
A high-performance BaTiO3-grafted-GO-laden poly(ethylene oxide)-based membrane as an electrolyte for all-solid lithium-batteries
N.
Angulakshmi
ab,
Goutam Prasanna
Kar
c,
Suryasarathi
Bose
 *c,
E. Bhoje
Gowd
*d,
Sabu
Thomas
b and
A. Manuel
Stephan
*a
*c,
E. Bhoje
Gowd
*d,
Sabu
Thomas
b and
A. Manuel
Stephan
*a
aCSIR – Central Electrochemical Research Institute, Karaikudi 630 006, India. E-mail: arulmanuel@gmail.com
bMahatma Gandhi University, Kottayam 686560, India
cMaterials Engineering Department, Indian Institute of Science, Bangalore-560012, India. E-mail: sbose@materials.iisc.ernet.in
dMaterials Science and Technology Division, CSIR-National Institute for Interdisciplinary Science and Technology, Trivandrum 695 019, India. E-mail: bhojegowd@gmail.com
First published on 29th July 2016
Abstract
Nanocomposite polymer electrolytes (NCPEs) comprising poly(ethylene oxide) (PEO), barium titanate-grafted-graphene oxide (BaTiO3-g-GO) and lithium bis(trifluoromethanesulfonyl imide) (LiTFSI) were prepared by a simple hot-press technique. An increase of two orders of magnitude in the ionic conductivity was achieved upon incorporation of BaTiO3-g-GO in the polymeric matrix even at 0 °C. The addition of BaTiO3-g-GO as a filler has significantly enhanced the thermal stability and mechanical integrity of the membrane. The BaTiO3-g-GO-laden membrane was found to have better interfacial properties with a lithium metal anode than the filler-free membrane. Charge–discharge studies revealed a stable cycling profile even at 5C-rate and it was found to be superior to those reported earlier.
1 Introduction
Lithium-ion batteries have revolutionized portable electronic devices such as laptop computers, mobile phones etc., in the last two decades.1,2 Also, immense efforts have been made to employ these storage systems for electric vehicles and stationary storage of renewable energies such as power grid applications. However, several important criteria such as energy density, power density, rate capability, cycle life, cost and above all safety aspects have to be addressed.3,4 The state-of-art lithium-ion batteries are composed of a carbonaceous anode and a lithium-transition metal oxide cathode separated by a porous polyolefin separator soaked in a non-aqueous liquid electrolyte.5,6 The lithium–polymer batteries, in which the non-aqueous liquid electrolyte is replaced by a solid polymer electrolyte, possess appealing properties such as high energy density, no-leakage of electrolyte and better safety.7–9 In particular, the lithium–polymer batteries have been the focus of many researchers due to the unique properties of lithium metal such as low atomic weight (6.99 g mol−1), and high electronegativity (−3.05 V vs. SHE) and specific capacity (3.76 A h g−1).10 Obviously, across the world numerous attempts have been made to identify suitable polymer electrolytes for applications in lithium batteries. Dry solid polymer electrolytes comprising a polymer host and a lithium salt (LiX) exhibit very low ionic conductivity (10−8 S cm−1) at 25 °C which makes them unsuitable for ambient temperature applications.10 Although gel polymer electrolytes offer high ionic conductivity, their poor interfacial properties and mechanical integrity remain as problem areas.11,12 The development of composite polymer electrolytes by the addition of inert fillers such as TiO2, SiO2, Al2O3etc., to enhance the mechanical and interfacial properties have been extensively studied in the last two decades.13,14 The electrochemical properties and charge–discharge characteristics of nanocomposite polymer electrolytes composed of nanochitin, MgAl2O4 and Mg, Cu and Al-based metal organic frameworks have been studied extensively by us.15–19 However, to our knowledge, most of the studies on composite polymer electrolytes have been focused on the effect of dispersed ceramic particles on the recrystallization kinetics of the PEO–polymer chains.9,20,21According to the effective-medium-theory (EMT) model, the non-conductive dispersoids in composite polymer electrolytes promote ionic conductivity due to the existence of a space-charge layer at the electrolyte/filler interface.22 A highly conductive layer is formed in the neighborhood of grain boundaries which is created by Lewis acid–base reactions. The permanent dipole of ferroelectric materials serves as a strong Lewis acid–base compared to the constituent atoms of non-ferroelectric ceramic inert fillers and provides a highly conductive layer by its strong interaction with other constituents of the polymer electrolyte.23
Takeda et al.24 reported the electrochemical properties of mixed-phase electrolytes comprising BaTiO3, PbTiO3 and LiNO3. The same group25 has reported the cycling behavior of a Li/PEO + BaTiO3 + LiTFSI/LiNi0.8Co0.2O2 cell; however, the cycling profile was found to be very poor even at 70 °C. The authors also reported the interfacial properties of BaTiO3-added PEO based electrolytes with different lithium salts.26
In this work, a unique hybrid nanoparticle consisting of BaTiO3 chemically coupled to graphene oxide (GO) sheets was used as the filler.27,28 By chemically coupling BaTiO3 onto GO, a high dielectric constant can be achieved through interfacial polarization. This has been well established by one of the authors.27,28 In the present study, this hybrid particle was incorporated in the PEO matrix to design polymer electrolytes for all-solid-state lithium-batteries. The membranes prepared by a conventional solvent casting method exhibit poor interfacial properties at the electrode/electrolyte interface which arises due to the presence of solvent molecules as impurities trapped by the high surface area inert fillers even after prolonged drying.29 In the present study, therefore, a simple hot-press technique was employed for the preparation of nanocomposite polymer electrolytes as reported earlier.29,30
2 Experimental procedure
2.1 Materials
PEO (Mol. wt 3 × 105) and BaTiO3 were procured from Sigma Aldrich and LiTFSI was obtained from Merck, (Germany). PVDF (Kynar-761, Mw = 440![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was provided by Arkema. All the solvents were procured from Sigma Aldrich.
000 g mol−1) was provided by Arkema. All the solvents were procured from Sigma Aldrich.
2.2 Synthesis of BaTiO3-g-GO
GO was synthesized by a modified Hummer's method. Briefly, graphite flakes were taken in a mixture of H2SO4 and H3PO4 (9:1 ratio) under ice cold conditions. KMnO4 was slowly added to the mixture maintaining a low temperature.31 After 24 h of stirring at room temperature, the mixture was added to cold DI H2O and 35% hydrogen peroxide (H2O2) (5–10 ml) was added drop wise until the solution turned yellow. The solution was filtered and the solid material was washed with DI H2O and diluted HCl and ethanol. The solid material was recovered and dried at room temperature.
Barium titanate (BT) nanoparticles were dispersed in H2O2 with the aid of probe sonication for 10–15 min at low temperature. The dispersed BT nanoparticles were refluxed in excess H2O2 at higher temperature (105 °C) for 5–6 h. As a result of this H2O2 treatment the inorganic nanoparticles are hydroxylated.32 The white powder was washed with deionized water (DI H2O) and then dried. Subsequently, the powder was refluxed with APTS at 80 °C for 24 h to introduce amine groups to form BT-NH2. These amine groups on BT nanoparticles were coupled to acid groups of GO in the presence of DCC resulting in the formation of BaTiO3-g-GO. The amount of GO was fixed at 0.1 wt% with respect to BT-NH2 nanoparticles. This synthesis process is illustrated in Scheme 1. The functionalization of GO with amine terminated BT particles was studied by FT-IR. Fig. 1a shows the FT-IR spectra of GO and BaTiO3-g-GO. GO contains several oxygen containing functional groups, which is well evident from the FT-IR spectra, such as, hydroxyl (OH) stretching at 3400 cm−1, carboxylic stretching at 1730 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching at 1625 cm−1, C–OH at 1220 cm−1 and C–O stretching at 1055 cm−1. Interestingly, the carboxylic stretching peaks at 1730 cm−1 disappear in BaTiO3-g-GO indicating the reaction between CO groups in GO and BT-NH2. Moreover, the transmission electron microscopy images of graphene oxide (Fig. 1b) and BaTiO3-g-GO (Fig. 1c) confirm this functionalization. The dielectric constants (ε′) of BaTiO3 increases upon conjugation with GO. To realize this, poly(vinylidene fluoride) (PVDF) composites were made using BaTiO3, GO and BaTiO3-g-GO. The dielectric constants of different PVDF composites are listed in Table 1. It is evident that the dielectric constant of PVDF increases with GO (0.1 wt%) and BaTiO3 (5 vol%) separately. The incorporation of BaTiO3-g-GO (5 vol%) further enhances the dielectric constant. Dipole polarization of GO and heterogeneity of the dielectric medium are attributed to this increment. This phenomenon is well reported in our earlier works.28
C stretching at 1625 cm−1, C–OH at 1220 cm−1 and C–O stretching at 1055 cm−1. Interestingly, the carboxylic stretching peaks at 1730 cm−1 disappear in BaTiO3-g-GO indicating the reaction between CO groups in GO and BT-NH2. Moreover, the transmission electron microscopy images of graphene oxide (Fig. 1b) and BaTiO3-g-GO (Fig. 1c) confirm this functionalization. The dielectric constants (ε′) of BaTiO3 increases upon conjugation with GO. To realize this, poly(vinylidene fluoride) (PVDF) composites were made using BaTiO3, GO and BaTiO3-g-GO. The dielectric constants of different PVDF composites are listed in Table 1. It is evident that the dielectric constant of PVDF increases with GO (0.1 wt%) and BaTiO3 (5 vol%) separately. The incorporation of BaTiO3-g-GO (5 vol%) further enhances the dielectric constant. Dipole polarization of GO and heterogeneity of the dielectric medium are attributed to this increment. This phenomenon is well reported in our earlier works.28
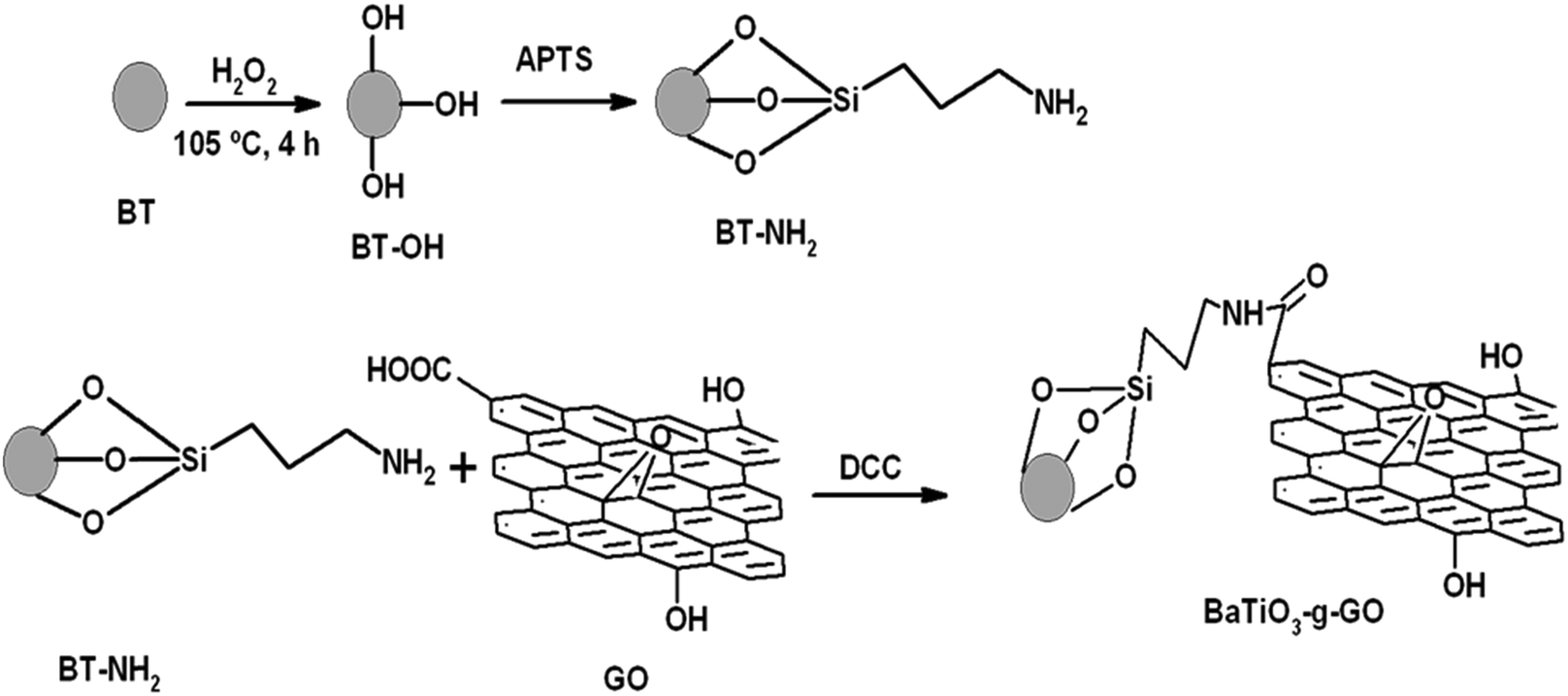 | ||
| Scheme 1 Steps of the synthesis of BT-NH2 and subsequently BaTiO3-g-GO. | ||
 | ||
| Fig. 1 (a) FT-IR spectra of GO and BaTiO3-g-GO. TEM micrographs of (b) GO and (c) BaTiO3-g-GO. | ||
| PVDF composites | ε′ |
|---|---|
| Neat | 13 |
| With 0.1 wt% GO | 14 |
| With 5 vol% BaTiO3 | 17 |
| With 5 vol% BaTiO3-g-GO (effective concentration of GO is 0.1 wt%) | 26 |
2.3 Preparation of NCPEs
PEO and LiTFSI were dried under vacuum for 48 h at 40 and 90 °C, respectively. BaTiO3-g-GO was also dried at 100 °C before use. NCPEs were prepared by dispersing BaTiO3-g-GO in polymeric solutions containing appropriate amounts of PEO and LiTFSI in anhydrous acetonitrile as displayed in Table 2. The resultant viscous solutions were cast as membranes. Upon evaporation of solvent, the resultant precursor was hot pressed under vacuum as described earlier.17–19 This procedure yielded homogenous and mechanically strong membranes with an average thickness of 40–60 microns and they were stored under vacuum at 50 °C for further characterization.| Sample | PEO (wt%) | BaTiO3-g-GO (wt%) | LiTFSI (wt%) |
|---|---|---|---|
| S1 | 95 | 0 | 5 |
| S2 | 93 | 2 | 5 |
| S3 | 85 | 10 | 5 |
| S4 | 80 | 10 | 10 |
| S5 | 75 | 10 | 15 |
2.4 Physico and electrochemical characterizations
The thermogravimetric analysis (TA Instruments, USA) has been performed between room temperature and 600 °C, while differential scanning calorimetry (TA instruments, Q200) was performed between −100 and +100 °C at heating and cooling rates of 10 °C min−1. The room temperature dielectric constant was measured using an Alpha-N Analyser, Novocontrol (Germany). X-ray diffraction measurements (SAXS and WAXS) were performed using an XEUSS SAXS/WAXS system by Xenocs. The Cu Kα radiation (wavelength λ = 1.54 Å) was collimated with an FOX2D mirror and two pairs of scatterless slits from Xenocs. The data were collected in the transmission mode geometry. The fiber diagrams were recorded using an image plate system (Mar 345 detector) and processed using Fit2D software. Silver behenate and silicon powder were used as calibrants to adjust the sample to detector distance for SAXS and WAXS, respectively.The ionic conductivity was measured by sandwiching the membrane between two stainless steel blocking electrodes (area 1 cm2) using an electrochemical impedance analyzer (Biologic, France) between the frequency range of 100 kHz and 50 mHz at various temperatures 0, 20, 30, 40, 50, 60 and 70 °C. The compatibility of the NCPE with the lithium metal anode was studied by assembling a symmetric cell composed of Li/NCPE/Li as a function of time under open circuit conditions at 70 °C. The lithium/NCPE interface was analyzed by FT-IR (FT-IR, ThermoNICOLET Corporation, Nexus Model-670) in the single internal reflection (SIR) mode. The tensile strength of the nanocomposite membrane was determined by using a tensile machine (Tinius Olsen) with a constant cross-head speed of 10 mm min−1. The sample was prepared according to ASTM standard D638.
The lithium iron phosphate (LiFePO4/C) cathode was synthesized in the form of nanostructured powder by a mild hydrothermal method as described by Meligrana et al.33 The composite cathode was prepared by blending 70% of the active material (LiFePO4/C) with 20% of acetylene black and 10% of PVDF as a binder in 1-methyl-2-pyrrolidone (NMP, Aldrich, USA) as reported earlier.17–19 The resultant slurry was coated on an aluminum foil current collector and was dried under vacuum at 120 °C for 5 h. Finally, a 2032-type coin cell was assembled with lithium metal as the anode and the NCPE as the electrolyte in an argon filled glove box (M Braun, Germany). The galvanostatic charge–discharge characteristics were measured (Arbin Instrument, USA) at different C-rates at 70 °C, setting the cut off voltages between 2.5 and 4.0 V versus Li/Li+.
3. Results and discussion
3.1 Thermal analysis
The influence of LiTFSI and BaTiO3-g-GO on the crystallization and melting behavior of PEO was studied by differential scanning calorimetry (DSC). Fig. 2a shows the DSC cooling thermograms of NCPEs with different concentrations of LiTFSI and BaTiO3-g-GO at a cooling rate of 10 °C min−1. All the samples showed exothermic peaks during cooling, corresponding to the crystallization of PEO. The LiTFSI containing PEO (sample S1) showed a melt crystallization temperature (Tc) of ca. 45 °C. With the addition of LiTFSI and BaTiO3-g-GO, the Tc was observed to be altered. The changes in crystallization and melting temperature are summarized in Table 3. With the addition of BaTiO3-g-GO, the Tc increased to 48.3 °C due to the increased crystallization rate of PEO resulting from the heterogeneous nucleation induced by BaTiO3-g-GO hybrid particles. On further increasing the concentration of LiTFSI, no change in the crystallization behavior of PEO was observed possibly due to agglomeration resulting in more ‘effective’ interactions between LiTFSI and PEO in the amorphous phase. The DSC cooling thermogram of sample S5 behaves anomalously due to the higher loading of both LiTFSI and BaTiO3-g-GO. Fig. 2b depicts the DSC second heating thermograms of NCPEs with different concentrations of LiTFSI and BaTiO3-g-GO at a heating rate of 10 °C min−1. The LiTFSI containing PEO (sample S1) exhibits a broad melting peak (melting temperature, Tm) at about 65 °C. With the addition of 2 and 10% of BaTiO3-g-GO, the Tm increased. This suggests that BaTiO3-g-GO nanoparticles contribute to the increase in melting temperature of PEO, which may be due to the change in the crystallization kinetics of PEO in the presence of BaTiO3-g-GO nanoparticles. On further increasing the concentration of LiTFSI, the Tm decreased. The reduction of Tm in the presence of excess LiTFSI shows that the interaction between the Li+ ions and ether O atoms of PEO. In order to further understand the interaction between the lithium salt and PEO, the relative percentage of crystallinity (Xc) was calculated for various nanocomposite polymer electrolytes using the following equation:where ΔHm is the enthalpy of fusion, w is the weight fraction of the NCPE and

 is the enthalpy of fusion of 100% crystalline PEO, which is taken as 213.7 J g−1. The degree of crystallinity is listed in Table 3. With the addition of BaTiO3-g-GO nanoparticles, the Xc increases moderately. However, at a higher concentration of LiTFSI (sample S5) a decrease in Xc was observed indicating an increase in the amorphous fraction in PEO. Higher concentrations of LiTFSI and BaTiO3-g-GO change the main chain dynamics of PEO due to interactions between Li+ ions and ether O atoms and the dipole–dipole interaction between BaTiO3-g-GO and PEO. This presumably results in an increase in the amorphous nature of the designed polymer electrolyte.
is the enthalpy of fusion of 100% crystalline PEO, which is taken as 213.7 J g−1. The degree of crystallinity is listed in Table 3. With the addition of BaTiO3-g-GO nanoparticles, the Xc increases moderately. However, at a higher concentration of LiTFSI (sample S5) a decrease in Xc was observed indicating an increase in the amorphous fraction in PEO. Higher concentrations of LiTFSI and BaTiO3-g-GO change the main chain dynamics of PEO due to interactions between Li+ ions and ether O atoms and the dipole–dipole interaction between BaTiO3-g-GO and PEO. This presumably results in an increase in the amorphous nature of the designed polymer electrolyte.
 | ||
| Fig. 2 DSC thermograms of NCPEs with different concentrations of LiTFSI and BaTiO3-g-GO obtained during (a) cooling from the melt and (b) reheating. | ||
| NCPE | Melting temperature (°C) | Crystallization temperature (°C) | X c (%) |
|---|---|---|---|
| S1 | 65 | 45.3 | 65.6 |
| S2 | 66.3 | 48.3 | 73.5 |
| S3 | 68.2 | 47.4 | 68.4 |
| S4 | 65 | 47.7 | 68.8 |
| S5 | 62.4 | 44.7 | 58.9 |
Fig. 3 exhibits the thermogravimetric traces of NCPEs with different concentrations of LiTFSI and BaTiO3-g-GO at a heating rate of 10 °C min−1 under nitrogen atmosphere. In all samples, the weight loss observed over a broad temperature range of 50–150 °C is attributed to the removal of moisture.34–36 For sample S1, the irreversible decomposition starts at 200 °C. However, with the addition of BaTiO3-g-GO, the thermal stability of the nanocomposites increased drastically as seen in samples S2 and S5. For instance, the decomposition started at 320 °C for sample S2. On the other hand, for sample S5, it occurred ca. 340 °C. This apparent increase in thermal stability of NCPEs is attributed to the interaction of PEO with BaTiO3-g-GO. We are speculating here that the radicals generated during the degradation of the PEO matrix were effectively trapped by the well dispersed BaTiO3-g-GO nanofillers and thus delayed the thermal degradation of the PEO matrix.
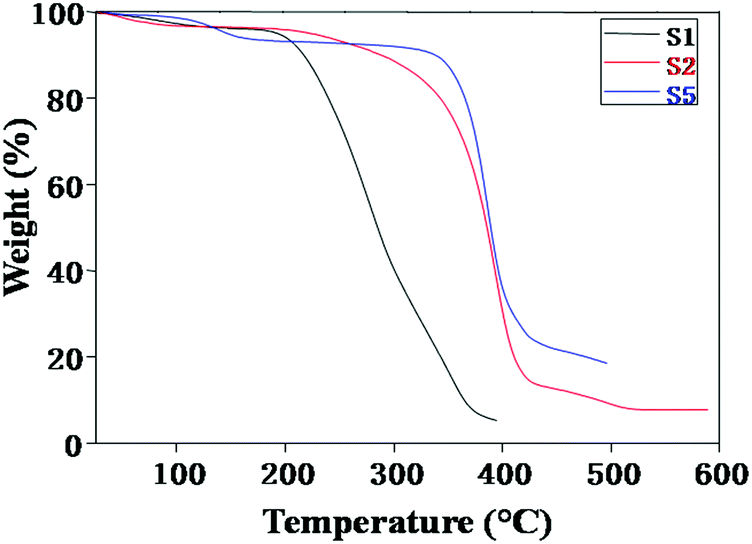 | ||
| Fig. 3 TGA thermograms of NCPEs with different concentrations of LiTFSI and BaTiO3-g-GO. | ||
3.2 Wide and small angle X-ray scattering studies
Recently, it has been demonstrated that semi-crystalline polymers are an ideal choice for designing polymer electrolytes. It was believed that the conductivity in polymer electrolytes was confined to the amorphous phase, where the segmental mobility of the polymer chains (above the glass transition temperature of the polymer) creates a disordered environment for better ion transport. However, recently it was also shown that the crystalline phase can offer even better ion transport and conductivity.37 Cheng and co-workers38 studied the effect of polymer crystallization on ion transport by fabricating a polymer single crystal electrolyte system with controlled crystal structure, crystallinity and orientation. In this work, wide and small angle X-ray scattering was used to understand the structure and morphology of the polymer electrolyte system.Fig. 4 shows the wide-angle X-ray diffraction patterns of nanocomposite polymer electrolytes containing different concentrations of LiTFSI and BaTiO3-g-GO. Both PEO and nanocomposites show intense reflections at 19.3° and 23.5°, which are assigned to the (120) and (032) reflections of the monoclinic crystal system.39–41 No change in the crystalline form of PEO was observed in the presence of nanofillers. The crystallinity of polymer nanocomposite electrolytes was calculated as the ratio of the area under the crystalline peaks to the total area under the X-ray diffraction pattern. The crystallinity estimated for various samples is summarized in Table 3. The crystallinity values estimated from the WAXD are in good agreement with the % crystallinity estimated using DSC thermograms.42 With the addition of LiTFSI and BaTiO3-g-GO (samples S2, S3, and S4) the degree of crystallinity increased moderately. However, higher amounts of LiTFSI and BaTiO3-g-GO in PEO (S5) increase the amorphous region in PEO as shown by a decrease in degree of crystallinity. These results clarified that low concentrations of nanofillers helped in increasing the crystallinity of PEO. However, when the amounts of LiTFSI and BaTiO3-g-GO increase, the amorphous content increases due to a more ‘effective’ interaction between PEO chains and Li+ and the dipole–dipole interaction between BaTiO3-g-GO and PEO.
 | ||
| Fig. 4 WAXD patterns of NCPEs with various concentrations of LiTFSI and BaTiO3-g-GO. | ||
Fig. 5 shows the Lorentz-corrected SAXS intensities for nanocomposite polymer electrolytes containing different concentrations of LiTFSI and BaTiO3-g-GO. Pure PEO shows a peak at 0.22 nm−1 (q1). For the pure PEO, additional scattering peaks up to the third order were clearly observed at high q and the ratios of the higher order peak positions can be assigned to 2:3 relative to the first order peak position. This integer ratio between the scattering peaks is characteristic of the periodic lamellar structure containing an alternate crystalline and amorphous microstructure. The first peak was used to calculate the long period L (the average sum of the crystalline lamellae thickness and the thickness of the amorphous part), using Bragg's law, where L = 2π/q1. The long period of pure PEO is 28 nm. No significant change in the lamellar structure was observed upon the addition of 5% of LiTFSI. On the other hand, polymer nanocomposite electrolytes containing LiTFSI and BaTiO3-g-GO show a weak peak in the q range (0.15–0.3 nm−1) corresponding to the PEO lamellar morphology. As discussed in the preceding sections (both DSC and WAXD), the nanocomposites have comparable levels of degree of crystallinity, but the contrast of the SAXS peak is weak for nanocomposites. It should be noted here that both LiTFSI and BaTiO3-g-GO are expected to reside in the amorphous regions of PEO and hence the overall electron density contrast may decreases as a result. This is illustrated in the graphical abstract. It is also worth mentioning here that the SAXS measurements were performed in the transmission geometry and some of the X-ray radiation was absorbed by BaTiO3-g-GO particles in the nanocomposites. In addition, the nanocomposite samples show strong scattering intensity at low q, which can be attributed to the scattering of well-dispersed BaTiO3-g-GO particles within the PEO matrix.
 | ||
| Fig. 5 Lorentz-corrected SAXS patterns of nanocomposite polymer electrolytes with different concentrations of LiTFSI and BaTiO3-g-GO. | ||
With the addition of a low concentration of BaTiO3-g-GO, these nanoparticles act as heterogeneous nucleating agents and increase both the crystallization rate and degree of crystallinity of PEO. However, on further increasing the concentration of BaTiO3-g-GO, no change in the crystallization behavior of PEO was observed due to agglomeration of the particles. At the same time, the interactions between BaTiO3-g-GO and PEO act as crosslinks to restrict the crystallization of PEO. Hence, the degree of crystallinity is reduced at a higher loading of BaTiO3-g-GO.
3.3 Tensile studies
Fig. 6 shows the stress–strain behavior of different NCPEs. The neat PEO shows a tensile strength of 1.75 MPa and a tensile modulus of 64.6 MPa. Upon addition of 5% LiTFSI (S1) we observed a dramatic increase in mechanical properties. The tensile strength increases to 154% and the tensile modulus increases to 207%. This could be because of enhanced interaction of electron lone pairs of O in PEO with the Li+ ions in LiTFSI. Similarly, in S3 with 5% LiTFSI and 2% BaTiO3-g-GO we observe better mechanical performance than neat PEO (S1). However, with further increase in LiTFSI and BaTiO3-g-GO, the tensile properties degrade as an excessive amount of inorganic nanoparticles affects the crystallinity.43 Moreover, the poor stress-transfer between inorganic nanoparticles and PEO decreases both tensile strength and modulus. | ||
| Fig. 6 Stress–strain behavior of NCPEs. | ||
3.4 Ionic conductivity
The variation of ionic conductivity as a function of inverse temperature at different concentrations of BaTiO3-g-GO and LiTFSI is displayed in Fig. 7. It is seen from this figure that the ionic conductivity increases with the increase in the BaTiO3-g-GO content and also with temperature. Irrespective of the concentrations of BaTiO3-g-GO and LiTFSI all curves show a remarkable change in the slope beyond 60 °C, which indicates the well-known transition of PEO from crystalline to amorphous phases. The incorporation of BaTiO3-g-GO in the polymeric matrix has significantly increased the ionic conductivity of the NCPE. An increase of two orders of magnitude in ionic conductivity is observed when the content of BaTiO3-g-GO is 10% (sample S5). Generally, for the NCPE, the ionic conductivity is not a linear function of filler concentration. At low concentrations, a diffusion effect takes place which tends to depress the ionic conductivity and is effectively opposed by specific interactions of Li+-ions with the filler surfaces, and thus promotes faster ionic transport. At a higher filler content, the conductivity is reduced due to the predominant dilution effect. Thus, the maximum ionic conductivity is achieved only in the concentration range between 5 and 15 wt%, however, it is mainly influenced by the nature of the filler. | ||
| Fig. 7 Ionic conductivity as a function of inverse temperature for different concentrations of NCPEs. | ||
Generally, Lewis acid groups of the added filler may compete with the Lewis acid lithium cations for the formation of complexes with the alkoxide of PEO-chains, as well as with the anions of the LiTFSI added.44 This results in structural modifications due to specific interactions of the polar surface groups on the inorganic filler. The Lewis acid–base interaction centers on the electrolyte species, and thus lowers the ionic coupling, and the salt dissolution is promoted by the formation of the “ion–filler complex”.17,19,44 In the present study, BaTiO3-g-GO has a Lewis acid center which can interact with the anions of the LiTFSI salt and these interactions reduce the crystallinity of the polymer host.44–46 This reduction in crystallinity could be the reason for the increased ionic conductivity. This claim was further supported by DSC and WAXS data.
3.5 Interfacial properties
Lithium is a highly reactive metal towards the electrolyte and/or residual impurities such as water. The nature of the passive layer at the lithium/electrolyte interface is complex and still a subject of debate. The nature of the surface layer formed at the interface depends on the ionic conductivity of the layer, type of lithium salt, solvent and temperature, etc.47–49 The Li/electrolyte interfacial properties play a vital role in terms of battery stability and cyclability. Fig. 8 depicts the time dependence of the interfacial resistance “Ri” for the symmetric cell Li/NCPE/Li measured at 70 °C. The value of interfacial resistance increases exponentially with time in the beginning and gets stabilized after 8 days for the BaTiO3-g-GO-free membrane. On the other hand, apparently, the value of “Ri” is lowered for the BaTiO3-g-GO-laden membrane. Although the value of “Ri” decreases from 1200 ohms to 280 ohms, after 96 hours the variation in the pattern is similar to that of the BaTiO3-g-GO-free membrane. The reduction in the value of “Ri” may be attributed to the entrapment of traces of residuals and impurities by ferrous ceramic fillers with permanent dipoles.23,24 Several reports are available on the cycling behavior of lithium–polymer cells with polymer nanocomposite electrolytes. But studies on the lithium/electrolyte interfaces are very few. Although techniques such as XPS, AES and EDAX are widely used to characterize the lithium/polymer electrolyte interface, they often destroy the lithium metal surface. The thickness of the solid electrolyte interface (SEI) was calculated here using the relationship,50,51| t = 2fmaxεrε0ARi | (1) |
 | ||
| Fig. 8 Variation of interfacial resistance “Ri” as a function of time. Inset: The Cole–Cole plot of the Li/NCPE (sample S5)/Li symmetric cell at different times at 70 °C. | ||
Fig. 9 illustrates the FT-IR spectrum of sample S5 (NCPE) and the lithium electrode surface upon contact with the NCPE for 24 h. It quite evident from the FT-IR spectrum that no discernible peak could be noticed on the lithium surface even after the membrane was in contact with the NCPE. However, the increase in interfacial resistance might have arisen from the presence of calcium compounds (<1%) that were present during the synthesis of PEO (to neutralize the catalyst and fumed silica which was added to increase the fluidity of PEO).29 All these constituents might react with lithium metal to increase the “Ri” values.
 | ||
| Fig. 9 FT-IR spectra of (a) sample S5 (NCPE) and (b) Li-surface upon contact with the NCPE. | ||
3.6 Charge–discharge studies
The typical galvanostatic charge–discharge cycling profile of the LiFePO4/NCPE/Li cell at 60 °C is depicted in Fig. 10a. It is clear from the figure, that the charge–discharge profile between 2.5 and 4.0 V vs. Li at 0.1C-rate has a well-defined plateau at 3.45 V vs. Li upon charging (de-lithiation) and about 3.3 V vs. Li upon discharging (lithiation) which is a characteristic behavior of the LiFePO4 electrode upon a reversible redox reaction with lithium.19,20 The charge–discharge plateau is highly reproducible and well defined. Fig. 10b shows the resulting plot of specific capacity as a function of the cycle number at different C-rates. In the initial charge–discharge processes, the cell delivered an initial discharge capacity of 134 mA h g−1 during its first 5 cycles with 90% Coulombic efficiency at 0.05C-rate. The Coulombic efficiency is increased to 98% after the first few cycles and remains stable even at higher C-rates which indicates good mechanical stability of the electrode during the Li+-ion intercalation/de-intercalation process, good interfacial contact, charge transport between the electrode and the polymer electrolyte and excellent reversibility after the initial surface reactions.19 At 1C-rate, the discharge capacity is reduced to 100 mA h g−1. It is noteworthy that the cell is able to be cycled even at 5C-rate which demonstrates the appreciable characteristics of the all-solid state electrolyte which provides fast lithium-ion exchange between the electrodes through the polymeric matrix. | ||
| Fig. 10 (a) Cycling profile of the LiFePO4/NCPE (sample S5)/Li cell at 60 °C. (b) Discharge capacity as a function of the cycle number for the LiFePO4/NCPE (sample S5)/Li cell at 70 °C. | ||
The reduction in the discharge capacity at higher current is a typical characteristic of the LiFePO4 material which is attributed to its low electronic conductivity and limited diffusion of Li+-ions into its structure that causes electrode polarization.52,53 A recent study also revealed that the increase in interfacial resistance which originates from parameters related to the electrode design such as thickness and density can cause capacity fading at higher rates.54 It is also obvious from the figure that the cell restores its specific capacity again at 0.1C rate from its 91st cycle indicating that the structural stability of the cathode material is retained.
It is important to note that even after 85 cycles the cell was able to deliver a discharge capacity of 120 mA h g−1 which restores the specific capacity at its 90th cycle (<3% decay in the discharge capacity). A similar observation was reported by Appetecchi et al.,53 who studied the cycling behavior of the PEO + LiCF3SO3 + SiO2 composite electrolyte at 90 °C and also observed a fade in capacity of 0.3 mA h g−1. In a similar way, Li and co-workers25,26 demonstrated the cycling profile of the Li/PEO + BaTiO3 + LiTFSI/LiNi0.2Co0.8O2 cell, however, the cell was cycled up to 50 cycles with a rapid fade in capacity. While comparing the present work with earlier reports [ref. 24, 25 and 43], a stable cycling and higher discharge capacity are achieved ever at lower temperature.
4 Conclusions
BaTiO3-g-GO was successfully synthesized and incorporated in PEO along with LiTFSI. The WAXS studies revealed that the crystallinity of PEO has increased at lower concentrations of nanofillers; however, at higher concentrations the amorphous content of PEO has increased due to the more ‘effective’ interaction between PEO chains and Li+ and the dipole–dipole interaction between BaTiO3-g-GO and PEO manifesting in more cross linking. The ionic conductivity, thermal stability and compatibility of the PEO-based electrolyte has increased upon the incorporation of BaTiO3-g-GO. The all-solid-state lithium polymer cell assembled by sandwiching the best performing nanocomposite electrolyte between the LiFePO4 cathode and the lithium metal anode delivered a discharge capacity approaching 110 mA h g−1 after prolonged cycling (up to 95 cycles) at 60 °C and at 1C-rate. This was found to be higher than those reported earlier for similar systems. Thus, these results warrant for an all-solid-state lithium–polymer cell conceived for safe application at elevated temperature.Acknowledgements
Dr N. A. L. gratefully acknowledges University Grants Commission, New Delhi for funding under Dr D. S. Kothari fellowship. Prof. S. Bose would like to acknowledge the support from DST, India.References
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 4, 2930–2946 CrossRef PubMed.
- E. Quartarone and P. Mustarelli, Chem. Soc. Rev., 2011, 40, 2525–2540 RSC.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- M. S. Whittigham, Chem. Rev., 2004, 104, 4271–4301 CrossRef.
- M. Winter and R. J. Bhod, Chem. Rev., 2004, 104, 4245–4260 CrossRef CAS PubMed.
- Z. Xue, D. He and X. Xie, Polym. Chem., 2015, 6, 1660–1687 RSC.
- A. Manuel Stephan, Eur. Polym. J., 2006, 21, 21–42 CrossRef.
- A. Manuel Stephan and K. S. Nahm, Polymer, 2006, 47, 5952–5964 CrossRef.
- R. Bouchet, S. Lascaud and M. Rosso, J. Electrochem. Soc., 2003, 150, A1385–A1389 CrossRef CAS.
- G. B. Appetecchi, F. Croce and B. Scrosati, Electrochim. Acta, 1990, 40, 991–997 CrossRef.
- A. Manuel Stephan, S. Gopukumar, N. G. Renganathan and M. A. K. Nathan, Eur. Polym. J., 2005, 41, 15–21 CrossRef.
- F. Croce, R. Curini, A. Martinelli, L. Perci, F. Ronci and B. Scrosati, J. Phys. Chem. B, 1999, 103, 10632–10638 CrossRef CAS.
- B. Scrosati, F. Croce and S. Panero, J. Power Sources, 2001, 100, 93–100 CrossRef CAS.
- N. Angulakshmi and A. Manuel Stephan, Electrochim. Acta, 2014, 127, 167–172 CrossRef CAS.
- N. Angulakshmi, S. Thomas, J. R. Nair, R. Bongiovanni, C. Gerbaldi and A. Manuel Stephan, J. Power Sources, 2013, 228, 294–299 CrossRef.
- N. Angulakshmi, M. A. K. Nathan, R. S. Kumar and A. Manuel Stephan, J. Phys. Chem. C, 2014, 118, 24240–24247 CAS.
- R. S. Kumar, M. Raja, M. A. K. Nathan and A. Manuel Stephan, RSC Adv., 2014, 4, 26171–26175 RSC.
- C. Gerbaldi, J. R. Nair, M. A. K. Nathan, R. S. Kumar, C. Ferrara, P. Mustarelli and A. Manuel Stephan, J. Mater. Chem. A, 2014, 2, 9948–9954 CAS.
- M. C. Borghini, M. Mastragostino, S. Passerini and B. Scrosati, J. Electrochem. Soc., 1995, 142, 2118–2121 CrossRef CAS.
- F. Capuano, F. Croce and B. Scrosati, J. Electrochem. Soc., 1991, 138, 1918–1922 CrossRef CAS.
- W. Nan and D. M. Smith, Mater. Sci. Eng., B, 1991, 10, 99–106 CrossRef.
- H. Y. Sun, H. J. Sohn, O. Yamamoto, Y. Takeda and N. Imanishi, J. Electrochem. Soc., 1999, 146, 1672–1676 CrossRef CAS.
- H. Y. Sun, Y. Takeda, N. Imanishi, O. Yamamoto and H. J. Sohn, J. Electrochem. Soc., 2000, 147, 2462–2467 CrossRef CAS.
- Q. Li, Y. Takeda, N. Imanishi, J. Yang, H. Y. Sun and O. Yamamoto, J. Power Sources, 2001, 97–98, 795–797 CrossRef CAS.
- Q. Li, H. Y. Sun, Y. Takeda, N. Imanishi, J. Yang and O. Yamamoto, J. Power Sources, 2002, 94, 201–205 CrossRef.
- G. P. Kar, S. Biswas, R. Rohini and S. Bose, J. Mater. Chem. A, 2015, 3, 7974–7985 CAS.
- M. Sharma, M. P. Singh, C. Srivastava, G. Madras and S. Bose, ACS Appl. Mater. Interfaces, 2014, 6, 21151–21160 CAS.
- J. H. Shin, F. Alessandrini and S. Passerini, J. Electrochem. Soc., 2005, 152, A283–A288 CrossRef CAS.
- A. Manuel Stephan, T. Prem Kumar, M. A. K. Nathan and N. Angulakshmi, J. Phys. Chem. B, 2009, 113, 1963–1971 CrossRef PubMed.
- P. Xavier, K. Sharma, K. Elayaraja, K. S. Vasu, A. K. Sood and S. Bose, RSC Adv., 2014, 4, 12376–12387 RSC.
- S. Biswas, G. P. Kar and S. Bose, J. Mater. Chem. A, 2015, 3, 12413–12426 CAS.
- C. Meligrana, C. Gerbaldi, A. Tuel, S. Bodoardo and N. Pennazi, J. Power Sources, 2006, 160, 516–522 CrossRef.
- R. Ribeiro, G. G. Silva and N. D. S. Mohallen, Electrochim. Acta, 2001, 46, 1679–1686 CrossRef CAS.
- T. Shodai, B. B. Owens, T. Ohtsuka and J. Yamaki, J. Electrochem. Soc., 1994, 141, 2978–2981 CrossRef CAS.
- S. P. Thomas, S. Thomas and S. Bandyopadhyay, J. Phys. Chem. C, 2009, 113, 97–104 CAS.
- Z. Gadjourova, Y. G. Andreev, D. P. Tunstall and P. G. Bruce, Nature, 2001, 412, 520–523 CrossRef CAS PubMed.
- S. Cheng, D. M. Smith and C. Y. Li, Macromolecules, 2014, 47, 3978–3986 CrossRef CAS.
- Y. Takahashi and H. Tadokoro, Macromolecules, 1973, 6, 672–675 CrossRef CAS.
- H. Tadokoro, Y. Chatani, T. Yoshihara, S. Tahara and S. Murahashi, Die Makromol. Chem., 1964, 73, 109–127 CrossRef CAS.
- J. M. Deitzel, J. D. Kleinmeyer, J. K. Hirvonen and N. C. Beck Tan, Polymer, 2001, 42, 8163–8170 CrossRef CAS.
- L. Zhu, S. Z. D. Cheng, B. H. Calhoun, Q. Ge, R. P. Quirk, E. L. Thomas, B. S. Hsiao, F. Yeh and B. Lotz, J. Am. Chem. Soc., 2000, 122, 5957–5967 CrossRef CAS.
- L. E. Nielsen, Mechanical Properties of Polymers, Van No strand Reinhold, New York, 1962 Search PubMed.
- F. Croce, L. Perci, B. Scrosati, F. S. Fiory, E. Plichta and M. A. Hendrickson, Electrochim. Acta, 2001, 46, 2457–2461 CrossRef CAS.
- W. Wieczorek, Z. Florjancyk and J. R. Stevens, Electrochim. Acta, 1995, 40, 2251–2258 CrossRef CAS.
- J. Przluski, M. Siekierski and W. Wieczorek, Electrochim. Acta, 1995, 40, 2101–2108 CrossRef.
- D. Fauteux, J. Electrochem. Soc., 1988, 135, 2231–2237 CrossRef CAS.
- D. Aurbach and A. Zaban, J. Electroanal. Chem., 1993, 348, 155–179 CrossRef CAS.
- A. M. Christie, A. L. Oleksiak and C. A. Vincent, Electrochim. Acta, 1995, 40, 2403–2411 Search PubMed.
- I. Ismail, A. Noda, A. Nishimoto and M. Watanabe, Electrochim. Acta, 2001, 46, 1595–1603 CrossRef CAS.
- A. L. Oleksiak, Solid State Ionics, 1999, 119, 205–209 CrossRef.
- B. Scrosati and B. Neat, Application of Electroactive Polymers, Chapman & Hall, London, 1993, p. 204 Search PubMed.
- G. B. Appetecchi, J. Hassoun, B. Scrosati, F. Croce, F. Cassel and M. Salomon, J. Power Sources, 2003, 124, 246–253 CrossRef CAS.
- J. H. Kim, S. C. Woo, M. S. Park, K. J. Kim, T. Yim, J. S. Kim and Y. J. Kim, J. Power Sources, 2013, 229, 190–197 CrossRef CAS.
| This journal is © the Partner Organisations 2017 |