
Flexible assembly of targeting agents on porous magnetic nano-cargos by inclusion complexation for accurate drug delivery†
Dian
Li
ab,
Luyan
Sun
a,
Yuting
Zhang
a,
Meng
Yu
a,
Jia
Guo
a and
Changchun
Wang
*a
aState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Laboratory of Advanced Materials, Fudan University, Shanghai, 200433, People's Republic of China. E-mail: ccwang@fudan.edu.cn; Fax: +86-21-65640293
bTechnical Center of China Tobacco Guangxi Industrial Co., Ltd, Guangxi, 530001, People's Republic of China
First published on 7th September 2016
Abstract
Efficient delivery of anticancer drugs to increase the intracellular drug concentration in targeted tissues is urgently needed in cancer chemotherapy, since clinical drugs usually have serious side effects on normal tissues. In this paper, we developed a new method to fabricate multi-functional porous magnetic nano-cargos by grafting PEG/folate onto the surface and storing doxorubicin in the pores of magnetic supraparticles for accurate delivery of anticancer drugs. The anticancer drug doxorubicin was fixed into the porous magnetic cores with acid-sensitive linkers, which can be broken in acidic intracellular environments or organelles; the superficial PEG chains on the magnetic cores not only enhanced the dispersion stability of the nano-cargos but also immobilized folate modified α-cyclodextrin by inclusion complexation, and the α-cyclodextrin derivatives could be flexibly replaced as needed. Remarkable proliferation inhibition of cancer cells and minor side effects on normal cells were achieved due to the controlled drug release manner of the nano-drug system, indicating that this kind of nano-cargo has great potential in cancer chemotherapy for personalized and accurate treatment.
Introduction
Integrating nanotechnology into drug delivery systems for cancer chemotherapy is a rapidly growing research field, and the major challenge that needs to be overcome urgently is how to deliver drugs to tumor tissues accurately and effectively.1–3 In recent decades, magnetic nanoparticles as drug carriers have attracted much attention because they can be easily directed by an external magnetic force to target sites and then effectively enter tumor tissues by an enhanced permeability and retention (EPR) effect, and can be traced by magnetic resonance imaging (MRI).1,4,5 The magnetic nanoparticles are usually designed with special structures, including hollow,6,7 rattle-like,8,9 porous10,11 and core–shell structures,12,13 which can provide a large space for storage of anti-cancer drugs. Recently, a new pathway has been developed to generate large inner pores, which can produce additional inner surface in magnetic supraparticles for storage of guest molecules.14 However, even though the magnetic guidance and EPR effect can direct the drug-loaded nano-cargos to tumorous tissues, unexpected drug release during circulation in the body and poor cellular internalization of cancer cells are still a challenge, which can reduce the dosage of the anti-cancer drug in the target site and restrict the efficacy of anti-cancer drugs.15,16An ideal drug carrier is expected to precisely control drug release in targeted cells while limiting drug leakage in circulation.17,18 Due to the special environmental conditions of targeted tissues, many published papers have designed stimuli-responsive drug carriers to control drug release.19–23 The pH-responsive design for controlling drug release is frequently used, because the acidity in different cells and different cellular compartments is varied. According to the pH gradient between blood/normal tissue (pH = 7.4), the tumor tissue extracellular environment (6.8) and the intracellular cytoplasm/endosome/lysosome (6.0–5.0),24–27 the pH switch can be set in the range 5.0–6.5 for precise drug release in cancer cells.
To overcome the problem of poor cellular internalization, some specific superficial drug carrier designs matched to the unique requirements of targeted cellular surfaces have been utilized for the enhancement of cellular internalization.28–32 According to the specific overexpressing antigens and receptors in cancer cells,1,33 the surface of drug carriers can also be anchored with targeting agents, such as special proteins (e.g., monoclonal antibodies),34,35 oligonucleotides (e.g., aptamers)36,37 and small molecules (e.g., folate),38,39 to increase the internalization efficacy in cancer cells. However, the targeting molecules are generally bound to the surface of the drug carrier by irreversible chemical covalent bonds, which can not be changed once anchored, thus the drug carriers can only match limited types of cells, and are not suitable for personalized and accurate treatment. Recently, inclusion complexation between PEG and α-cyclodextrin, as a reversible special secondary interaction, has attracted much attention because this pre-complex can be reconstructed by other guest molecules.40–42 Jiang et al. reported a polymeric hydrogel consisting of PEG and α-cyclodextrin (α-CD) formed by inclusion complexation. When azopyridinium was introduced, the hydrogel quickly turned to liquid due to the PEG being replaced by azopyridinium in the inclusion complex.43,44 This result indicates that reversible grafting on the surface of drug carriers by inclusion complexation could be an efficient new method for superficial modification.
In this paper, we designed a new kind of multifunctional nano-cargo, with a combination of high magnetic responsiveness, flexible immobilization of functional molecules, and excellent pH-responsive drug release. The porous magnetic nanoparticles in the core acted as a framework to conjugate DOX through acid-sensitive chemical bonds in the pores, and folate-modified α-CD was anchored on the outer PEG shell by inclusion complexation. The superficial folate-modified PEG chains not only enhance the stability of the particles in solution, but also increase the cell uptake ability in folate-acceptor overexpressing HeLa cells. Additionally, the acidic intracellular environment of HeLa cells can break the acid-sensitive chemical bond to release DOX for chemotherapy. This new kind of multi-responsive drug delivery system exhibited very high cytotoxicity to HeLa cells and minor side effects on normal cells (HEK 293T cells) compared with free DOX, showing great potential for accurate treatment of tumours.
Experimental section
Materials
Iron(III) chloride hexahydrate (FeCl3·6H2O), ammonium acetate (NH4OAc), sodium hydroxide (NaOH), ethylene glycol (EG), anhydrous pyridine, 4-dimethylaminopyridine (DMAP), anhydrous ethanol, folate (FA) and anhydrous dimethyl sulfoxide were purchased from Shanghai Chemical Company and used as received. Rhodamine-B, anhydride, 4-toluene sulfonyl chloride (TsCl), N,N-dicyclohexylcarbodiimide (DCC), fluorescein isothiocyanate (FITC), glutathione (GSH), α-cyclodextrin, 2,3-dimethylmaleic N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC·HCl), N-hydroxysuccinimide (NHS), 2,3-dimethylmaleic anhydride, succinic anhydride and tetrahydrophthalic anhydride were purchased from Aladdin (Shanghai, China) and used as received. Hydrazine (NH2NH2·H2O) and azobisisobutyronitrile (AIBN) were purchased from Sinopharm Chemical Reagent Corp and used as received. Methyl mercaptoacetate and NH2 modified PEG (Mw ∼ 2000) were purchased from Aldrich. Agarose was purchased from Gene Tech (Shanghai) Company. Doxorubicin (DOX) hydrochloride was purchased from Beijing Hua Feng United Technology CO., Ltd (Beijing, China) and used as received. Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin G, streptomycin, and trypsinase were obtained from GIBCO BRL (Grand Island, NY) and used as received. Deionized water was used in all experiments. All other chemicals were commercially available and used without further purification.Preparation of PEG grafted magnetic supraparticles (MSP-PEG)
The magnetic supraparticles (MSPs) stabilized by agarose were prepared using a solvothermal method.11 The obtained MSPs could be grafted with PEG by two steps: firstly, 0.5 mmol (1.0 g) –NH2 modified PEG (Mw ∼ 2000) and 1.2 equivalents of succinic anhydride were dissolved in 5 mL dichloromethane, and the mixture was vigorously stirred for 5 h in a 25 mL round-bottom flask at room temperature after the addition of 1.2 equivalent triethylamine (TEA). The crude product was purified by precipitation in diethyl ether. Secondly, 200 mg of the obtained –COOH modified PEG and 100 mg DCC were dissolved in 10 mL DMF with stirring for 2 h. The solution was transferred to a three-necked flask containing 40 mL MSP dispersion (2.5 mg mL−1), followed by addition of a trace amount of DMAP (5 mg) for catalysis, and the reaction was allowed to proceed for 24 h with stirring. The resulting product was separated by magnet and washed five times with ethanol. All the products were obtained after lyophilizing under vacuum for 3 days.Preparation of porous MSP-PEG (p-MSP-PEG)
The porous MSP-PEG (p-MSP-PEG) was synthesized by an etching method.16 MSP-PEG (50 mg) was ultrasonically dispersed in a 100 mL three-necked bottle containing 50 mL DMF, and then 1.6 mL hydrazine and 0.4 mL methyl mercaptoacetate were added. After N2 sweeping for 30 min, the mixed solution was reacted at 80 °C for 1 h. The reaction was terminated with cold ethanol. The product was separated by magnet and the black supernatant was removed. After washing with ethanol and water three times each, the obtained p-MSP-PEG was dried under vacuum for 24 h.Preparation of DOX-loaded p-MSP-PEG (DOX@p-MSP-PEG)
DOX was loaded into the p-MSP-PEG via an acidic stimulus-responsive chemical bond over two steps. Firstly, 20 mg of the prepared p-MSP-PEG was dispersed in 20 mL anhydrous DMF, and then excess 2,3-dimethylmaleic anhydride (80 mg) was added to the dispersion, followed by adding trace DMAP (5 mg) as catalyst. After reacting for 6 h, the product was purified by magnetic separation and water rinsing. In the second step, all the products were re-dispersed in 20 mL DMF containing 50 mg EDC and 50 mg NHS, and then allowed to react for 4 h. After that, 20 mg DOX and 100 μL TEA were added, and the reaction was maintained at room temperature for 24 h. The resulting product was collected by magnet, and washed with DMF and PBS buffer (pH = 7.4) until the supernatant became transparent, to ensure that the free DOX had been thoroughly removed. The final DOX-loaded p-MSP-PEG (DOX@p-MSP-PEG) was lyophilized under vacuum for 3 days. As for the samples of SA-DOX@p-MSP-PEG and THPA-DOX@p-MSP-PEG, these were prepared by the same method, but the 2,3-dimethylmaleic anhydride was switched for succinic anhydride or tetrahydrophthalic anhydride.Synthesis of folate modified α-cyclodextrin (α-CD-FA)
The folate modified α-cyclodextrin (α-CD-FA) was synthesized by two steps, with reference to previous reports.45 For the first step, 1.5022 g α-cyclodextrin was dissolved in 10 mL pyridine with an ice-water bath, and then 5 mL 0.262 g mL−1 4-toluene sulfonyl chloride (TsCl) was added dropwise to the solution over 1 h. After reacting for another 1 h, water was added to terminate the reaction, and the solution became transparent. When the solution was transferred into acetone, a white precipitate was produced, which could be collected by centrifugation (8000 rpm). The activated α-cyclodextrin (α-CD-OTs) was obtained by vacuum drying. For the second step, 100 mg α-CD-OTs and 6 mg amino-modified folate38 were mixed in 15 mL water. After the pH was altered to 10 with 0.1 M NaOH (aq.), the solution was allowed to react with stirring overnight. The solution was dialyzed with a dialysis tube (cutoff Mn = 1000) in water to remove the surplus amino-modified folate, and the resulting yellow product was lyophilized under vacuum for 3 days.Preparation of folate modified DOX@p-MSP-PEG (DOX@p-MSP-PEG-FA)
The folate modified DOX@p-MSP-PEG was obtained by inclusion complexation between PEG and the α-cyclodextrin. The DOX@p-MSP-PEG (25 mg) was dispersed in 20 mL pH = 7.4 PBS buffer containing 2 mg α-CD-FA, and the solution was stirred at 0 °C for 6 h. The resulting mixture was rinsed with pH = 7.4 PBS buffer until the supernatant became colorless, followed by lyophilizing under vacuum for 3 days. As for the SA-DOX@p-MSP-PEG-FA and THPA-DOX@p-MSP-PEG-FA, these were prepared by the same method, but the DOX@p-MSP-PEG was switched for SA-DOX@p-MSP-PEG or THPA-DOX@p-MSP-PEG.In vitro cytotoxicity experiments
The in vitro cytotoxicity of DOX@p-MSP-PEG-FA, SA-DOX@p-MSP-PEG-FA, THPA-DOX@p-MSP-PEG-FA and free DOX was determined using HeLa cells; meanwhile, the in vitro cytotoxicity of DOX@p-MSP-PEG-FA and free DOX was determined with HEK 293T cells using the CCK8 method.32 Specifically, 100 μL of cells were seeded in a 96-well flat culture plate at a density of 1 × 104 cells per well and subsequently incubated for 24 h to allow attachment. Samples with different DOX concentrations (0.1, 0.25, 0.5, 1.0, 2.5, 5.0, 10 μg mL−1) in DMEM were then added to each group (three wells) for an additional 24-hour incubation. After removing the previous nutrient solution, the cells were incubated in 110 μL of DMEM containing 10 μL of CCK8 solution for 1 h. The absorbance of the suspension was measured at 450 nm on an ELISA reader and the cell viability was calculated by means of the following formula: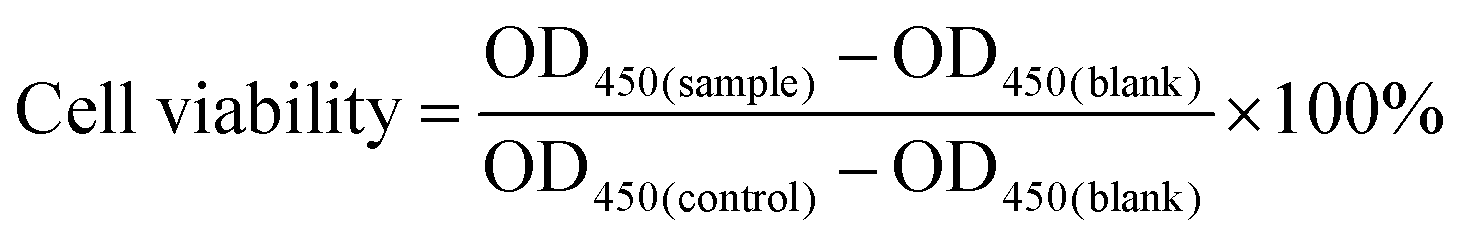
The in vitro cytotoxicity of DOX@p-MSP-PEG-FA with α-CD or folate was obtained by adding α-CD or folate to DMEM to form the 0.1 mg mL−1 solution.
Confocal laser scanning microscope (CLSM) experiments
HeLa cells and HEK 293T cells were separately incubated in φ – 15 mm thin bottom culture chambers at a density of 2 × 105 cells per chamber and subsequently incubated for 24 h to allow attachment. Each sample (2 mL) with 0.5 μg mL−1 DOX in DMEM (calculated by loaded rate) was then added to the φ – 15 mm thin bottom culture chamber for an additional incubation. After a certain incubation time, the cells were washed with DMEM and PBS three times, followed by 0.001 mg mL−1 Hoechst 33342 staining for 20 min. After Hoechst 33342 was removed by PBS washing, the samples were observed by CLSM with excitation wavelengths of 405 nm and 542 nm.Flow cytometer (FCM) experiments
HeLa cells and HEK 293T cells were separately incubated in 6-well flat culture plates at a density of 1 × 105 cells per well and subsequently incubated for 24 h to allow attachment. Each sample (2 mL) with 0.5 μg mL−1 DOX in DMEM (calculated by loaded rate) was then added to each well for an additional incubation. After a certain incubation time, the cells were washed with DMEM and PBS three times. The cells were separated by digestion of pancreatic enzyme and then immobilized by paraformaldehyde. After washing with PBS to remove the surplus paraformaldehyde, the cells were re-dispersed in 1 mL PBS in a flow cell tube for the flow cytometer experiment. The excitation wavelength was 488 nm and the emission wavelength was 580 nm.Characterization
Transmission electron microscopy (TEM) images were collected on an H-600 (Hitachi, Japan) transmission electron microscope at an accelerating voltage of 75 kV. High-resolution transmission electron microscopy (HRTEM) images were taken on a JEM-2010 (JEOL, Japan) transmission electron microscope at an accelerating voltage of 200 kV. Samples dispersed at an appropriate concentration were cast onto a carbon-coated copper grid. Magnetic characterization was carried out on a Model 6000 physical property measurement system (Quantum Design, USA) at 300 K. Powder X-ray diffraction (XRD) patterns were obtained using an X'Pert Pro (Panalytical, Netherlands) diffraction meter with Cu Kα radiation at λ = 0.154 nm operating at 40 kV and 40 mA. Fourier transform infrared (FT-IR) spectra were recorded on a Magna-550 (Nicolet, USA) spectrometer where the samples were dried and mixed with KBr to be compressed onto a plate for measurement. Thermogravimetric (TG) analysis data were obtained on a Pyris-1 (Perkin Elmer, USA) thermal analysis system under nitrogen flow and at a heating rate of 20 °C min−1 from 100 to 800 °C. The fluorescence emission spectra were recorded on an RF-5301PC spectrometer with an excitation wavelength of 365 nm (Shimadzu, Japan). Ultraviolet-visible (UV-vis) absorption spectra were recorded on a UV-3150 (Shimadzu, Japan) ultraviolet-visible spectrophotometer. Confocal laser scanning microscopy (CLSM) images were obtained using an FV1000 confocal laser scanning microscope (Olympus, Tokyo, Japan). ICP atomic emission spectrometry was performed using a P-4010 (Hitachi, Japan) spectrometer. Flow cytometer analysis was performed on a FACS Aria II (Becton-Dickinson, San Jose, CA, USA), with an excitation wavelength of 488 nm and an emission wavelength of 580 nm.Results and discussion
Preparation of the multifunctional magnetic nano-cargos for targeted delivery of DOX
Starting from magnetic supraparticles (MSP) stabilized by agarose, PEG modified magnetic supraparticles (MSP-PEG) were prepared through the reaction between the carboxyl group of PEG and the hydroxyl group of agarose. Then, the MSP-PEG was etched by the reported redox-etching method to form porous PEG modified magnetic supraparticles (p-MSP-PEG).38 The agarose on the inner surface could be further modified by a series of chemical reactions (Scheme 1b), and the anti-cancer drug doxorubicin could be conjugated into the pores of p-MSP-PEG via an acid-sensitive chemical bond (DOX@p-MSP-PEG), which could be broken apart in an acidic environment to recover the original structure of DOX (Scheme 1c). Finally, to endow the DOX@p-MSP-PEG with targeting capabilities, folate modified α-cyclodextrin (CD-FA) could be grafted onto the surface of DOX@p-MSP-PEG by inclusion complexation between PEG and α-CD (Scheme 1a).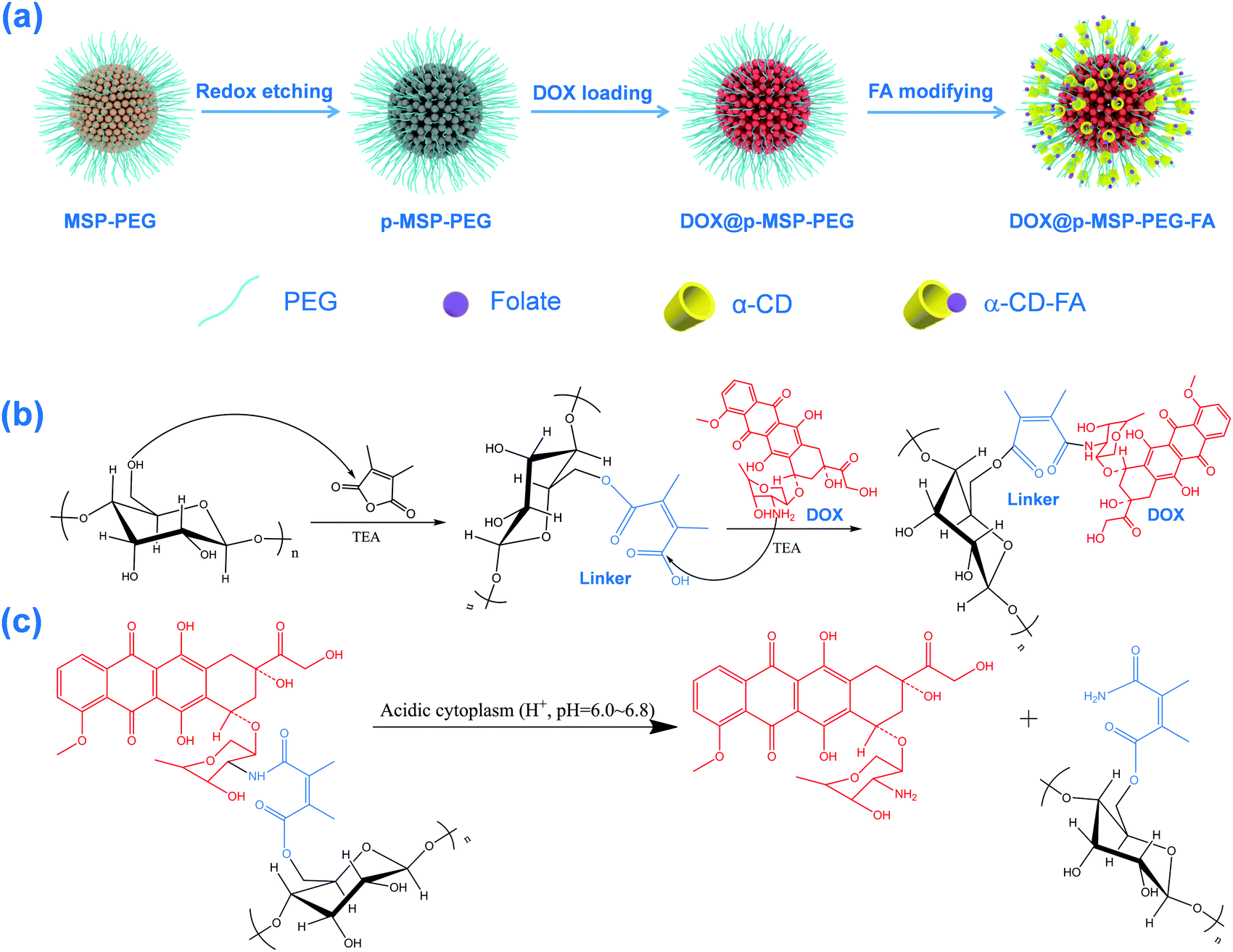 | ||
| Scheme 1 (a) The preparation procedure for DOX-loaded FA-targeting porous magnetic supraparticles (MSP); (b) the grafting of DOX to the agarose chain on the surface of the porous magnetic supraparticles; (c) the cleavage mechanism of the acid-sensitive linker between DOX and the agarose chain. | ||
The structure of the p-MSP-PEG was characterized by TEM and SEM (Fig. 1a and b). From these figures, we found that the p-MSP-PEG had uniform shape with a diameter of approximately 160 nm, and obvious mesoporous cavities existed in the particles. The porosity of p-MSP-PEG was investigated by the Brunauer–Emmett–Teller (BET) method (Fig. 1c). Its BET surface area was 165.6 m2 g−1 and the total pore volume was 0.45 cm3 g−1. The p-MSP-PEG also has the same acidic degradation ability as its precursor MSP, and can be quickly dissolved in pH = 5.0 buffer solution (Fig. 1d). The powder XRD results showed that the p-MSP-PEG still retained good crystallinity with high magnetization (67.4 emu g−1) and superparamagnetic properties (Fig. 1e and f).
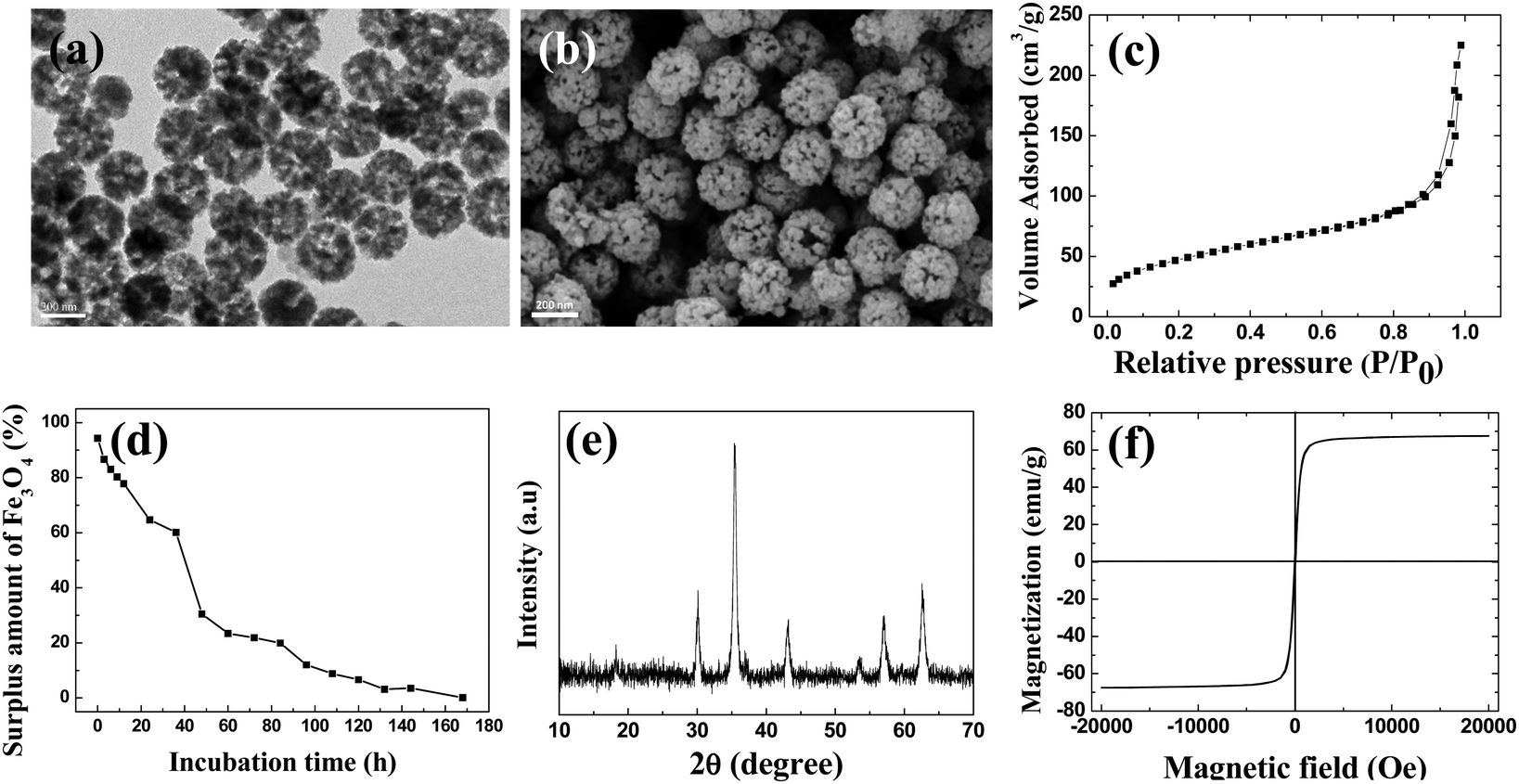 | ||
| Fig. 1 (a) TEM image and (b) SEM image of p-MSP-PEG carriers; (c) BET curve of p-MSP-PEG; (d) the surplus amount of p-MSP-PEG in solution from different incubation times in acidic buffer solution (pH = 5.0); (e) XRD spectrum of p-MSP-PEG; (f) VSM curve of p-MSP-PEG. | ||
Since the etching process does not influence the agarose chains, we could use 2,3-dimethylmaleic anhydride as a specific linker to connect agarose and anti-cancer drug DOX. The result was proved by FT-IR (Fig. S1a, ESI†), which showed that the peaks corresponding to the C–O bond in the 1050–1200 cm−1 range intensified after PEG modification, demonstrating that PEG was grafted onto the MSP successfully. When 2,3-dimethylmaleic anhydride was used as the acid-sensitive linker, DOX was covalently bonded to the surface of the p-MSP-PEG, and the emerging peak at 1720 cm−1 indicated the formation of an ester between agarose and the linker. Meanwhile, the new peak at 1580 cm−1 illustrated the formation of an ester bond between DOX and the linker, which was evidence to prove that DOX was loaded into p-MSP-PEG (DOX@p-MSP-PEG) successfully. To measure the loaded amount of DOX, TGA measurements were conducted (Fig. S1b, ESI†), and the drug loading amount was calculated to be about 25 wt%. It should be mentioned that the superficial agarose was mostly grafted by PEG, so the DOX ought to be loaded in the inner cavity of p-MSP-PEG due to the outer hydroxyl groups having previously reacted with PEG.
In order to improve the targeting ability of the DOX@p-MSP-PEG carriers, folate was bridge-linked to the DOX@p-MSP-PEG carriers by the inclusion complexation of PEG and folate modified α-CD. First, folate modified α-CD (α-CD-FA) was synthesized through a succession of synthetic steps as described in the Preparation section. After DOX@p-MSP-PEG was treated with α-CD-FA, we found the fluorescence spectrum of folate in the sample of DOX@p-MSP-PEG-FA (Fig. S2a, ESI†), illustrating that the folate had been grafted onto the nano-cargos as designed. Interestingly, when we put DOX@p-MSP-PEG-FA into α-CD solution and re-collected the nano-cargos from the solution, we found that the fluorescence of the nano-cargos disappeared, which resulted from the quick exchange when the DOX@p-MSP-PEG-FA encountered a large amount of free α-CD, and α-CD-FA could be quickly replaced by free α-CD due to the reversibility of the inclusion complexation between PEG and α-CD. If we put the above nano-cargos (after α-CD treatment) into α-CD-FA solution, the fluorescence of the nano-cargos appeared again. This result demonstrated that the α-CD-FA on the surface can be easily replaced by other α-CD derivatives, and also confirmed the flexible immobilization manner of the targeting molecules (Fig. S2b, ESI†). Therefore, p-MSP-PEG as a targeting drug delivery platform can be designed to load different targeting molecules using α-CD modified with different targeting agents.
As an acid responsive linker, the chemical bond of 2,3-dimethyl succinamide could control the release of DOX, as it quickly breaks up at low pH (Scheme 1c).27,38 To evaluate the acid responsive ability, we chose four pH values to investigate the drug release profile of DOX@p-MSP-PEG-FA (Fig. 2). At pH = 7.4, only a small amount of DOX was released from the nano-cargos, because the neutral medium could not break up the chemical linker easily. When the pH was shifted to 6.0, half of the DOX was released from the nano-cargos within 72 h. The more acidic the solution, the faster the DOX release, and over 90 wt% DOX could be released at pH = 4.0 over 72 h. This pH-sensitive release manner can be ascribed to the fact that, once the pH of the solution is shifted to 6.0, the 2,3-dimethyl succinamide bond can be hydrolyzed to restore the amino and carboxyl groups, and the anti-cancer drug DOX is released into the solution quickly. Meanwhile, with the decrease in pH, the p-MSP-PEG starts to degrade as well, so the released DOX in the pores can escape quickly from the nano-cargos. Therefore, DOX@p-MSP-PEG-FA as a drug delivery system can trap DOX tightly in neutral media such as plasma or normal tissues, while quickly releasing DOX at lower pH sites like cancer tissues.
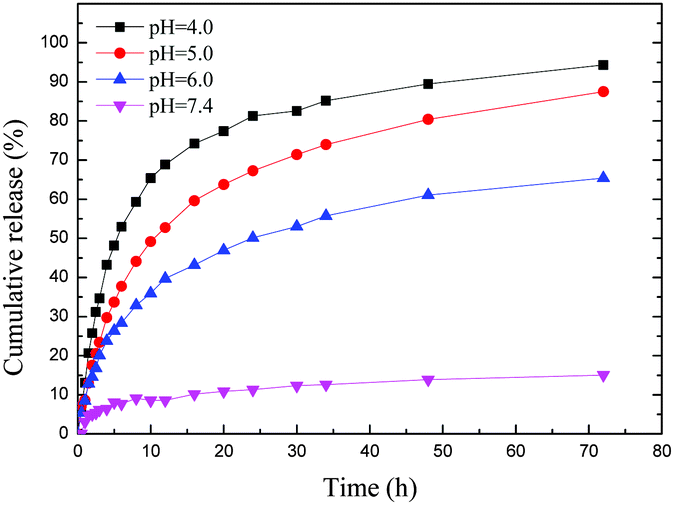 | ||
| Fig. 2 The DOX release profile of DOX@p-MSP-PEG-FA in different pH buffer solutions. | ||
Cell uptake experiments for different nano-cargos
In order to test the targeting properties, the in vitro cell uptake behavior of DOX@p-MSP-PEG with folate on the surface was investigated. When HeLa cells were incubated with DOX@p-MSP-PEG-FA for 2.5 h, the strong fluorescence of DOX could be detected by confocal laser scanning microscopy (CLSM), illustrating that the DOX@p-MSP-PEG-FA nano-cargos had been internalized by the HeLa cells (Fig. S3, ESI†). Meanwhile, when we put the free folate in the incubated solution, the fluorescence of DOX in the HeLa cells became weaker over the same incubation period, because the free folate competed against the DOX@p-MSP-PEG-FA nano-cargos (Fig. 3a) for the folate acceptors. Interestingly, when α-CD was added to the incubated solution, the fluorescence of DOX in the HeLa cells became weaker too, which demonstrated that the targeting ability of the nano-cargos was influenced by the replacement of α-CD-FA by α-CD. As mentioned before, folate was fixed on the surface of the nano-cargos by inclusion complexation, and could be replaced when the free α-CD entered the incubated solution, which decreased the amount of α-CD-FA on the surface of the magnetic nano-cargos. Meanwhile, increasing the amount of free folate in the incubated solution could also reduce the internalization of the nano-cargos by the HeLa cells for similar reasons.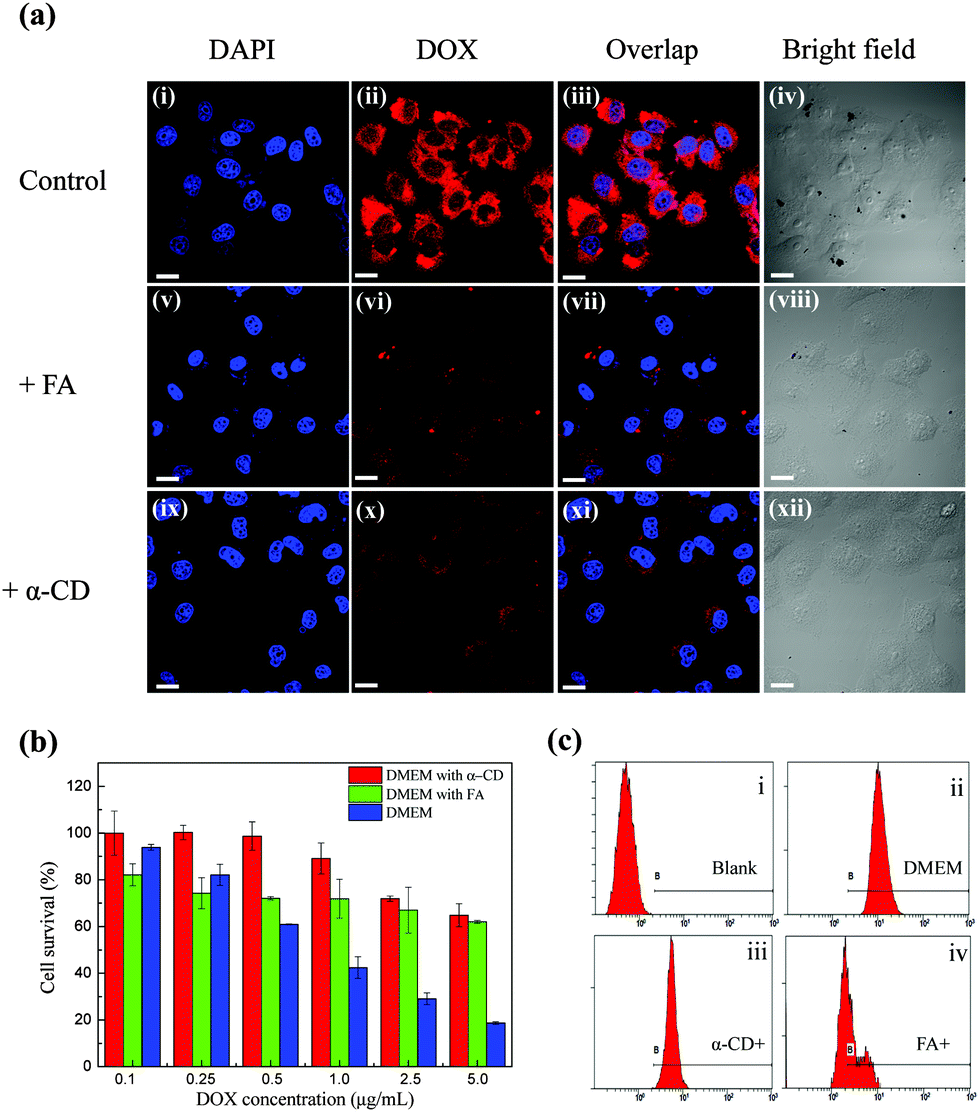 | ||
| Fig. 3 (a) CLSM images of HeLa cells incubated with (i–iv) DOX@p-MSP-PEG-FA dispersion, (v–viii) DOX@p-MSP-PEG-FA + folate dispersion and (ix–xii) DOX@p-MSP-PEG-FA + α-CD dispersion. All scale bars are 20 μm; (b) the cell survival of HeLa cells at different concentrations of DOX@p-MSP-PEG-FA in DMEM, DMEM + free folate and DMEM + free α-CD after 24 h incubation; (c) flow cytometer graphs of HeLa cells incubated in (i) DMEM + 2 μg mL−1 DOX@p-MSP-PEG-FA, (ii) DMEM, (iii) DMEM + 1 μg mL−1 α-CD, and (iv) DMEM + 1 μg mL−1 folate. The excitation wavelength is 520 nm. | ||
Moreover, the flow cytometer (FCM) results further confirmed that the efficacy of DOX delivered to cells decreased with the addition of free folate or α-CD to the incubated solution (Fig. 3c). The lower drug delivery efficacy could reduce the inhibitive effects on cancer cells (Fig. 3b). From the above results, we found that the FCM coincided with the CLSM results very well. The surface-anchored folate could induce quick cell uptake, which matches the original design for the targeting drug delivery system.
Drug release properties of different kinds of DOX-loaded nano-cargos in HeLa cells
Following cellular internalization, another key issue is whether the covalently conjugated DOX molecules can be efficiently released. As an experimental control, we synthesized two other types of DOX@p-MSP-PEG-FA with different linkers (Fig. S4, ESI†), in which 2,3-dimethylmaleic anhydride (DMMA) was replaced with succinic anhydride (SA) and tetrahydrophthalic anhydride (THPA). For the cell uptake test, each of the samples was incubated with HeLa cells at the same DOX dosage. According to the CLSM results (Fig. 4a), strong red fluorescence was detected in HeLa cells with the DMMA-DOX sample after 24 h incubation, demonstrating that DOX could rapidly escape from the nano-cargos into the cytoplasm. In sharp contrast, the SA-DOX and THPA-DOX samples showed very low fluorescence intensities in cells, which implied that the DOX could not be released from the nano-cargos due to the non-acid-sensitive bonds. Compared with the linker constructed by DMMA, the linkers constructed by SA or THPA did not show obvious acid-responsive behaviour. Even in the acidic cellular environment, DOX could not break free of the linkers and escape into the cytoplasm. The weak fluorescence intensity observed in the SA-DOX and THPA-DOX samples after 24 h incubation can be attributed to the stability of the p-MSP, which could only degrade gradually under such conditions. | ||
| Fig. 4 (a) CLSM images of HeLa cells incubated with (i–iv) DOX@p-MSP-PEG-FA in DMEM, (v–viii) SA-DOX@p-MSP-PEG-FA in DMEM and (ix–xii) THPA-DOX@p-MSP-PEG-FA in DMEM for 24 h. All scale bars are 20 μm; (b) the cell survival of HeLa cells at different concentrations of THPA-DOX@p-MSP-PEG-FA (THPA-DOX), SA-DOX@p-MSP-PEG-FA (SA-DOX) and DMMA-DOX@p-MSP-PEG-FA (DMMA-DOX) after 24 h incubation; (c) flow cytometer graphs of HeLa cells incubated in (i) DMEM, (ii) DMEM + 2 μg mL−1 DMMA-DOX, (iii) DMEM + 2 μg mL−1 THPA-DOX and (iv) DMEM + 2 μg mL−1 SA-DOX. The excitation wavelength is 520 nm. | ||
The DMMA-DOX sample (DMMA-DOX@p-MSP-PEG-FA) showed the best inhibitive efficacy compared to the other two types of nano-cargos in the cytotoxicity results (Fig. 4b). Combined with the FCM data in Fig. 4c, we concluded that the better cytotoxicity of DMMA-DOX@p-MSP-PEG-FA could be attributed to the quick release of DOX in the targeted cells.
Proliferation inhibition of different types of cells by DOX-loaded nano-cargos
Due to the rapid cell uptake in the targeted cells and the controlled drug release ability, DOX@p-MSP-PEG-FA should exhibit excellent proliferation inhibition of the targeted cells.27 To verify the effectiveness of DOX@p-MSP-PEG-FA for selectively inhibiting specific cells, we utilized two typical types of cells, HeLa cells and HEK 293T cells. The HeLa cells represented model cells with over-expressing folate acceptors and an acidic cellular environment, while the HEK 293T cells represented normal cells without folate acceptors and with a neutral cellular environment.38,46 After the DOX@p-MSP-PEG-FA was incubated with these two types of cells for 24 h, the CLSM results showed strong fluorescence in HeLa cells but weak fluorescence in HEK 293T cells (Fig. S5, ESI†). For further statistical and quantitative analysis, FCM measurements were carried out (Fig. S6, ESI†). Compared with the same dosage of free DOX, the DOX@p-MSP-PEG-FA showed a similar fluorescence intensity in HeLa cells throughout the whole incubation time, but this was obviously different in HEK 293T cells. This demonstrated that DOX@p-MSP-PEG-FA exhibited different behaviour when it encountered different cells. The intensity of DOX fluorescence in HEK 293T cells increased slowly with prolonged incubation time; this result could be ascribed to the slow cellular uptake rate and limited DOX release in the cytoplasm, which would be preferable for normal cells during chemotherapy of tumor cells.Meanwhile, the CCK-8 assay was used to verify the in vitro cytotoxicity of DOX@p-MSP-PEG-FA towards HeLa and HEK 293T cells, and the results are shown in Fig. 5. At first, DOX@p-MSP-PEG-FA showed similar cell proliferation inhibition of HeLa cells compared with free DOX. When the incubation time was prolonged over 48 h, the cell proliferation inhibition of DOX@p-MSP-PEG-FA surmounted that of free DOX, demonstrating that more nano-cargos were internalized and more DOX was delivered into the cells. However, compared with free DOX, DOX@p-MSP-PEG-FA showed little cell proliferation inhibition of HEK 293T cells across the whole range of drug dosage and incubation time, which matched the former measurements very well. Taken together, it can be concluded that the DOX@p-MSP-PEG-FA drug delivery system not only enhances cell proliferation inhibition of folate-acceptor over-expressing cancer cells, but also reduces the side effects on normal cells.
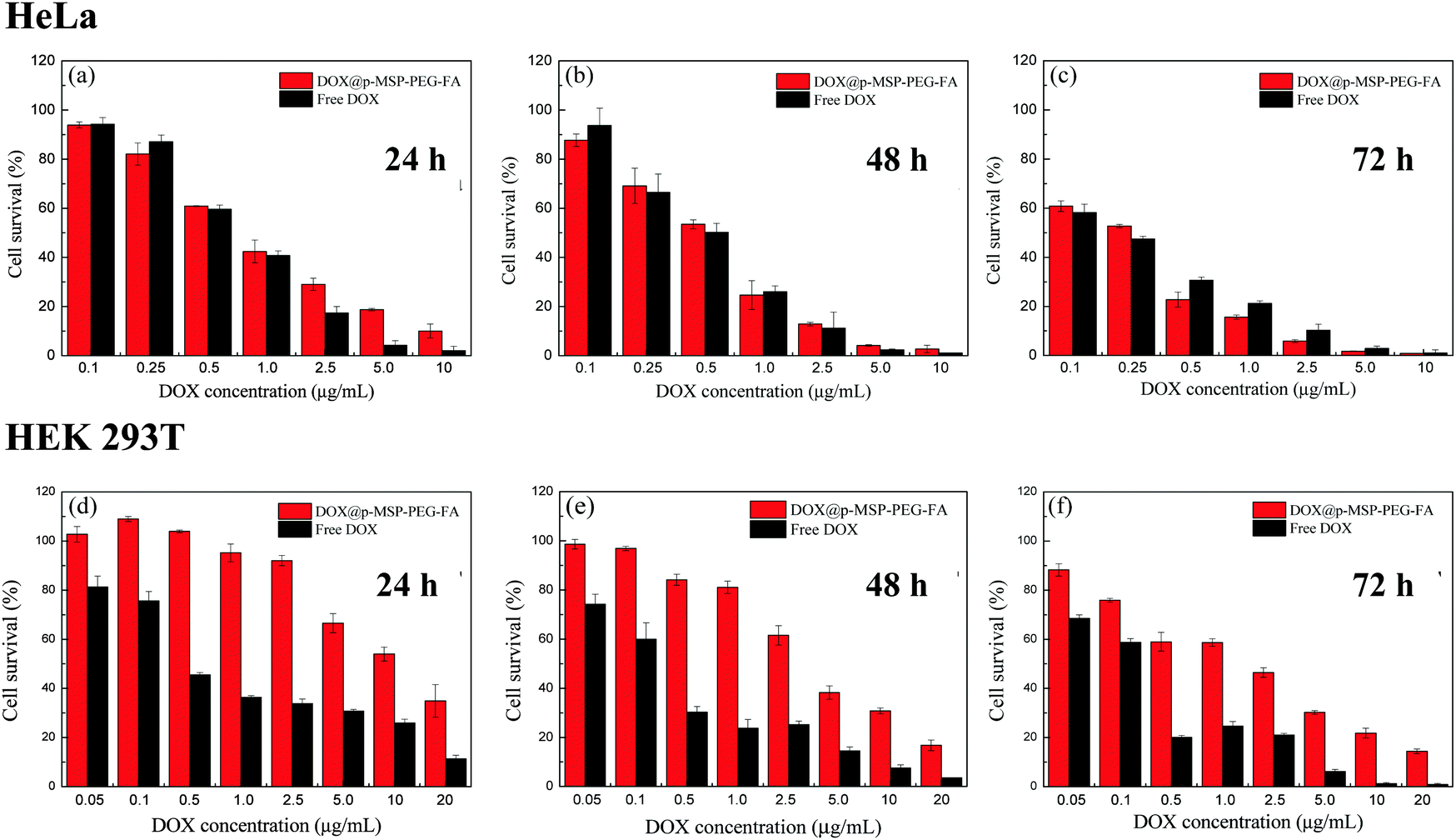 | ||
| Fig. 5 Survival of HeLa cells at eight different concentrations of DOX@p-MSP-PEG-FA and free DOX after incubation for (a) 24 h, (b) 48 h and (c) 72 h; survival of HEK 293T cells at eight different concentrations of DOX@p-MSP-PEG-FA and free DOX after incubation for (d) 24 h, (e) 48 h and (f) 72 h. | ||
Conclusions
This work demonstrated the fabrication of a new type of anti-cancer drug delivery system (nano-cargo) on the basis of flexible immobilization of the targeting agent by inclusion complexation. The nano-cargos provide a large inner surface for the covalent conjugation of DOX, while having a PEG-modified shell for anchoring of α-CD-FA by inclusion complexation, which can easily modulate the targeting agent amount and type as required. The superficial FA endowed the magnetic nano-cargos with rapid cellular uptake in folate-acceptor over-expressing cells. With pH sensitive linkers between agarose and DOX, the drug-loaded magnetic nano-cargos showed enhanced proliferation inhibition to HeLa cells, while low cytotoxicity was exhibited towards HEK 293T cells. The synergistic effect of the targeting and controlled release capabilities of the nano-cargos showed robust potential for actual tumor chemotherapy.Acknowledgements
This work was supported by the National Science Foundation of China (Grant No. 21474017), State Key Project of Research and Development (Grant No. 2016YFC1100300) and the Science and Technology Commission of Shanghai (Grant No. 13JC1400500). D. L. and L. S. contributed equally to this work.Notes and references
- C. L. Weaver, J. M. LaRosa, X. L. Luo and X. T. Cui, ACS Nano, 2014, 8(2), 1834 CrossRef CAS PubMed.
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872 CrossRef CAS PubMed.
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16 CrossRef CAS PubMed.
- V. P. Torchilin, Pharm. Res., 2006, 24, 1 CrossRef PubMed.
- K. Cheng, S. Peng, C. J. Xu and S. H. Sun, J. Am. Chem. Soc., 2009, 131, 10637 CrossRef CAS PubMed.
- D. Li, J. Tang, J. Guo, S. L. Wang, D. Chaudhary and C. C. Wang, Chem. – Eur. J., 2012, 18, 16517 CrossRef CAS PubMed.
- Q. L. S. Fang, H. Xuan, W. Q. Jiang and X. L. Gong, Adv. Funct. Mater., 2011, 21, 1902 CrossRef CAS.
- P. Yang, F. Wang, X. F. Luo, Y. T. Zhang, J. Guo, W. B. Shi and C. C. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 12581 CAS.
- B. Luo, S. Xu, A. Luo, W. R. Wang, S. L. Wang, J. Guo, Y. Lin, D. Y. Zhao and C. C. Wang, ACS Nano, 2011, 5, 1428 CrossRef CAS PubMed.
- D. Li, J. Tang, C. Wei, J. Guo, S. L. Wang, D. Chaudhary and C. C. Wang, Small, 2012, 8, 2690–2698 CrossRef CAS PubMed.
- H. J. Qiu, B. Cui, G. M. Li, J. H. Yang, H. X. Peng, Y. S. Wang, N. N. Li, R. C. Gao, Z. G. Chang and Y. Y. Wang, J. Phys. Chem. C, 2014, 118, 14929 CAS.
- D. Li, Y. T. Zhang, S. Jin, J. Guo, H. F. Gao and C. C. Wang, J. Mater. Chem. B, 2014, 2, 5187 RSC.
- D. Li, Y. T. Zhang, M. Yu, Q. An, J. Guo, J. Q. Lu and C. C. Wang, Chem. Commun., 2015, 51, 1908 RSC.
- T. Etrych, M. Jelinkova, B. Rihova and K. Ulbrich, J. Controlled Release, 2001, 73, 89–102 CrossRef CAS PubMed.
- K. Ulbrich and V. Subr, Adv. Drug Delivery Rev., 2004, 56, 1023 CrossRef CAS PubMed.
- J. W. Cui, Y. Yan, Y. J. Wang and F. Caruso, Adv. Funct. Mater., 2012, 22, 4718–4723 CrossRef CAS.
- Q. Zhang, X. L. Wang, P. Z. Li, K. T. Nguyen, X. J. Wang, Z. Luo, H. C. Zhang, N. S. Tan and Y. L. Zhao, Adv. Funct. Mater., 2014, 24, 2450–2461 CrossRef CAS.
- H. Meng, M. Xue, T. Xia, Y. L. Zhao, F. J. Tamanoi, F. Stoddart, J. I. Zink and A. E. Nel, J. Am. Chem. Soc., 2010, 132, 12690 CrossRef CAS PubMed.
- C. Park, K. Lee and C. Kim, Angew. Chem., Int. Ed., 2009, 48, 1275 CrossRef CAS PubMed.
- A. Schlossbauer, J. Kecht and T. Bein, Angew. Chem., Int. Ed., 2009, 48, 3092 CrossRef CAS PubMed.
- R. Tang, C. S. Kim, D. J. Solfiell, S. Rana, R. Mout, E. M. Velázquez-Delgado, A. Chompoosor, Y. Jeong, B. Yan, Z. J. Zhu, C. Kim, J. A. Hardy and V. M. Rotello, ACS Nano, 2013, 7, 6667 CrossRef CAS PubMed.
- Y. J. Pan, Y. Y. Chen, D. R. Wang, C. Wei, J. Guo, D. R. Lu, C. C. Chu and C. C. Wang, Biomaterials, 2012, 33, 6570 CrossRef CAS PubMed.
- J. Z. Du, T. M. Sun, W. J. Song, J. Wu and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 3621 CrossRef CAS PubMed.
- Q. Zhang, F. Liu, K. T. Nguyen, X. Ma, X. J. Wang and B. G. Xing, Adv. Funct. Mater., 2012, 22, 5144 CrossRef CAS.
- D. Li, Y. T. Zhang, P. Yang, M. Yu, J. Guo, J. Q. Lu and C. C. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 12329 CAS.
- J. Z. Du, X. J. Du, C. Q. Mao and J. Wang, J. Am. Chem. Soc., 2011, 133, 17560 CrossRef CAS PubMed.
- P. S. Low, W. A. Henne and D. D. Doorneweerd, Acc. Chem. Res., 2008, 41, 120 CrossRef CAS PubMed.
- Y. A. Zhong, F. H. Meng, C. Deng and Z. Y. Zhong, Biomacromolecules, 2014, 15, 1955 CrossRef CAS PubMed.
- S. E. Baek, K. H. Lee, Y. S. Park, D. K. Oh, S. Oh, K. S. Kim and D. E. Kim, J. Controlled Release, 2014, 196, 234 CrossRef CAS PubMed.
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2012, 64, 24 CrossRef.
- Y. Y. Yuan, C. Q. Mao, X. J. Du, J. Z. Du, F. Wang and J. Wang, Adv. Mater., 2012, 24, 5476 CrossRef CAS PubMed.
- E. Wagner, Expert Opin. Biol. Ther., 2007, 7, 587 CrossRef CAS PubMed.
- J. F. Stefanick, J. D. Ashley and B. Bilgicer, ACS Nano, 2013, 7, 8115 CrossRef CAS PubMed.
- J. Ahn, Y. Miura, N. Yamada, T. Chida, X. Y. Liu, A. Kim, R. Sato, R. Tsumura, Y. Koga and M. Yasunaga, Biomaterials, 2015, 39, 23 CrossRef CAS PubMed.
- S. S. Oh, B. F. Lee, F. A. Leibfarth and H. T. Soh, J. Am. Chem. Soc., 2014, 136, 15010 CrossRef CAS PubMed.
- R. W. Wang, G. Z. Zhu, L. Mei, Y. Xie, H. B. Ma, M. Ye, F. L. Qing and W. H. Tan, J. Am. Chem. Soc., 2014, 136, 2731 CrossRef CAS PubMed.
- D. Li, Y. T. Zhang, M. Yu, J. Guo, D. Chaudhary and C. C. Wang, Biomaterials, 2013, 34, 7913 CrossRef CAS PubMed.
- E. Vlashi, L. E. Kelderhouse, J. E. Sturgis and P. S. Low, ACS Nano, 2013, 7, 8573 CrossRef CAS PubMed.
- Z. Wang and Y. Chen, Macromolecules, 2007, 40, 3402–3407 CrossRef CAS.
- M. Guo, M. Jiang, S. Pispas, W. Yu and C. Zhou, Macromolecules, 2008, 41, 9744–9749 CrossRef CAS.
- G. S. Chen and M. Jiang, Chem. Soc. Rev., 2011, 40, 2254 RSC.
- X. Liao, G. Chen, X. Liu, W. Chen, F. Chen and M. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4409 CrossRef CAS PubMed.
- J. Liu, G. Chen, M. Guo and M. Jiang, Macromolecules, 2010, 43, 8086 CrossRef CAS.
- A. R. Khan, P. Forgo, K. J. Stine and V. T. D'Souza, Chem. Rev., 1998, 98, 1977 CrossRef CAS PubMed.
- D. Li, Y. T. Zhang, R. M. Li, J. Guo, C. C. Wang and C. B. Tang, Small, 2015, 11(18), 2200 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00049e |
| This journal is © the Partner Organisations 2017 |