of 2565 F g−1 at 1 A g−1 and maintain 70% of the capacitance with the current density increasing 10 times.149 Hu and co-workers fabricated porous NiO/Ni(OH)2 composite (PNC) encapsulated in 3D interconnected PEDOT nanoflowers on metal wires (Cu–Ni alloy) through a mild electrochemical route. The surface of PNC is coated with a protective thin layer of steady PEDOT, which can improve the electronic conductivity and structural stability of the NiO/Ni(OH)2/PEDOT composites. The 3D flower-like nanostructure achieves a high specific capacitance of 404.1 mF cm−2 at a current density of 4 mA cm−2 and a long-term cycling stability with 82.2% capacitance retention after 1000 cycles. Moreover, a fiber-shaped, flexible, all-solid-state asymmetric supercapacitor was fabricated with the as-prepared NiO/Ni(OH)2/PEDOT composite as the positive electrode, the ordered mesoporous carbon (CMK-3) fiber as the negative electrode and a polymer gel as the electrolyte, and it delivered a high output voltage of 1.5 V, a high specific capacitance of 31.6 mF cm−2 and a high specific energy density of 0.011 mW h cm−2.150
3.2.1.4 Conducting polymer/other metal oxide–hydroxide composites. Many other metal oxide/hydroxide materials, such as vanadium oxides (V2O5), hematite (α-Fe2O3) and cobalt monoxide (CoO), have also been investigated and incorporated with conducting polymers as electrode materials for supercapacitors. V2O5 has attracted intense attention for its variable oxidation states, surface/bulk redox reactions and layered structures, which facilitate efficient ion diffusion.94,151 Liu and co-workers synthesized large surface area V2O5–PANI composite nanowires using an electrodeposition method, which can promote the effective contact of electrochemical active materials with the electrolyte and achieve a high specific capacitance of 412 F g−1 at 4.5 mA cm−2.127 α-Fe2O3 has also been explored as an anode electrode material for asymmetric supercapacitors because of its high theoretical specific capacitance, low cost, non-toxicity and abundance. The hybridization of α-Fe2O3 with conducting polymers holds great promise to improve the capacitance and cycling stability of α-Fe2O3. Li and co-workers designed a high-performance asymmetric supercapacitor with highly ordered 3D α-Fe2O3@PANI core–shell nanowire arrays as the anode and PANI nanorods grown on carbon cloths as the cathode, and it exhibited a high volumetric capacitance of 2.02 mF cm−3 and an excellent cycling life with a capacitance retention of 95.77% after 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.92 CoO, another electrochemically active material, has also been studied for pseudocapacitive energy storage by elaborate fabrication with conducting polymers to address the issues of capacitance and stability. Liu and co-workers designed a well-aligned CoO@PPy nanowire array grown on nickel foam as a pseudocapacitive electrode. The assembled asymmetric supercapacitor exhibited a high specific capacitance of 2223 F g−1 and outstanding cycling stability because of the shortened ion diffusion channels and the highly conductive nanowire arrays.152
000 cycles.92 CoO, another electrochemically active material, has also been studied for pseudocapacitive energy storage by elaborate fabrication with conducting polymers to address the issues of capacitance and stability. Liu and co-workers designed a well-aligned CoO@PPy nanowire array grown on nickel foam as a pseudocapacitive electrode. The assembled asymmetric supercapacitor exhibited a high specific capacitance of 2223 F g−1 and outstanding cycling stability because of the shortened ion diffusion channels and the highly conductive nanowire arrays.152
Nickel cobaltite (NiCo2O4) has been reported to have better electrochemical activity and conductivity with an outstanding specific capacitance of 1400 F g−1 at a sweep rate of 25 mV s−1 compared to pure Co3O4 and NiO.153–155 As exhibited in Fig. 6, Xia and co-workers reported 3D NiCo2O4@PPy coaxial nanowire arrays (NWAs) on carbon textiles for high-performance flexible supercapacitors, where the mesoporous NiCo2O4 nanowire arrays serve as the highly capacitive “core” and the uniformly coated PPy nanospheres act as the highly conductive “shell.” The ordered NiCo2O4 mesoporous NWAs can shorten ion transport pathways, and the highly conductive PPy nanospheres can facilitate ion diffusion, which lead to a high specific capacitance, enhanced rate capability and cycling stability. Moreover, a flexible asymmetric supercapacitor device was successfully fabricated using the as-obtained NiCo2O4@PPy and activated carbon as electrodes, which exhibit a high energy density (58.8 W h kg−1 at 365 W kg−1) and outstanding cycling life with ∼82.9% capacitance retention after 5000 cycles.150
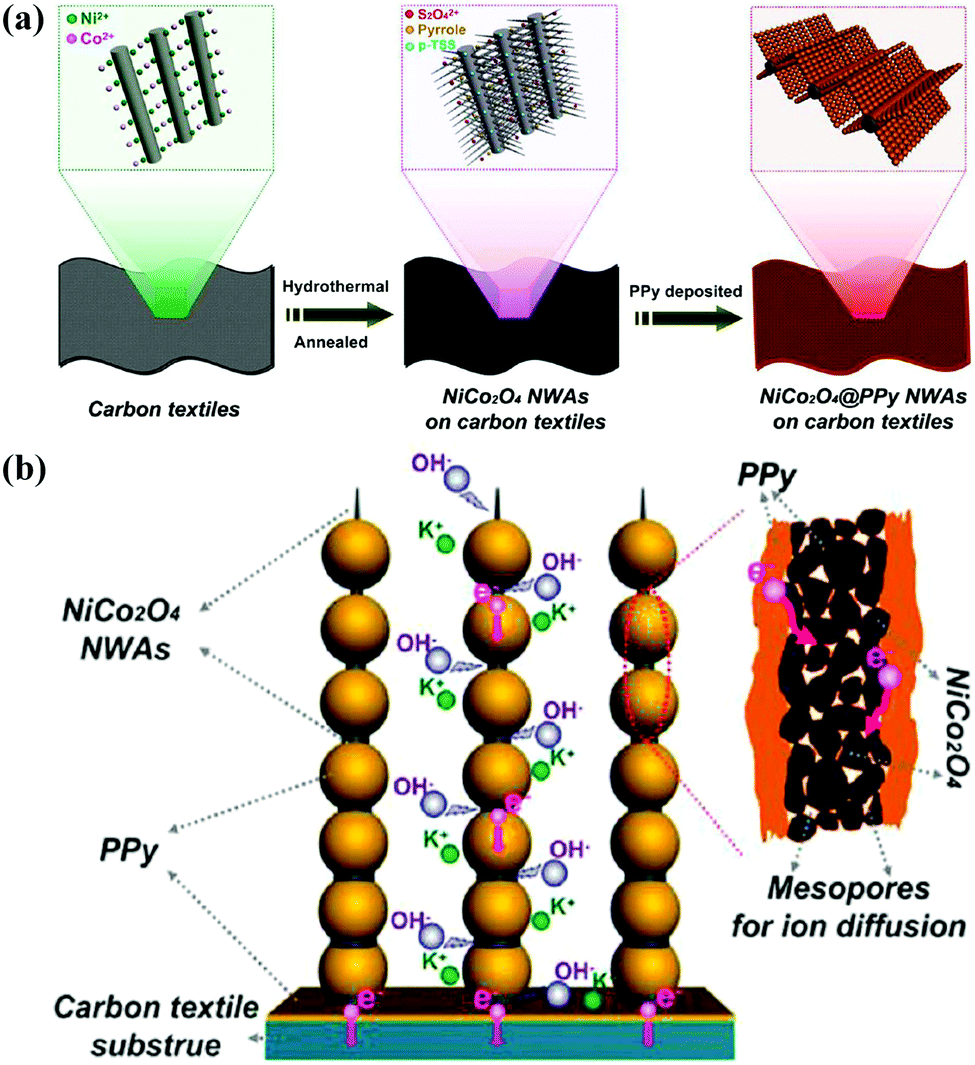 | ||
| Fig. 6 (a) Schematic illustration of the fabrication process of hierarchical mesoporous NiCo2O4@PPy NWAs on carbon textiles. (b) Schematic representation of rechargeable supercapacitor based on NiCo2O4@PPy NWAs on carbon textiles. Reprinted with permission from ref. 150 Copyright 2015 American Chemical Society. | ||
Among the metal sulfides, MoS2 is expected to have excellent electrochemical capacitive properties because of its double layer charge storage as well as the range of oxidation states of Mo, from +2 to +6, which can guarantee its pseudocapacitive characteristics. Liu and co-workers adopted an in situ oxidative polymerization method to prepare a 3D tubular MoS2/PANI nanostructure with PANI nanowire arrays vertically aligned on the external and internal surfaces of 3D tubular MoS2, which serves as both the active materials in the electrochemical reaction and framework to provide more pathways for insertion and extraction of ions. The as-prepared MoS2/PANI composites not only achieve an enhanced specific capacitance of 552 F g−1 at 0.5 A g−1, but they also have an excellent rate capability of 82% from 0.5 to 30 A g−1.163 Furthermore, it has been reported that octahedral (1T) phase MoS2 sheets exhibit a high electrical conductivity, large surface area, and unique surface chemical characteristics, making them an attractive substrate for the growth of nanostructured conducting polymers.164–166 As shown in Fig. 7, Xu and co-workers synthesized a MoS2/PANI@C hierarchical composite by directly depositing PANI on mono-layered MoS2 sheets containing 72% 1T-phase MoS2 followed by a carbon shell coating. The multiple synergistic effects between PANI and MoS2 as well as the carbon coating led to an excellent energy storage performance with a superb capacitance of 678 F g−1 and remarkable cycling stability with an 80% retention after 10000 cycles.167
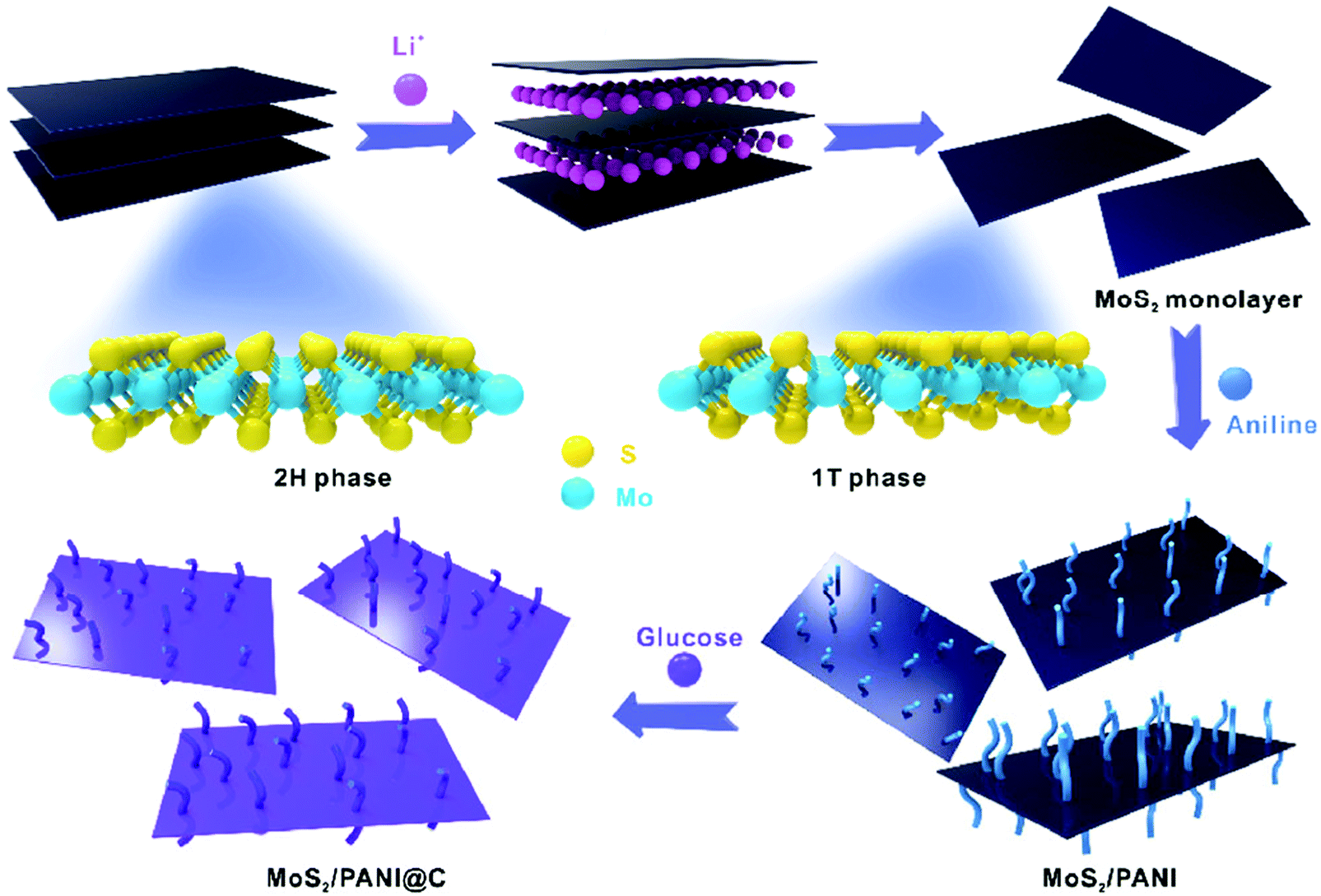 | ||
| Fig. 7 Schematic diagram of the MoS2/PANI@C process for the synthesis of carbon-encapsulated PANI based on MoS2 monolayer nanosheets. Reprinted with permission from ref. 167 Copyright 2016 Springer. | ||
 | ||
| Fig. 8 (a) Schematic illustration for the fabrication of PPy@PANI DNTAs. (b) Nanotube array architecture, double-walled structure, and high conductivity in electrode provide ion and electron “highways” and high utilization rate of electrode. (c) Specific capacitance values of PPy@/PANI DNTAs, PPy nanotube arrays and PANI nanotube arrays as a function of cycle number at 10 mV s−1. Reprinted with permission from ref. 175 Copyright 2014 The Royal Society of Chemistry. | ||
Since the improvement of the specific capacitance as well as the cyclic stability of conducting polymer electrodes has become an urgent problem, in Section 3.2 we summarized and suggested several types of pseudocapacitive materials, such as metal oxides, metal hydroxides and metal sulfides, that might replace carbon materials as excellent candidates for creating composites with conducting polymers for high-performance supercapacitors.
3.3 Microstructural control for conducting polymer composites
In general, the microstructure of electrode materials plays a critical role in the final performance of the supercapacitors.176 Therefore, to sufficiently utilize the high capacitance of conducting polymers, the microstructure of the conducting polymer composites should be rationally designed and controlled. As will be shown later, three models can be conveniently categorized based on the microstructural control of the as-fabricated conducting polymer composites: (i) anchored model, (ii) wrapped model, and (iii) sandwiched model. The anchored model demonstrates that the carbon materials, metal oxides and other supporting materials play a supporting role for subsequent immobilizations of conducting polymers. In the wrapped model, conducting polymers are tightly wrapped within the carbon material, metal oxide and other nanomaterial shells. The sandwiched model consists of sandwiched conducting polymer layers and inserted other parts. Between each sandwiched layer of the conducting polymers, an internal skeleton shell and a cladding layer forms an interconnected framework. This section will mainly focus on the three models mentioned above to describe the effects of the microstructural control of conducting polymer composites on their electrochemically capacitive performances.Direct coating of conducting polymers including PANI, PPy and PEDOT on rGO via an in situ polymerization was achieved by Zhao and co-workers.183 In this work, the conducting polymer composites showed a superior electrochemical performance to that of pristine PANI. For instance, the specific capacitance of the rGO–PANI composite reaches 361 F g−1 at a current density of 0.3 A g−1. More than 80% of the initial capacitance is retained after 1000 cycles, which is much better than that of pristine PANI. The good capacitive performance of these composites is derived from the good electrical conductivity of the composites. Conducting polymer-anchored CNTs have also been widely studied.184–195 Gupta and co-workers prepared PANI/SWNT composites by in situ electrochemical deposition of PANI onto SWNTs. In this work, the microstructure of the PANI/SWNT composites strongly influences their specific capacitance. The highest specific capacitance (463 F g−1) is obtained in the optimized condition of 73 wt% PANI deposited on the SWNTs.188
2D nanostructured composites such as metal oxides and metal sulfides have good capacitive properties because of their sheet-like morphology, and they are expected to exhibit a large surface area and excellent capacitive performance.196–199 What is more, the transition metal center may exhibit a range of oxidation states, offering its pseudocapacitive characteristics. Conducting polymer-anchored metal oxides and metal sulfides mainly involve vanadium oxide (V2O5), molybdenum oxide (MoO3) and molybdenum disulfide (MoS2) as substrates to improve their specific capacitive and long-term cycling stability.199–206 Wang and co-workers have prepared highly conductive composites of MoS2/PANI via the direct intercalation of an aniline monomer into MoS2. This architecture is also beneficial for improving the capacitance and cycling stability of MoS2/PANI composite electrodes. In comparison with a specific capacitance of 131 F g−1 and 42% retention of capacitance over 600 cycles in the PANI electrode, the MoS2/PANI composite electrode shows a specific capacitance of 390 F g−1 and 86% retention of capacitance over 1000 cycles. The MoS2/PANI composite electrodes provide enhanced capacitive performances that synergistically combine pseudocapacitance of the PANI and the electrochemical double layer capacitance from the MoS2.199
It is worth mentioning that conducting polymers with a morphology of vertically aligned nanowire arrays anchored on supporting materials have emerged as ideal electrode materials for supercapacitors.40,207,208 This unique nanostructure has several advantages as an electrode material. Firstly, each nanowire is electrically connected with the supporting materials, making full use of all the nanowires to boost the capacitive performance. Secondly, the nanowire arrays allow for efficient charge transport and reduce the ion-transport length. Thirdly, there are spaces between the aligned nanowires that can accommodate large volume changes during the charge and discharge cycles without fractures.30 For instance, PANI nanowire arrays on MoS2,209 ordered bimodal mesoporous carbon,210 GO,68 rGO modified graphite electrode,211 as well as PPy nanowire arrays on carbon fibers212 and GO213 were also intensely studied. Xu and co-workers successfully prepared hierarchical composites of PANI nanowire arrays vertically grown on GO sheets.68 The as-fabricated PANI–GO composites exhibited a morphology-dependent capacitance performance, and the specific capacitance of PANI/GO was as high as 555 F g−1 at a condition of 0.05 M aniline. After 2000 charge and discharge cycles, the PANI/GO composite still had 92% of its initial capacitance, while pristine PANI held only 74% of its initial capacitance. The enhanced stability of the PANI/GO composite is ascribed to the synergistic effect of the GO and PANI nanowire arrays. The strong π–π stacking interactions between the GO and PANI nanowire arrays allowed the composites to be electrochemically stable. Moreover, the GO sheets and vertically aligned nanowire arrays can undertake some mechanical deformation during charge and discharge cycles, which also prevents PANI from agglomerating, and is beneficial to the improved stability.
Inspired by the findings that graphene sheets can be decorated onto positively charged colloids via electrostatic interaction-induced self-assembly,225 Zhou and co-workers reported the fabrication of graphene-wrapped PANI nanofibers by a direct assembly of the positively charged PANI nanofibers with negatively charged GO, followed by the reduction of GO by hydrazine. The graphene-wrapped PANI nanofibers displayed a high specific capacitance of over 250 F g−1 and improved cycling stability with a capacitance retention of 74% over 1000 cycles.70 Liu and co-workers fabricated PANI hollow sphere (PANI-HS)@electrochemical reduced graphene oxide (ERGO) composites with core–shell structures via a solution-based co-assembly process (demonstrated in Fig. 9).221 The unique nanostructures, such as porous and hollow structures, provide particular advantages to capacitive performances because of the enlarged surface area and shortened diffusion lengths for both mass and ion transport.226–228 The PANI-HS@ERGO composites show a specific capacitance of 614 F g−1 at a current density of 1 A g−1. The capacitance decreases by 10% after 500 charge and discharge cycles, suggesting a good cycling stability. The prominent properties are because of the synergistic effects of the two components of PANI-HS and ERGO.221
 | ||
| Fig. 9 Schematic illustration of the preparation of PANI-HS@ERGO composites. Reprinted with permission from ref. 221 Copyright 2013 American Chemical Society. | ||
The sandwiched model can be constructed by electrostatic interaction, hydrogen bonding, van der Waals forces, and charge transfer complexes.229–236 In particular, layer-by-layer approaches via the sequential adsorption of different components have drawn substantial attention because of their features including the construction of sandwiched conducting polymer composites with tailored composition and surface functionality. Graphene, GO, CNTs and some metal oxides have been utilized to construct conducting polymer composites with a sandwiched configuration.229–241 Zhao and co-workers prepared conducting polymer-pillared GO sheets based on the electrostatic interactions between negatively charged GO sheets and positively charged surfactant micelles (as illustrated in Fig. 10). The cationic surfactant micelles might adsorb on the surfaces of the GO sheets via electrostatic interaction, and then a GO-surfactant multilayer structure, in which the surfactant micelles located between the GO sheets can be formed. The subsequent polymerization of the monomers solubilized in the hydrophobic cores of the surfactant micelles will be predominant. Finally, a layered GO structure sandwiched with conducting polymers will be formed by the removal of the surfactants. In this work, the GO/PPy composite shows a high capacitance of 500 F g−1, indicating an excellent electrochemical capacitive performance of a sandwiched conducting polymer composite. The advantages of the composite materials are as follows: (1) the exfoliated GO sheets are beneficial for the attachment of the conducting polymer on both sides; (2) the mechanical stability of the composites during the charge and discharge processes would be enhanced because of the closely-packaged sandwiched nanostructure; (3) the combination of GO with conducting polymer pillars would effectively reduce the dynamic resistance of electrolyte ions.240
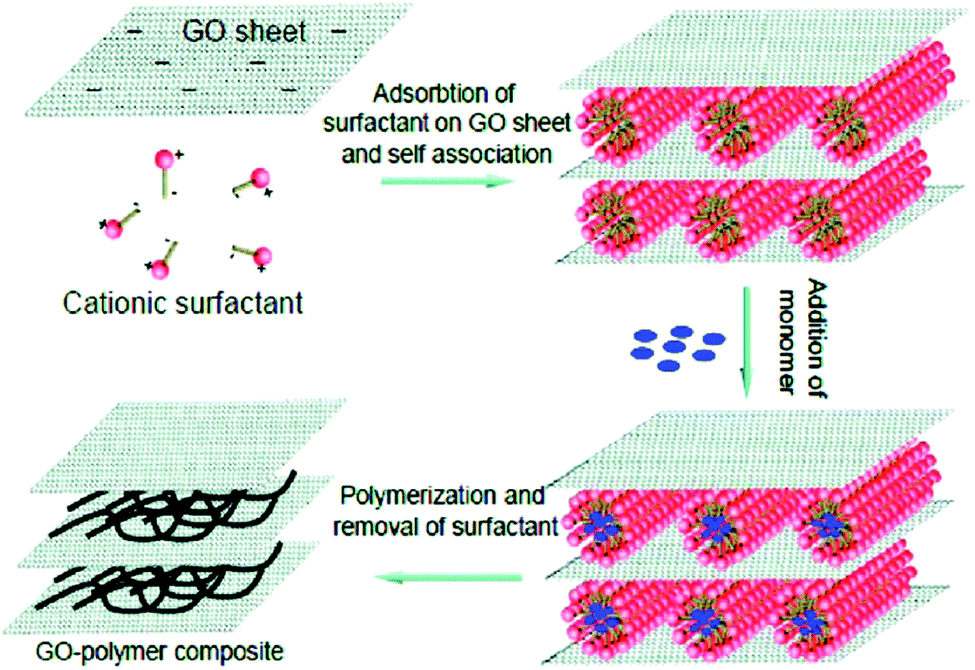 | ||
| Fig. 10 Schematic illustration of the formation process of GO/PPy composite. Reprinted with permission from ref. 240 Copyright 2010 American Chemical Society. | ||
To date, three structural models for conducting polymer composites have been developed. These nanostructured conducting polymer composites can combine the synergistic effects of conducting polymers and corresponding components together. Thus, they would possess a larger specific capacitance and better long-term cyclic and rate stability than neat conducting polymers and corresponding components with a simple nanostructure.
4. Outlook and conclusion
In this review, we presented a comprehensive overview of conducting polymer composites as advanced electrode materials for supercapacitors. The main problems that neat conducting polymer electrodes encounter are that they have poor rate and cyclic stability. The drawbacks of conducting polymers can be minimized by effective combination of conducting polymers with other materials to fabricate composite electrodes. Incorporation of the composite nanostructures in fabrication of the conducting polymer composites ensures the composite electrodes have a largely improved energy density and excellent cycling stability. However, at the present stage, the capacitances of the conducting polymer composites achieved are insignificant compared to their theoretical values. Despite the enhanced performance after the composition of conducting polymers with metal oxides/hydroxides/sulfides, there is still plenty of room to further improve the capacitance properties. In addition, although some reports have been reviewed, the design and synthesis of ternary conducting polymer composite electrodes for supercapacitors are still far from being thoroughly explored. Such hybrid nanostructures in a multiphase system could be more trustworthy for better-working supercapacitors. In order to take advantage of both energy storage mechanisms of electrochemical double layer capacitance and pseudocapacitance, other carbon materials, for example cheap carbon materials, may be used to ultimately boost the specific capacitance, energy density and power density. Again, the nature of the conducting polymers gives them considerable structural diversity, high flexibility and excellent durability. Moreover, the electrochromic properties of conducting polymers may be expected as a special advantage for their potential applications in smart supercapacitors. With a high interest in conducting polymer composites for the last decade and the current vigorous research developments, conducting polymer composite electrodes are expected to play a momentous part in flexible, smart and economical energy storage applications in the future.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21504012, 51125011 and 51433001) and the Fundamental Research Funds for the Central Universities of China (No. 16D110617).Notes and references
- B. E. Conway, J. Electrochem. Soc., 1991, 138, 1539–1548 CrossRef CAS.
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- A. Burke, J. Power Sources, 2000, 91, 37–50 CrossRef CAS.
- K. H. An, W. S. Kim, Y. S. Park, J.-M. Moon, D. J. Bae, S. C. Lim, Y. S. Lee and Y. H. Lee, Adv. Funct. Mater., 2001, 11, 387–392 CrossRef CAS.
- Y. Zhang, H. Feng, X. Wu, L. Wang, A. Zhang, T. Xia, H. Dong, X. Li and L. Zhang, Int. J. Hydrogen Energy, 2009, 34, 4889–4899 CrossRef CAS.
- H. Chen, T. N. Cong, W. Yang, C. Tan, Y. Li and Y. Ding, Prog. Nat. Sci., 2009, 19, 291–312 CrossRef CAS.
- J. Y. Lee, K. Liang, K. H. An and Y. H. Lee, Synth. Met., 2005, 150, 153–157 CrossRef CAS.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- H. Yoo, S. K. Sul, Y. Park and J. Jeong, IEEE Trans. Ind. Appl., 2008, 44, 108–114 CrossRef.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- B. E. Conway, V. Birss and J. Wojtowicz, J. Power Sources, 1997, 66, 1–14 CrossRef CAS.
- M.-S. Wu and P.-C. J. Chiang, Electrochem. Solid-State Lett., 2004, 7, A123–A126 CrossRef CAS.
- W. Sugimoto, H. Iwata, Y. Murakami and Y. Takasu, J. Electrochem. Soc., 2004, 151, A1181–A1187 CrossRef CAS.
- X. Dong, W. Shen, J. Gu, L. Xiong, Y. Zhu, H. Li and J. Shi, J. Phys. Chem. B, 2006, 110, 6015–6019 CrossRef CAS PubMed.
- C. Peng, S. Zhang, D. Jewell and G. Z. Chen, Prog. Nat. Sci., 2008, 18, 777–788 CrossRef CAS.
- V. K. Thakur, G. Ding, J. Ma, P. S. Lee and X. Lu, Adv. Mater., 2012, 24, 4071–4096 CrossRef CAS PubMed.
- C. S. Park, C. Lee and O. S. Kwon, Polymers, 2016, 8, 249–266 CrossRef.
- F. S. Omar, N. Duraisamy, K. Ramesh and S. Ramesh, Biosens. Bioelectron., 2016, 79, 763–775 CrossRef CAS PubMed.
- S. Prakash, T. Chakrabarty, A. K. Singh and V. K. Shahi, Biosens. Bioelectron., 2013, 41, 43–53 CrossRef CAS PubMed.
- S. Sivakkumar and R. Saraswathi, J. Power Sources, 2004, 137, 322–328 CrossRef CAS.
- L. Shao, J.-W. Jeon and J. L. Lutkenhaus, Chem. Mater., 2011, 24, 181–189 CrossRef.
- Y.-H. Huang and J. B. Goodenough, Chem. Mater., 2008, 20, 7237–7241 CrossRef CAS.
- G. Liu, S. Xun, N. Vukmirovic, X. Song, P. Olalde-Velasco, H. Zheng, V. S. Battaglia, L. Wang and W. Yang, Adv. Mater., 2011, 23, 4679–4683 CrossRef CAS PubMed.
- Y. E. Miao, W. Fan, D. Chen and T. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 4423–4428 CAS.
- H. Talbi, P. E. Just and L. H. Dao, J. Appl. Electrochem., 2003, 33, 465–473 CrossRef CAS.
- X. Li and B. Wei, Nano Energy, 2013, 2, 159–173 CrossRef CAS.
- A. B. Fuertes, G. Lota, T. A. Centeno and E. Frackowiak, Electrochim. Acta, 2005, 50, 2799–2805 CrossRef CAS.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS.
- K. Wang, H. P. Wu, Y. N. Meng and Z. X. Wei, Small, 2014, 10, 14–31 CrossRef CAS PubMed.
- L. Z. Fan, Y. S. Hu, J. Maier, P. Adelhelm, B. Smarsly and M. Antonietti, Adv. Funct. Mater., 2007, 17, 3083–3087 CrossRef CAS.
- G. Yu, X. Xie, L. Pan, Z. Bao and Y. Cui, Nano Energy, 2013, 2, 213–234 CrossRef CAS.
- M. N. Hyder, S. W. Lee, F. Ç. Cebeci, D. J. Schmidt, Y. Shao-Horn and P. T. Hammond, ACS Nano, 2011, 5, 8552–8561 CrossRef CAS PubMed.
- G. Lota, K. Fic and E. Frackowiak, Energy Environ. Sci., 2011, 4, 1592–1605 CAS.
- J. Liu, J. Sun and L. Gao, J. Phys. Chem. C, 2010, 114, 19614–19620 CAS.
- J. Ge, G. Cheng and L. Chen, Nanoscale, 2011, 3, 3084–3088 RSC.
- M. R. Arcila-Velez, R. K. Emmett, M. Karakaya, R. Podila, K. P. Diaz-Orellana, A. M. Rao and M. E. Roberts, Synth. Met., 2016, 215, 35–40 CrossRef CAS.
- M. N. Hyder, S. W. Lee, F. C. Cebeci, D. J. Schmidt, Y. Shao-Horn and P. T. Hammond, ACS Nano, 2011, 5, 8552–8561 CrossRef CAS PubMed.
- X. B. Yan, Z. J. Han, Y. Yang and B. K. Tay, J. Phys. Chem. C, 2007, 111, 4125–4131 CAS.
- N. Hui, F. L. Chai, P. P. Lin, Z. L. Song, X. T. Sun, Y. N. Li, S. Y. Niu and X. L. Luo, Electrochim. Acta, 2016, 199, 234–241 CrossRef CAS.
- J. L. Yu, W. B. Lu, S. P. Pei, K. Gong, L. Y. Wang, L. H. Meng, Y. D. Huang, J. P. Smith, K. S. Booksh, Q. W. Li, J. H. Byun, Y. Oh, Y. S. Yan and T. W. Chou, ACS Nano, 2016, 10, 5204–5211 CrossRef CAS PubMed.
- N. K. Sidhu and A. C. Rastogi, Mater. Chem. Phys., 2016, 176, 75–86 CrossRef CAS.
- K. Y. Shi, X. Pang and I. Zhitomirsky, J. Appl. Polym. Sci., 2015, 132, 9 Search PubMed.
- N. Lachman, H. P. Xu, Y. Zhou, M. Ghaffari, M. Lin, D. Bhattacharyya, A. Ugur, K. K. Gleason, Q. M. Zhang and B. L. Wardle, Adv. Mater. Interfaces, 2014, 1, 6 Search PubMed.
- V. H. R. de Souza, M. M. Oliveira and A. J. G. Zarbin, J. Power Sources, 2014, 260, 34–42 CrossRef.
- P. Pattananuwat, K. Wang, M. Tagaya and T. Kobayashi, Chem. Lett., 2014, 43, 1155–1157 CrossRef CAS.
- W. L. Wu, Y. F. Li, L. Q. Yang, Y. X. Ma and X. Yan, Synth. Met., 2014, 193, 48–57 CrossRef CAS.
- S. Lagoutte, P. H. Aubert, M. Pinault, T. V. Francois, M. Mayne-L'Hermite and C. Chevrot, Electrochim. Acta, 2014, 130, 754–765 CrossRef CAS.
- Z. Q. Niu, P. S. Luan, Q. Shao, H. B. Dong, J. Z. Li, J. Chen, D. Zhao, L. Cai, W. Y. Zhou, X. D. Chen and S. S. Xie, Energy Environ. Sci., 2012, 5, 8726–8733 CAS.
- Y. Zhou, H. P. Xu, N. Lachman, M. Ghaffari, S. Wu, Y. Liu, A. Ugur, K. K. Gleason, B. L. Wardle and Q. M. Zhang, Nano Energy, 2014, 9, 176–185 CrossRef CAS.
- C.-M. Yoon, D. Long, S.-M. Jang, W. Qiao, L. Ling, J. Miyawaki, C.-K. Rhee, I. Mochida and S.-H. Yoon, Carbon, 2011, 49, 96–105 CrossRef CAS.
- B. L. Fletcher, E. D. Hullander, A. V. Melechko, T. E. McKnight, K. L. Klein, D. K. Hensley, J. L. Morrell, M. L. Simpson and M. J. Doktycz, Nano Lett., 2004, 4, 1809–1814 CrossRef CAS.
- K. L. Klein, A. V. Melechko, T. E. McKnight, S. T. Retterer, P. D. Rack, J. D. Fowlkes, D. C. Joy and M. L. Simpson, J. Appl. Phys., 2008, 103, 061301 CrossRef.
- J. Jang, J. Bae, M. Choi and S. H. Yoon, Carbon, 2005, 43, 2730–2736 CrossRef CAS.
- B. Genorio, W. Lu, A. M. Dimiev, Y. Zhu, A.-R. O. Raji, B. Novosel, L. B. Alemany and J. M. Tour, ACS Nano, 2012, 6, 4231–4240 CrossRef CAS PubMed.
- L. Li, A.-R. O. Raji, H. Fei, Y. Yang, E. L. Samuel and J. M. Tour, ACS Appl. Mater. Interfaces, 2013, 5, 6622–6627 CAS.
- R.-R. Bi, X.-L. Wu, F.-F. Cao, L.-Y. Jiang, Y.-G. Guo and L.-J. Wan, J. Phys. Chem. C, 2010, 114, 2448–2451 CAS.
- L. Li, A. R. O. Raji, H. L. Fei, Y. Yang, E. L. G. Samuel and J. M. Tour, ACS Appl. Mater. Interfaces, 2013, 5, 6622–6627 CAS.
- N. A. Kumar and J. B. Baek, Chem. Commun., 2014, 50, 6298–6308 RSC.
- M. T. Pettes, H. Ji, R. S. Ruoff and L. Shi, Nano Lett., 2012, 12, 2959–2964 CrossRef CAS PubMed.
- Y. Ge, C. Y. Wang, K. W. Shu, C. Zhao, X. T. Jia, S. Gambhir and G. G. Wallace, RSC Adv., 2015, 5, 102643 RSC.
- L. F. Lai, L. Wang, H. P. Yang, N. G. Sahoo, Q. X. Tam, J. L. Liu, C. K. Poh, S. H. Lim, Z. X. Shen and J. Y. Lin, Nano Energy, 2012, 1, 723–731 CrossRef CAS.
- N. A. Kumar, H.-J. Choi, Y. R. Shin, D. W. Chang, L. Dai and J.-B. Baek, ACS Nano, 2012, 6, 1715–1723 CrossRef CAS PubMed.
- J. Yan, L. P. Yang, M. Q. Cui, X. Wang, K. J. J. Z. Chee, V. C. Nguyen, V. Kumar, A. Sumboja, M. Wang and P. S. Lee, Adv. Energy Mater., 2014, 4, 1400781 CrossRef.
- S. Huang, L. L. Ren, J. Guo, H. Zhu, C. Zhang and T. X. Liu, Carbon, 2012, 50, 216–224 CrossRef CAS.
- C. Vallés, P. Jiménez, E. Munoz, A. M. Benito and W. K. Maser, J. Phys. Chem. C, 2011, 115, 10468–10474 Search PubMed.
- L. Q. Xu, Y. L. Liu, K. G. Neoh, E. T. Kang and G. D. Fu, Macromol. Rapid Commun., 2011, 32, 684–688 CrossRef CAS PubMed.
- J. J. Xu, K. Wang, S. Z. Zu, B. H. Han and Z. X. Wei, ACS Nano, 2010, 4, 5019–5026 CrossRef CAS PubMed.
- M. Q. Sun, G. C. Wang, C. Y. Yang, H. Jiang and C. Z. Li, J. Mater. Chem., 2015, 3, 3880–3890 RSC.
- S. P. Zhou, H. M. Zhang, Q. Zhao, X. H. Wang, J. Li and F. S. Wang, Carbon, 2013, 52, 440–450 CrossRef CAS.
- X. Q. Yang, A. R. Liu, Y. W. Zhao, H. J. Lu, Y. J. Zhang, W. Wei, Y. Li and S. Q. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 23731–23740 CAS.
- N. A. Kumar, H. J. Choi, Y. R. Shin, D. W. Chang, L. M. Dai and J. B. Baek, ACS Nano, 2012, 6, 1715–1723 CrossRef CAS PubMed.
- Y. Song, J. L. Xu and X. X. Liu, J. Power Sources, 2014, 249, 48–58 CrossRef CAS.
- C. J. Raj, B. C. Kim, W. J. Cho, W. G. Lee, S. D. Jung, Y. H. Kim, S. Y. Park and K. H. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 13405–13414 CAS.
- A. Ghosh and Y. H. Lee, ChemSusChem, 2012, 5, 480–499 CrossRef CAS PubMed.
- X. H. Zhou, L. F. Li, S. M. Dong, X. Chen, P. X. Han, H. X. Xu, J. H. Yao, C. Q. Shang, Z. H. Liu and G. L. Cui, J. Solid State Electrochem., 2012, 16, 877–882 CrossRef CAS.
- Y. Zhao, K. Watanabe, R. Nakamura, S. Mori, H. Liu, K. Ishii and K. Hashimoto, Chem. – Eur. J., 2010, 16, 4982–4985 CrossRef CAS PubMed.
- G. X. Qu, J. L. Cheng, X. D. Li, D. M. Yuan, P. N. Chen, X. L. Chen, B. Wang and H. S. Peng, Adv. Mater., 2016, 28, 3646–3652 CrossRef CAS PubMed.
- M. K. Liu, S. X. He, W. Fan, Y. E. Miao and T. X. Liu, Compos. Sci. Technol., 2014, 101, 152–158 CrossRef CAS.
- W. Fan, Y. E. Miao, Y. P. Huang, W. W. Tjiu and T. X. Liu, RSC Adv., 2015, 5, 9228–9236 RSC.
- W. Fan, Y. Y. Xia, W. W. Tjiu, P. K. Pallathadka, C. B. He and T. X. Liu, J. Power Sources, 2013, 243, 973–981 CrossRef CAS.
- X. Xie, M. Ye, L. Hu, N. Liu, J. R. McDonough, W. Chen, H. N. Alshareef, C. S. Criddle and Y. Cui, Energy Environ. Sci., 2012, 5, 5265–5270 CAS.
- H.-Y. Tsai, C.-C. Wu, C.-Y. Lee and E. P. Shih, J. Power Sources, 2009, 194, 199–205 CrossRef CAS.
- E. Raymundo-Pinero, K. Kierzek, J. Machnikowski and F. Béguin, Carbon, 2006, 44, 2498–2507 CrossRef CAS.
- M. Seredych, D. Hulicova-Jurcakova, G. Q. Lu and T. J. Bandosz, Carbon, 2008, 46, 1475–1488 CrossRef CAS.
- Z. Zhou and X.-F. Wu, J. Power Sources, 2013, 222, 410–416 CrossRef CAS.
- E. Ra, E. Raymundo-Piñero, Y. Lee and F. Béguin, Carbon, 2009, 47, 2984–2992 CrossRef CAS.
- Y. Hou, Y. Cheng, T. Hobson and J. Liu, Nano Lett., 2010, 10, 2727–2733 CrossRef CAS PubMed.
- Z. Zhou and X.-F. Wu, J. Power Sources, 2014, 262, 44–49 CrossRef CAS.
- B. Anothumakkool, A. T. A. Torris, S. N. Bhange, M. V. Badiger and S. Kurungot, Nanoscale, 2014, 6, 5944–5952 RSC.
- L. J. Bian, F. Luan, S. S. Liu and X. X. Liu, Electrochim. Acta, 2012, 64, 17–22 CrossRef CAS.
- H. Yang, H. Xu, M. Li, L. Zhang, Y. Huang and X. Hu, ACS Appl. Mater. Interfaces, 2016, 8, 1774–1779 CAS.
- X. Li, X. Zang, Z. Li, X. Li, P. Li, P. Sun, X. Lee, R. Zhang, Z. Huang and K. Wang, Adv. Funct. Mater., 2013, 23, 4862–4869 CAS.
- J. X. Zhu, L. J. Cao, Y. S. Wu, Y. J. Gong, Z. Liu, H. E. Hoster, Y. H. Zhang, S. T. Zhang, S. B. Yang, Q. Y. Yan, P. M. Ajayan and R. Vajtai, Nano Lett., 2013, 13, 5408–5413 CrossRef CAS PubMed.
- X. Zang, Q. Chen, P. Li, Y. He, X. Li, M. Zhu, X. Li, K. Wang, M. Zhong and D. Wu, Small, 2014, 10, 2583–2588 CrossRef CAS PubMed.
- H. Kashani, L. Y. Chen, Y. Ito, J. H. Han, A. Hirata and M. W. Chen, Nano Energy, 2016, 19, 391–400 CrossRef CAS.
- M. Arulepp, L. Permann, J. Leis, A. Perkson, K. Rumma, A. Janes and E. Lust, J. Power Sources, 2004, 133, 320–328 CrossRef CAS.
- Y. Gogotsi, A. Nikitin, H. Ye, W. Zhou, J. E. Fischer, B. Yi, H. C. Foley and M. W. Barsoum, Nat. Mater., 2003, 2, 591–594 CrossRef CAS PubMed.
- L. L. Zhang, S. Li, J. T. Zhang, P. Z. Guo, J. T. Zheng and X. S. Zhao, Chem. Mater., 2010, 22, 1195–1202 CrossRef CAS.
- J. Shen, C. Yang, X. Li and G. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 8467–8476 CAS.
- X. Lu, D. Hui, S. Yang, H. Liang, L. Zhang, L. Shen, Z. Fang and X. Zhang, Electrochim. Acta, 2011, 56, 9224–9232 CrossRef CAS.
- W. Fan, Y. E. Miao, L. S. Zhang, Y. P. Huang and T. X. Liu, RSC Adv., 2015, 5, 31064–31073 RSC.
- C. Zhang, W. W. Tjiu and T. X. Liu, Polym. Chem., 2013, 4, 5785–5792 RSC.
- Q. Cheng, J. Tang, N. Shinya and L. C. Qin, J. Power Sources, 2013, 241, 423–428 CrossRef CAS.
- M. K. Liu, Y. E. Miao, C. Zhang, W. W. Tjiu, Z. B. Yang, H. S. Peng and T. X. Liu, Nanoscale, 2013, 5, 7312–7320 RSC.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- M. Mastragostino, C. Arbizzani and F. Soavi, Solid State Ionics, 2002, 148, 493–498 CrossRef CAS.
- J.-H. Sung, S.-J. Kim and K.-H. Lee, J. Power Sources, 2003, 124, 343–350 CrossRef CAS.
- A. Rudge, J. Davey, I. Raistrick, S. Gottesfeld and J. P. Ferraris, J. Power Sources, 1994, 47, 89–107 CrossRef CAS.
- V. Gupta and N. Miura, Mater. Lett., 2006, 60, 1466–1469 CrossRef CAS.
- K. S. Ryu, K. M. Kim, N.-G. Park, Y. J. Park and S. H. Chang, J. Power Sources, 2002, 103, 305–309 CrossRef CAS.
- M. Kalaji, P. Murphy and G. Williams, Synth. Met., 1999, 102, 1360–1361 CrossRef CAS.
- S. Ghosh, T. Maiyalagan and R. N. Basu, Nanoscale, 2016, 8, 6921–6947 RSC.
- H. Choi and H. Yoon, Nanomaterials, 2015, 5, 906–936 CrossRef CAS.
- R. Ramachandran, S. M. Chen and G. G. Kumar, Int. J. Electrochem. Sci., 2015, 10, 10355–10388 CAS.
- H. Yoon, M. Choi, K. J. Lee and J. Jang, Macromol. Res., 2008, 16, 85–102 CrossRef CAS.
- J. Wang and D. Zhang, Adv. Polym. Technol., 2013, 32, E323–E368 CrossRef CAS.
- A. C. Rastogi, S. B. Desu and R. K. Sharma, Electrochim. Acta, 2008, 53, 7690–7695 CrossRef.
- K. Lota, V. Khomenko and E. Frackowiak, J. Phys. Chem. Solids, 2004, 65, 295–301 CrossRef CAS.
- S. M. Chen, R. Ramachandran, V. Mani and R. Saraswathi, Int. J. Electrochem. Sci., 2014, 9, 4072–4085 Search PubMed.
- M. E. Abdelhamid, A. P. O'Mullane and G. A. Snook, RSC Adv., 2015, 5, 11611–11626 RSC.
- C. Arbizzani, M. Mastragostino and L. Meneghello, Electrochim. Acta, 1996, 41, 21–26 CrossRef CAS.
- C. D. Lokhande, D. P. Dubal and O. S. Joo, Curr. Appl. Phys., 2011, 11, 255–270 CrossRef.
- X. Lang, A. Hirata, T. Fujita and M. Chen, Nat. Nanotechnol., 2011, 6, 232–236 CrossRef CAS PubMed.
- Z. S. Wu, D. W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H. M. Cheng, Adv. Funct. Mater., 2010, 20, 3595–3602 CrossRef CAS.
- D.-D. Zhao, S.-J. Bao, W.-J. Zhou and H.-L. Li, Electrochem. Commun., 2007, 9, 869–874 CrossRef CAS.
- M. H. Bai, L. J. Bian, Y. Song and X. X. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 12656–12664 CAS.
- S. Li, D. Wu, C. Cheng, J. Wang, F. Zhang, Y. Su and X. Feng, Angew. Chem., Int. Ed., 2013, 52, 12105–12109 CrossRef CAS PubMed.
- K. H. Chang, C. C. Hu, K. H. Chang and C. C. Hu, Electrochem. Solid-State Lett., 2004, 7, 466–469 CrossRef.
- L.-M. Huang, H.-Z. Lin, T.-C. Wen and A. Gopalan, Electrochim. Acta, 2006, 52, 1058–1063 CrossRef CAS.
- X. Li, Y. Wu, F. Zheng, M. Ling and F. Lu, Solid State Commun., 2014, 197, 57–60 CrossRef CAS.
- S. Cho, M. Kim and J. Jang, ACS Appl. Mater. Interfaces, 2015, 7, 10213–11227 CAS.
- H. S. Nam, K. M. Kim, H. K. Sang, B. C. Kim, G. G. Wallace and J. M. Ko, Polym. Bull., 2012, 68, 553–560 CrossRef CAS.
- R. Y. Song, J. H. Park, S. R. Sivakkumar, H. K. Sang, J. M. Ko, D. Y. Park, S. M. Jo and Y. K. Dong, J. Power Sources, 2007, 166, 297–301 CrossRef CAS.
- R. Liu, J. Duay, T. Lane and L. S. Bok, Phys. Chem. Chem. Phys., 2010, 12, 4309–4316 RSC.
- P. R. Deshmukh, R. N. Bulakhe, S. N. Pusawale, S. D. Sartale and C. D. Lokhande, RSC Adv., 2015, 5, 28687–28695 RSC.
- J. Zang, S. J. Bao, C. M. Li, H. Bian, X. Cui, Q. Bao, C. Q. Sun, J. Guo and K. Lian, J. Phys. Chem. C, 2008, 112, 14843–14847 CAS.
- W. Yao, H. Zhou and Y. Lu, J. Power Sources, 2013, 241, 359–366 CrossRef CAS.
- C. Wang, Y. Zhan, L. Wu, Y. Li and J. Liu, Nanotechnology, 2014, 25, 305401 CrossRef PubMed.
- A. H. Gemeay, I. A. Mansour, R. G. El-Sharkawy and A. B. Zaki, Eur. Polym. J., 2005, 41, 2575–2583 CrossRef CAS.
- F. Meng, X. Yan, Y. Zhu and P. Si, Nanoscale Res. Lett., 2013, 8, 1–8 CrossRef PubMed.
- X. Zhang, L. Ji, S. Zhang and W. Yang, J. Power Sources, 2007, 173, 1017–1023 CrossRef CAS.
- R. Liu and S. B. Lee, J. Am. Chem. Soc., 2008, 130, 2942–2943 CrossRef CAS PubMed.
- Y. Hu, Y. V. Tolmachev and D. A. Scherson, J. Electroanal. Chem., 1999, 468, 64–69 CrossRef CAS.
- J.-W. Lang, L.-B. Kong, W.-J. Wu, M. Liu, Y.-C. Luo and L. Kang, J. Solid State Electrochem., 2009, 13, 333–340 CrossRef CAS.
- A. C. Sonavane, A. I. Inamdar, D. S. Dalavi, H. P. Deshmukh and P. S. Patil, Electrochim. Acta, 2010, 55, 2344–2351 CrossRef CAS.
- C. Bora, A. Kalita, D. Das, S. K. Dolui and P. K. Mukhopadhyay, Polym. Int., 2014, 63, 445–452 CrossRef CAS.
- W. Ji, J. Ji, X. Cui, J. Chen, D. Liu, H. Deng and Q. Fu, Chem. Commun., 2015, 51, 7669–7672 RSC.
- B. Sun, X. He, X. Leng, Y. Jiang, Y. Zhao, H. Suo and C. Zhao, RSC Adv., 2016, 6, 43959–43963 RSC.
- D. Kong, W. Ren, C. Cheng, Y. Wang, Z. Huang and H. Y. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 21334–21346 CAS.
- C. Z. Wu, F. Feng and Y. Xie, Chem. Soc. Rev., 2013, 42, 5157–5183 RSC.
- C. Zhou, Y. Zhang, Y. Li and J. Liu, Nano Lett., 2013, 13, 2078–2085 CrossRef CAS PubMed.
- T. Y. Wei, C. H. Chen, H. C. Chien, S. Y. Lu and C. C. Hu, Adv. Mater., 2010, 22, 347–351 CrossRef CAS PubMed.
- Z. Wu, Y. Zhu and X. Ji, J. Mater. Chem., 2014, 2, 14759–14772 RSC.
- N. Jabeen, Q. Xia, M. Yang and H. Xia, ACS Appl. Mater. Interfaces, 2016, 8, 6093–6100 CAS.
- C.-H. Lai, M.-Y. Lu and L.-J. Chen, J. Mater. Chem., 2012, 22, 19–30 RSC.
- G. A. Muller, J. B. Cook, H.-S. Kim, S. H. Tolbert and B. Dunn, Nano Lett., 2015, 15, 1911–1917 CrossRef CAS PubMed.
- S. J. Bao, C. M. Li, C. X. Guo and Y. Qiao, J. Power Sources, 2008, 180, 676–681 CrossRef CAS.
- F. Tao, Y. Q. Zhao, G. Q. Zhang and H. L. Li, Electrochem. Commun., 2007, 9, 1282–1287 CrossRef CAS.
- J. Q. Yang, X. C. Duan, Q. Qin and W. J. Zheng, J. Mater. Chem., 2013, 1, 7880–7884 RSC.
- H. Peng, G. Ma, K. Sun, J. Mu, H. Wang and Z. Lei, J. Mater. Chem., 2013, 2, 3303–3307 RSC.
- S. Peng, L. Fan, C. Wei, H. Bao, H. Zhang, W. Xu and J. Xu, Cellulose, 2016, 1–13 Search PubMed.
- L. Ren, G. Zhang, Y. Zhe, L. Kang, X. Hua, S. Feng, Z. Lei and Z. H. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 28294–28302 Search PubMed.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313–318 CrossRef CAS PubMed.
- G. Minoli, Science, 2013, 341, 1502–1505 CrossRef PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- C. Yang, Z. X. Chen, I. Shakir, Y. X. Xu and H. B. Lu, Nano Res., 2016, 9, 951–962 CrossRef CAS.
- T. Liu, L. Finn, M. Yu, H. Wang, T. Zhai, X. Lu, Y. Tong and Y. Li, Nano Lett., 2014, 14, 2522–2527 CrossRef CAS PubMed.
- D. P. Dubal, S. V. Patil, G. S. Gund and C. D. Lokhande, J. Alloys Compd., 2013, 552, 240–247 CrossRef CAS.
- H. Mi, X. Zhang, X. Ye and S. Yang, J. Power Sources, 2008, 176, 403–409 CrossRef CAS.
- L. Wen, H. Ping, S. Zhang, F. Dong and Y. Ma, J. Power Sources, 2014, 266, 347–352 CrossRef.
- A. Q. Zhang, Y. Zhang, L. Z. Wang and X. F. Li, Polym. Compos., 2011, 32, 1–5 CrossRef CAS.
- J. Wang, Y. Xu, X. Chen and X. Du, J. Power Sources, 2007, 163, 1120–1125 CrossRef CAS.
- Y. Xu, J. Wang, W. Sun and S. Wang, J. Power Sources, 2006, 159, 370–373 CrossRef CAS.
- Z. L. Wang, X. J. He, S. H. Ye, Y. X. Tong and G. R. Li, ACS Appl. Mater. Interfaces, 2014, 6, 642–647 CAS.
- C. Wei, P. S. Lee and Z. Xu, RSC Adv., 2014, 4, 31416–31423 RSC.
- H. L. Cao, X. F. Zhou, Y. M. Zhang, L. Chen and Z. P. Liu, J. Power Sources, 2013, 243, 715–720 CrossRef CAS.
- Z. Y. Gao, F. Wang, J. L. Chang, D. P. Wu, X. R. Wang, X. Wang, F. Xu, S. Y. Gao and K. Jiang, Electrochim. Acta, 2014, 133, 325–334 CrossRef CAS.
- X. L. Li, H. F. Song, Y. L. Zhang, H. Wang, K. Du, H. Y. Li, Y. Yuan and J. M. Huang, Int. J. Electrochem. Sci., 2012, 7, 5163–5171 CAS.
- L. Mao, K. Zhang, H. S. O. Chan and J. S. Wu, J. Mater. Chem., 2012, 22, 80–85 RSC.
- P. Sekar, B. Anothumakkool and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 7661–7669 CAS.
- D. J. Shi, C. J. Wei, F. Duan and M. Q. Chen, Integr. Ferroelectr., 2015, 161, 76–84 CrossRef CAS.
- J. T. Zhang and X. S. Zhao, J. Phys. Chem. C, 2012, 116, 5420–5426 CAS.
- C. M. Chang, C. J. Weng, C. M. Chien, T. L. Chuang, T. Y. Lee, J. M. Yeh and Y. Wei, J. Mater. Chem., 2013, 1, 14719–14728 RSC.
- M. G. Deng, B. C. Yang and Y. D. Hu, J. Mater. Sci., 2005, 40, 5021–5023 CrossRef CAS.
- B. Dong, B. L. He, C. L. Xu and H. L. Li, Mater. Sci. Eng., B, 2007, 143, 7–13 CrossRef CAS.
- V. Gupta and N. Miura, Electrochim. Acta, 2006, 52, 1721–1726 CrossRef CAS.
- V. Gupta and N. Miura, J. Power Sources, 2006, 157, 616–620 CrossRef CAS.
- C. Peng, J. Jin and G. Z. Chen, Electrochim. Acta, 2007, 53, 525–537 CrossRef CAS.
- S. R. Sivakkumar, W. J. Kim, J. A. Choi, D. R. MacFarlane, M. Forsyth and D. W. Kim, J. Power Sources, 2007, 171, 1062–1068 CrossRef CAS.
- Y. K. Zhou, B. L. He, W. J. Zhou, J. Huang, X. H. Li, B. Wu and H. I. Li, Electrochim. Acta, 2004, 49, 257–262 CrossRef CAS.
- K. H. An, K. K. Jeon, J. K. Heo, S. C. Lim, D. J. Bae and Y. H. Lee, J. Electrochem. Soc., 2002, 149, A1058–A1062 CrossRef CAS.
- M. Raicopol, A. Pruna and L. Pilan, J. Chem., 2013, 7, DOI:10.1155/2013/367473.
- J. Wang, Y. L. Xu, F. Yan, J. B. Zhu, J. P. Wang and F. Xiao, J. Solid State Electrochem., 2010, 14, 1565–1575 CrossRef CAS.
- M. L. Wang, Y. Q. Gao, Q. Sun and J. W. Zhao, J. Electrochem. Soc., 2014, 161, B297–B304 CrossRef CAS.
- V. Kumar and P. S. Lee, J. Phys. Chem. C, 2015, 119, 9041–9049 CAS.
- Z. Y. Luo, Y. H. Zhu, E. H. Liu, T. T. Hu, Z. P. Li, T. T. Liu and L. C. Song, Mater. Res. Bull., 2014, 60, 105–110 CrossRef CAS.
- H. S. Nam, K. M. Kim, S. H. Kim, B. C. Kim, G. G. Wallace and J. M. Ko, Polym. Bull., 2012, 68, 553–560 CrossRef CAS.
- H. J. Tang, J. Y. Wang, H. J. Yin, H. J. Zhao, D. Wang and Z. Y. Tang, Adv. Mater., 2015, 27, 1117–1123 CrossRef CAS PubMed.
- X. Li, Y. J. Wu, F. Zheng, M. Ling and F. H. Lu, Solid State Commun., 2014, 197, 57–60 CrossRef CAS.
- Y. Liu, B. H. Zhang, Y. Q. Yang, Z. Chang, Z. B. Wen and Y. P. Wu, J. Mater. Chem., 2013, 1, 13582–13587 RSC.
- J. F. Zang, S. J. Bao, C. M. Li, H. J. Bian, X. Q. Cui, Q. L. Bao, C. Q. Sun, J. Guo and K. R. Lian, J. Phys. Chem. C, 2008, 112, 14843–14847 CAS.
- M. H. Bai, T. Y. Liu, F. Luan, Y. Li and X. X. Liu, J. Mater. Chem., 2014, 2, 10882–10888 RSC.
- L. J. Bian, H. L. He and X. X. Liu, RSC Adv., 2015, 5, 75374–75379 RSC.
- P. R. Deshmukh, R. N. Bulakhe, S. N. Pusawale, S. D. Sartale and C. D. Lokhande, RSC Adv., 2015, 5, 28687–28695 RSC.
- K. J. Huang, L. Wang, Y. J. Liu, H. B. Wang, Y. M. Liu and L. L. Wang, Electrochim. Acta, 2013, 109, 587–594 CrossRef CAS.
- C. X. Hu, S. J. He, S. H. Jiang, S. L. Chen and H. Q. Hou, RSC Adv., 2015, 5, 14441–14447 RSC.
- K. Wang, J. Y. Huang and Z. X. Wei, J. Phys. Chem. C, 2010, 114, 8062–8067 CAS.
- J. X. Zhu, W. P. Sun, D. Yang, Y. Zhang, H. H. Hoon, H. Zhang and Q. Y. Yan, Small, 2015, 11, 4123–4129 CrossRef CAS PubMed.
- Y. F. Yan, Q. L. Cheng, Z. J. Zhu, V. Pavlinek, P. Saha and C. Z. Li, J. Power Sources, 2013, 240, 544–550 CrossRef CAS.
- L. Xia, J. F. Xia and Z. H. Wang, RSC Adv., 2015, 5, 93209–93214 RSC.
- Z. H. Huang, Y. Song, X. X. Xu and X. X. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 25506–25513 CAS.
- F. Chen, P. Liu and Q. Zhao, Electrochim. Acta, 2012, 76, 62–68 CrossRef CAS.
- X. Cai, Q. Zhang, S. Wang, J. Peng, Y. Zhang, H. Ma, J. Li and M. Zhai, J. Mater. Sci., 2014, 49, 5667–5675 CrossRef CAS.
- H. Cao, X. Zhou, Y. Zhang, L. Chen and Z. Liu, J. Power Sources, 2013, 243, 715–720 CrossRef CAS.
- A. Chandrasoma, R. Grant, A. E. Bruce and M. R. M. Bruce, J. Power Sources, 2012, 219, 285–291 CrossRef CAS.
- C.-M. Chang, C.-J. Weng, C.-M. Chien, T.-L. Chuang, T.-Y. Lee, J.-M. Yeh and Y. Wei, J. Mater. Chem., 2013, 1, 14719–14728 RSC.
- B. Chaoqing, Y. Aishui and W. Haoqing, Electrochem. Commun., 2009, 11, 266–269 CrossRef.
- T. Chau, R. Singhal, D. Lawrence and V. Kalra, J. Power Sources, 2015, 293, 373–379 CrossRef.
- C. Chen, W. Fan, Q. Zhang, T. Ma, X. Fu and Z. Wang, J. Appl. Polym. Sci., 2015, 132, 42290–42294 Search PubMed.
- W. Fan, C. Zhang, W. W. Tjiu, K. P. Pramoda, C. B. He and T. X. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 3382–3391 CAS.
- M. C. Liu, X. L. Wu, C. L. Chen, Q. Wang, T. Wen and X. K. Wang, Sci. Adv. Mater., 2013, 5, 1686–1693 CrossRef CAS.
- H. Jiang, J. Ma and C. Z. Li, J. Mater. Chem., 2012, 22, 16939–16942 RSC.
- J. Y. Lei, Z. Q. Jiang, X. F. Lu, G. D. Nie and C. Wang, Electrochim. Acta, 2015, 176, 149–155 CrossRef CAS.
- S. A. Ju, K. Kim, J. H. Kim and S. S. Leet, ACS Appl. Mater. Interfaces, 2011, 3, 2904–2911 CAS.
- H. Chen, S. X. Zhou, M. Chen and L. M. Wu, J. Mater. Chem., 2012, 22, 25207–25216 RSC.
- J. S. Chen, Z. Y. Wang, X. C. Dong, P. Chen and X. W. Lou, Nanoscale, 2011, 3, 2158–2161 RSC.
- X. H. Tang, Z. H. Liu, C. X. Zhang, Z. P. Yang and Z. L. Wang, J. Power Sources, 2009, 193, 939–943 CrossRef CAS.
- C. X. Guo, G. Yilmaz, S. C. Chen, S. F. Chen and X. M. Lu, Nano Energy, 2015, 12, 76–87 CrossRef CAS.
- G. Q. Han, Y. Liu, E. J. Kan, J. Tang, L. L. Zhang, H. H. Wang and W. H. Tang, RSC Adv., 2014, 4, 9898–9904 RSC.
- Q. L. Hao, X. F. Xia, W. Lei, W. J. Wang and J. S. Qiu, Carbon, 2015, 81, 552–563 CrossRef CAS.
- J. W. Jeon, S. R. Kwon and J. L. Lutkenhaus, J. Mater. Chem., 2015, 3, 3757–3767 RSC.
- Y. Li, H. R. Peng, G. C. Li and K. Z. Chen, Eur. Polym. J., 2012, 48, 1406–1412 CrossRef CAS.
- X. B. Liu, N. Wen, X. L. Wang and Y. Y. Zheng, ACS Sustainable Chem. Eng., 2015, 3, 475–482 CrossRef CAS.
- J. Luo, Y. Z. Chen, Q. Ma, R. Liu and X. Y. Liu, J. Mater. Chem. C, 2014, 2, 4818–4827 RSC.
- R. Oraon, A. De Adhikari, S. K. Tiwari and G. C. Nayak, ACS Sustainable Chem. Eng., 2016, 4, 1392–1403 CrossRef CAS.
- J. J. Cong, Y. Z. Chen, J. Luo and X. Y. Liu, J. Solid State Chem., 2014, 218, 171–177 CrossRef CAS.
- W. Q. Dai, L. Ma, M. Y. Gan, S. Y. Wang, X. W. Sun, H. H. Wang, H. N. Wang and T. Zhou, Mater. Res. Bull., 2016, 76, 344–352 CrossRef CAS.
- T. X. Wang, L. Yuan, G. Z. Liang and A. J. Gu, Appl. Surf. Sci., 2015, 359, 754–765 CrossRef CAS.
- L. L. Zhang, S. Y. Zhao, X. N. Tian and X. S. Zhao, Langmuir, 2010, 26, 17624–17628 CrossRef CAS PubMed.
- S. P. Zhou, H. M. Zhang, X. H. Wang, J. Li and F. S. Wang, RSC Adv., 2013, 3, 1797–1807 RSC.
| This journal is © the Partner Organisations 2017 |