DOI:
10.1039/C6QM00031B
(Research Article)
Mater. Chem. Front., 2017,
1, 512-520
Triphenylamine-based donor–π–acceptor organic phosphors: synthesis, characterization and theoretical study†
Received
25th April 2016
, Accepted 24th July 2016
First published on 7th September 2016
Abstract
We have designed and synthesized donor (D) and acceptor (A) phosphors [D and A refer to the electron-donating and electron-withdrawing moieties, respectively] as yellow-emitting organic phosphors. The photophysical and electrochemical properties of the phosphors have been studied in detail. The UV-vis spectra of the organic phosphors show multiple absorption bands (UV to near UV region, due to π–π* transitions of the conjugated chains), and all the phosphors show yellowish-green/yellow (556, 576, 594 nm) emission with appropriate Commission Internationale de I'Eclairage (CIE) color coordinate values. In contrast, a bathochromic shift was observed in the solid-state emission spectra for the one donor–two acceptors and one donor–three acceptors due to the π–π interaction and aggregation induced emission. Electrochemical studies were carried out to find the HOMO, LUMO and band gap of the phosphors, and these were verified by density functional theory (DFT) calculations. By using time dependent-DFT (TD-DFT) calculations, singlet and triplet energy levels were also calculated. In addition, solvatochromism studies were also carried out and the results were interpreted using the Lippert–Mataga equation. White light can be realized from the currently synthesized organic yellow phosphors integrated with blue LEDs or blue fluorophores.
Introduction
Phosphors are active components in light-emitting diodes (LEDs), and have attracted widespread interest over the decades. White LED & OLED (WLEDs, solid state lighting) technology has advanced the trading-in of existing less efficient and environmentally benign technologies such as incandescent and fluorescent lamps.1 In comparison with traditional lighting, the InGaN-based WLEDs have many advantages in energy efficiency, long lifetime, compactness, environment friendliness and designable features.2 The wavelength converting phosphor materials play a vital role for the creation of white light from LEDs (solid-state lighting). Several approaches are available to mimic white light from LEDs: first, a combination of an InGaN-based blue LED chip with a yellow phosphor (e.g., YAG:Ce3+-based materials); second, a blue LED chip combined with a green and a red emitting phosphor instead of single yellow emitting phosphor.3,4 These two phosphors absorb partially blue light from the InGaN chip and convert into green and red photons and produce white light by complementary color mixing. Research efforts have been directed for over a decade on the improvement of existing convertors or to find new wavelength convertors for potential applications in WLEDs. The concept of using remote phosphors is exciting, i.e. the combination of the near UV or blue LED chip and a RBG or yellow phosphor at an appropriate distance.5 This technology (remote-type phosphor-LEDs) will help to overcome some high-tech limitations including thermal quenching of organic phosphors, light extraction efficiency and light distribution within remote-type designed packaging. However, most of the efforts have been faithful to inorganic phosphors such as YAG:Ce3+, M2Si5N8 (M = Ca, Sr, Ba), Sr[LiAl3N4]:Eu2+, BaLa2Si2S8:Eu2+, K2TiF6:Mn4+ and Ca5(SiO4)2F2:Eu2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6,7, in which the key concepts are yet to be improved for practical applications, i.e. high cost, difficulties in color tuning and non-uniform dispersion.8 In contrast, organic color conversion phosphor materials offer significant advantages, such as low manufacturing cost and structural flexibility, but their use as white light sources is limited by their generally lower efficiency when compared with their inorganic LED counterparts. One of the interesting paths to develop WLEDs is through a hybrid inorganic/organic LED architecture, where a blue emissive inorganic LED is coated with an organic material that has an absorbance band aligned with the emission wavelength of the inorganic LED structure.9–11 The organic phosphor material acts as an energy down-converter for the inorganic LEDs, converting some of the emitted high energy blue photons to lower energy yellowish red light. The mixing of both blue and yellowish red delivers a high quality white output (warm white light). The material system of choice for the inorganic LED is the III-nitride alloy system, which can emit light from the ultraviolet (UV), through the visible to the infrared spectrum.12 Such hybrid inorganic/organic LED architectures offer the potential to combine the advantages of both technologies, for example, the excellent electronic properties of inorganic substrates and the broad, tunable emission of organic phosphors.13–16 Additionally, employing an organic phosphor material in place of traditional phosphors in hybrid LEDs would avoid an industry dependency on costly rare-earth containing materials for the development of solid-state lighting.17 The higher response speed of the organic materials compared to the existing phosphors offers additional advantages which are used in visible light communications.18,19 The design and synthesis of highly efficient organic phosphors are key determinants in the performance of the hybrid white LEDs. Organic phosphors typically possess strong absorptions in the near UV to blue region (where the LED emission occurs), which are caused by π–π* transitions. It is possible to tune the emission color gamut by changing the molecular design and modifying the structure of the organic phosphors.20
6,7, in which the key concepts are yet to be improved for practical applications, i.e. high cost, difficulties in color tuning and non-uniform dispersion.8 In contrast, organic color conversion phosphor materials offer significant advantages, such as low manufacturing cost and structural flexibility, but their use as white light sources is limited by their generally lower efficiency when compared with their inorganic LED counterparts. One of the interesting paths to develop WLEDs is through a hybrid inorganic/organic LED architecture, where a blue emissive inorganic LED is coated with an organic material that has an absorbance band aligned with the emission wavelength of the inorganic LED structure.9–11 The organic phosphor material acts as an energy down-converter for the inorganic LEDs, converting some of the emitted high energy blue photons to lower energy yellowish red light. The mixing of both blue and yellowish red delivers a high quality white output (warm white light). The material system of choice for the inorganic LED is the III-nitride alloy system, which can emit light from the ultraviolet (UV), through the visible to the infrared spectrum.12 Such hybrid inorganic/organic LED architectures offer the potential to combine the advantages of both technologies, for example, the excellent electronic properties of inorganic substrates and the broad, tunable emission of organic phosphors.13–16 Additionally, employing an organic phosphor material in place of traditional phosphors in hybrid LEDs would avoid an industry dependency on costly rare-earth containing materials for the development of solid-state lighting.17 The higher response speed of the organic materials compared to the existing phosphors offers additional advantages which are used in visible light communications.18,19 The design and synthesis of highly efficient organic phosphors are key determinants in the performance of the hybrid white LEDs. Organic phosphors typically possess strong absorptions in the near UV to blue region (where the LED emission occurs), which are caused by π–π* transitions. It is possible to tune the emission color gamut by changing the molecular design and modifying the structure of the organic phosphors.20
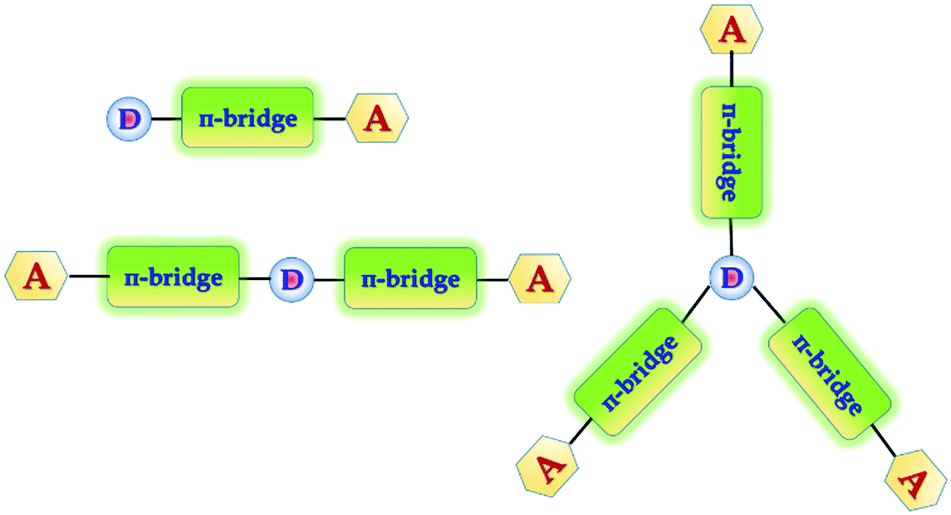
In general, white emission in OLEDs is typically generated from multiple emitters to cover a wide spectral window, e.g. the whole visible region from 380 to 680 nm. Recent prototypes of WOLEDs have even benefited from favorable features, such as homogenous large-area emission, and being lightweight and ultra-thin. In the case of lighting applications, WOLEDs are expected to achieve low energy consumption, long lifetimes and low cost to compete with other light sources such as fluorescent tubes and inorganic LEDs. In order to improve the efficiency as well as the color purity of the WOLEDs so they can be considered for commercial applications, a lot of effort has been made, which includes more emphasis on developing efficient and stable materials. To date, the three primary color strategy (mixing of RGB) is superior because it is easier to reach white light with Commission Internationale de I'Eclairage (CIE) coordinates, due to simultaneous presence of the three primary colors. However, the efficiency of devices with primary colors is low due to high operational voltages and unexpected spectral variations due to a recombination zone shift.21 In general, aggregation induced emission (AIE) is another process which is associated with chromophore aggregation.22 In this process weakly luminescent chromophores are induced to emit efficiently in the aggregated state.23 This phenomenon is useful towards efficient solid-state light emitters, bioprobes, chemosensors, and smart materials.22 To further improve the efficiencies of the devices as well as the color purity, it is necessary to develop novel blue and yellowish-orange emitters with high luminescence efficiencies. In order to accomplish the above stated advantages, we have designed and synthesized a group of 1-phenylethanone decorated triphenylamine conjugated organic phosphors for white OLEDs. In general, most of the sensitizers that have been designed and developed have a D–π-bridge–A dyad structure.24 Triphenylamine, coumarin, and indoline25 are examples of electron donating building blocks that provide high stability and are suitable for device fabrication. Triphenylamine is used as the molecule core to construct branched molecules due to its excellent electron donating ability (D) and variable star-shaped structure.26 In spite of the large availability of donating groups, the selection of acceptors remains fairly short. During our search for acceptor structures, we came across a previous report on dibenz[a,c]phenazine (DBP) derivatives,27 which demonstrated their high light absorptivity and their suitable frontier orbital energy levels for application in dye sensitized solar cells (DSSCs). The systems with extended π-electrons and small reorganization energy provide a good route to enhance charge separation lifetimes, and DBP is an interesting choice as a fused D–A core for organic phosphors. Keeping this in mind we have designed and synthesized one donor–one acceptor (D–π-bridge–A), one donor–two acceptors (A–π-bridge–D–π-bridge–A) and one donor–three acceptors (D–π-bridge–A(–π-bridge–D)–π-bridge–D) groups, where D and A refer to the electron-donating and electron-withdrawing moieties, respectively. Therefore, a target molecule with high intense light emission under near UV excitation is expected (Fig. 1).
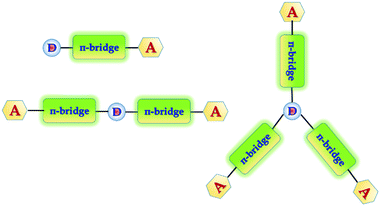 |
| | Fig. 1 Designed donor–(π-bridge)–acceptor molecules. | |
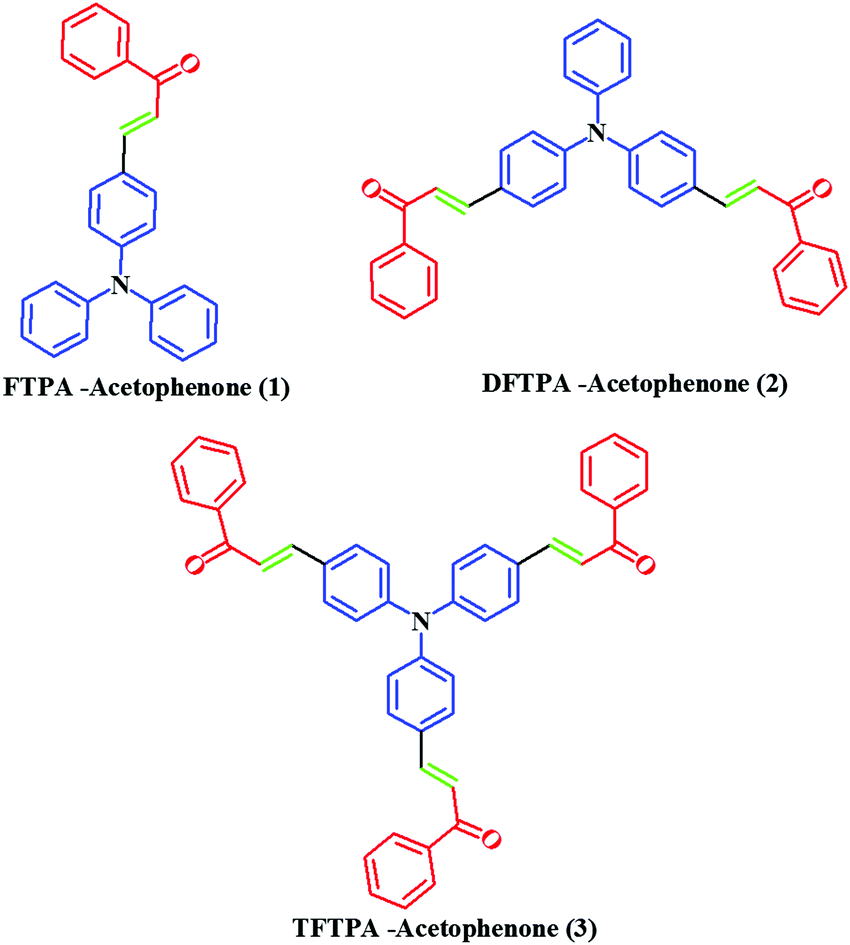
In this study, we report on the synthesis and characterization of triphenylamine-based phosphors with multiple electron acceptors of 1-phenylethanone. In the molecular design, (E)-3-(4-(diphenylamino)phenyl)-1-phenylprop-2-en-1-one (FTPA-acetophenone = formyl triphenylamine-acetophenone) is the basic model,32 in which the TPA unit was connected to one molecule of 1-phenylethanone. In the second case, 1-phenylethanone was introduced to the adjacent phenyl ring of FTPA, to obtain DFTPA-acetophenone (diformyl triphenylamine-acetophenone) that has two electron acceptors. Similarly, in the third case 1-phenylethanone was attached to the third phenyl ring of FTPA, yielding the structure of TFTPA-acetophenone (triformyl triphenylamine-acetophenone). The introduction of multiple electron acceptors to the adjacent phenyl ring of TPA-based phosphors would have a significant influence on the efficiency and brightness of the LEDs. The phosphors were characterized by proton and carbon NMR, DTA-TGA analysis and powder X-ray diffraction. The photophysical studies were also carried out by using UV-visible and photoluminescence spectroscopy (including solvatochromism), and verified the spectral data theoretically (density functional theory (DFT) and TD-DFT). The electrochemical studies were carried out to find the energy gap (HOMO–LUMO gap), and were also verified by DFT calculations. From the emission spectral values the Commission International del'Eclairage (CIE) values were also calculated by using MATLAB software (Fig. 2).
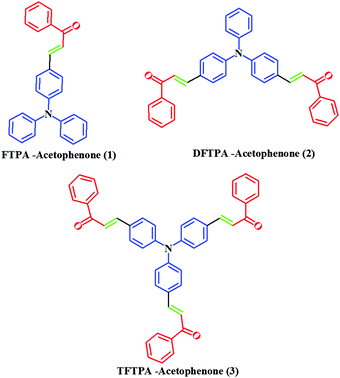 |
| | Fig. 2 Chemical structure of TPA-substituted acetophenones. | |
Experimental section
General information
1H NMR spectra were measured on a BRUKER AV 400 Avance-III (400 MHz) instrument with tetramethylsilane as the internal standard. The absorption and photoluminescence excitation and emission spectra of the target compound were measured using a SHIMADZU UV-2450 spectrophotometer and a HORIBA FLUOROMAX – 4P spectrophotometer, respectively. The absolute quantum yields of the synthesized phosphors were determined by using quinine sulfate in 1 M H2SO4 (Φ = 0.546). Thermogravimetric analysis (TGA) was performed on a NETZSCH – Germany thermal analyzer under a nitrogen atmosphere at a heating rate of 10 °C min−1. Cyclic voltammetry experiments were performed in dimethylformamide solution containing 0.1 M tert-butylammonium perchlorate using Ag/AgCl as the reference electrode at a scan rate of 100 mV s−1 using an AUTOLAB 302N Modular potentiostat. Elemental analysis of the synthesized phosphors was measured by using an Elementar Analysen Systeme, Germany/Vario EL instrument. The CIE color chromaticity coordinates of the phosphor were calculated from the emission spectral values by using MATLAB software. X-ray powder diffraction (XRD) spectra of the phosphors were measured using Cu-Kα1 radiation (Rigaku, ULTIMA IV) in the 2θ range from 5 to 60°. All the measurements were carried out at room temperature (RT).
Computational details
The molecules under study were first optimized in the gas phase using density functional theory and the Becke three parameter Lee–Yang–Parr (B3LYP)28 form for the exchange–correlation potential and the 6-31G(d,p) basis set. All the structures were found to be in the minima of the potential energy surface as the normal mode of frequencies were all positive. After that we performed the UV-vis spectra calculations using time dependent density functional theory (TD-DFT)29 with gas phase optimized geometries. It is expected that the geometry of the molecule will change in the solvent phase in comparison to that of the gas phase, and hence the optimization of the geometries in the solvent phase has also been carried out. We found that there is no such prominent change in the geometries of the molecules. As the UV-vis experiment was done in the solution phase, we also calculated the UV-vis spectra in the solvent phase by using the polarizable continuum model (PCM)30 approach within the TD-DFT methodology. Singlet and triplet energy calculations were performed by using the TD-SCF and B3LYP/6-31G(P) approach. All calculations were carried out using the Gaussian09 W31 and GaussianView suite of programs.
Materials
All reagents were used as purchased without any further purification. All operations involving air-sensitive reagents were performed under a dry nitrogen atmosphere. 4-Formyltriphenylamine (FTPA), 4,4′-diformyltriphenylamine (DFTPA) and 4,4′,4′′-triformyltriphenylamine (TFTPA) were prepared according to literature procedures,27 and the target organic phosphors were synthesized by using the aldol condensation procedure.32 All the phosphor materials are soluble in common organic solvents, such as toluene, tetrahydrofuran (THF), dichloromethane (DCM), chloroform, acetonitrile, acetone, N,N-dimethylformamide (DMF) and methanol (MeOH), but are insoluble in water.
Synthesis of FTPA.
In a clean-dried two neck round bottom flask dimethylformamide (DMF) (1.71 mL) was taken and added dropwise to phosphorous oxychloride (2.05 mL, 22.04 mmol) at 5 °C. After 30 min, triphenylamine (TPA) (2 g, 8.163 mmol) in DMF 22 (15 mL) was added dropwise to the reaction mixture at the same temperature. The resulting mixture was stirred overnight at RT, then poured into water and neutralized with diluted sodium hydroxide solution (up to pH 6), then extracted with chloroform and washed with brine solution followed by drying with sodium sulphate. The crude product was washed with a diethyl ether:hexane (3:7) mixture solvent and recrystallized in ethanol to obtain a pure (90%) pale yellow colored fine solid. 1H NMR data (CDCl3, 400 MHz): δ = 9.82 (s, 1H), 7.70 (d, 2H), 7.38–7.34 (m, 2H), 7.25–7.24 (m, 2H), 7.20–7.15 (m, 6H), 7.03 (d, 2H). 13C NMR data (CDCl3, 100 MHz): δ = 153.3, 146.1, 131.3, 129.7, 129.0, 126.3, 125.1, 119.3.
Synthesis of DFTPA.
Phosphorus oxychloride (3.74 mL, 39.98 mmol) was added dropwise at 0 °C under N2 to DMF (4.08 mL, 27.95 mmol) and the reaction mixture was stirred for 1 h. Triphenylamine (1 g, 4.0816 mmol) was added, and the resulting mixture was stirred at 105 °C for 4 h. After cooling to RT, the mixture was poured into ice water slowly, whereby a brown solid precipitated out. The solid was filtered, and washed with water. The crude product was purified by column chromatography (CH2Cl2:Hex = 1:3) to yield (81%) a yellow solid. 1H NMR data (CDCl3, 400 MHz): δ = 9.90 (s, 2H), 7.77–7.80 (d, 4H), 7.39–7.43 (t, 2H), 7.25–7.29 (t, 1H), 7.20–7.21 (d, 6H). 13C NMR data (CDCl3, 100 MHz): δ = 190.56, 151.20, 132.56, 131.53, 131.34, 128.80, 127.09, 124.54, 122.77.
Synthesis of TFTPA.
Phosphorus oxychloride (8.01 mL, 85.71 mmol) was added dropwise at 0 °C under N2 to DMF (4.69 mL, 60.44 mmol) and the reaction mixture was stirred for 1 h. Triphenylamine (1 g, 4.0816 mmol) was added, and the resulting mixture was stirred at 105 °C for 4 h. After cooling to RT, the mixture was poured into ice water slowly, whereby a brown solid precipitated out. The solid was filtered, and washed with water. The crude product was purified by column chromatography (CH2Cl2:Hex = 1:3) to yield (38%) a bright yellow solid. 1H NMR data: δ = 9.96 (s, 3H), 7.77–7.87 (d, 6H), 7.14–7.277 (d, 6H). 13C NMR data (CDCl3, 100 MHz): δ = 191.31, 152.14, 133.73, 132.04, 125.48.
Synthesis of phosphor material.
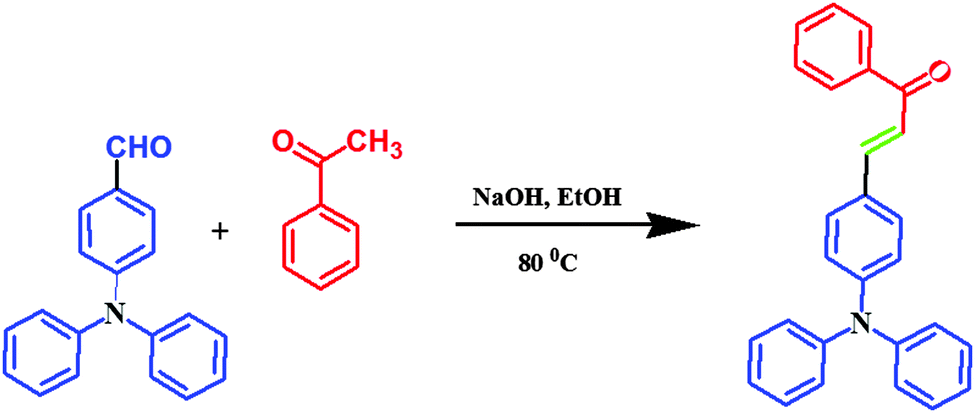
Aldehyde (1.831 mmol, 1 eq.) and NaOH (1.831 mmol, 1 eq.) were added into a mixture of 30 mL of water and 25 mL of ethanol, then 1-phenylethanone (3.663 mmol, 2 eq.) was added. The mixture was heated and stirred at 90 °C for 4 h. After cooling, the mixture was filtered and washed with plenty of water and then dried at RT to produce a yellow powder with a yield of 80% (Scheme 1).
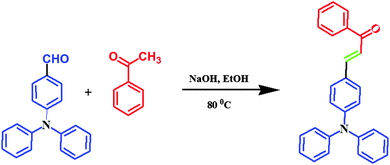 |
| | Scheme 1 Synthesis of TPA substituted yellow phosphors. | |
FTPA-acetophenone.
1H NMR data: δ = 8.01 (d, 2H), 7.77 (d, 1H), 7.58–7.54 (t, 1H), 7.51–7.48 (m, 4H), 7.39 (d, 1H), 7.33–7.30 (t, 4H), 7.18–7.09 (m, 6H), 7.04 (d, 2H). 13C NMR data (CDCl3, 100 MHz): δ = 195.26, 143.29, 137.13, 134.39, 134.16, 133.20, 133.05, 132.46, 130.14, 128.78, 126.20, 124.06. Anal. calcd for C27H21NO: C, 86.37; H, 5.64; N, 3.73; O, 4.26. Found: C, 86.27; H, 5.59; N, 3.71; O, 4.23.
DFTPA-acetophenone.
1H NMR data: δ = 8.02–8.04 (d, 2H), 7.74–7.84 (m, 4H), 7.58–7.61 (m, 2H), 7.54–7.56 (m, 3H), 7.47–7.48 (d, 1H), 7.37–7.44 (m, 3H), 7.18–7.21 (m, 9H), 7.13–7.16 (m, 2H). 13C NMR data (CDCl3, 100 MHz): δ = 195.29, 148.67, 137.43, 137.33, 136.00, 135.87, 134.82, 134.48, 133.30, 133.10, 131.73, 130.94, 129.32, 128.02, 127.41, 125.88. Anal. calcd for C36H27NO2: C, 85.52; H, 5.38; N, 2.77; O, 6.33. Found: C, 85.49; H, 5.32; N, 2.71; O, 6.30.
TFTPA-acetophenone.
1H NMR data: δ = 8.031–8.049 (d, 6H), 7.77–7.87 (m, 4H), 7.59–7.69 (m, 6H), 7.48–7.64 (m, 8H), 7.27 (d, 2H), 7.19–7.21 (t, 3H), 7.11 (d, 2H). 13C NMR data (CDCl3, 100 MHz): δ = 194.92, 142.98, 137.35, 136.06, 134.58, 134.51, 132.75, 129.13, 128.06, 127.21, 126.30. Anal. calcd for C45H33NO3: C, 85.01; H, 5.23; N, 2.20; O, 7.55. Found: C, 85.10; H, 5.16; N, 2.15; O, 7.51.
Results and discussion
Thermal properties
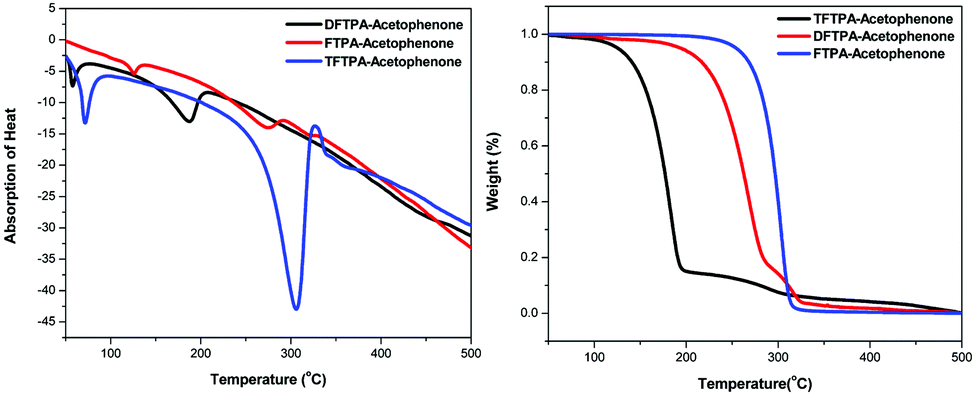
Differential thermal analysis (DTA) and thermogravimetric analysis (TGA) of the phosphors was carried out simultaneously by employing a NETZSCH thermal analyzer. The sample was heated at a rate of 10 °C min−1 in a nitrogen atmosphere. The TG-DTA curves of synthesized phosphors are depicted in Fig. 3. In the DTA curve of FTPA-acetophenone, the first endothermic peak is observed at 274 °C and is attributed to the meting point of the sample. Another important observation is that there is no phase transition until the material melts, and this enhances the temperature range for its utility in LED applications. The presence of water of crystallization in the molecular structure is indicated by the presence of the weight loss near 125 °C. Further, there is no decomposition near the melting point. This ensures the suitability of the material for possible application in LEDs, where the phosphor materials are required to withstand high temperatures (∼125 °C). The weight loss starts at around 138 °C and the weight loss corresponding to decomposition of the phosphor is observed at 350 °C, which takes place over a large temperature range (138–350 °C) where almost all the gaseous fragments like carbon dioxide and ammonia may be liberated. The TGA reveals exactly the same changes shown by DTA.
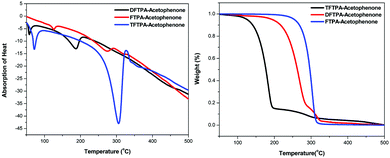 |
| | Fig. 3 (a) DTA curves of yellow phosphors. (b) TGA curves of yellow phosphors. | |
The second endothermic peak in the DTA curve shows that the phosphor material is fully decomposed at 318 °C. The sharpness of the endothermic peak shows the good degree of crystallinity of the grown sample. The weight loss temperatures of the DFTPA-acetophenone and TFTPA-acetophenone phosphors are 57, 375 °C and 70, 450 °C, respectively. The temperatures of decomposition (Td: corresponding to 5% weight loss) of DFTPA-acetophenone and TFTPA-acetophenone are 187 °C and 306 °C, respectively. However, the corresponding temperature ranges of the DFTPA-acetophenone and TFTPA-acetophenone phosphors with the same weight loss are 57–375 °C and 70–450 °C, based on the thermal properties of the synthesized phosphors, it conclude that the synthesized phosphors can be used for the remote phosphor based LEDs.33
Powder XRD studies
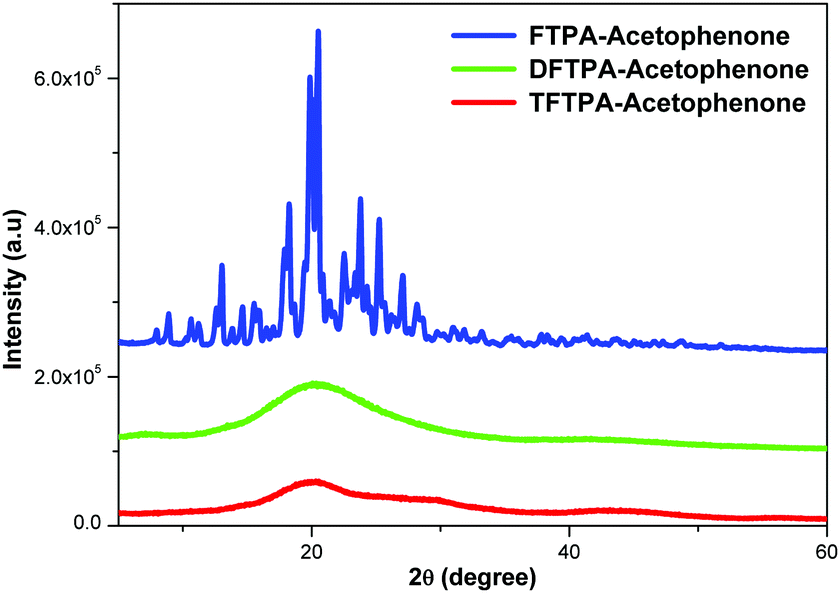
XRD patterns of the synthesized yellow phosphors are shown in Fig. 4. The FTPA-acetophenone is shown to be of crystalline nature, whereas the diffraction peaks of DFTPA and TFTPA-acetophenone evidence an amorphous phase. It clearly indicates that with the successive addition of acceptor groups to the TPA moiety, the crystalline nature of the FTPA-acetophenone changes to an amorphous nature. In general, amorphous luminophores are highly required to fabricate solution processable OLEDs, wherein the luminophore should not be crystalline under the device operating conditions. In general, amorphous behavior is achieved by the introduction of bulky substituents that hinder the packing of the molecules. The currently synthesized phosphors are potential candidates for solution processable white organic light emitting diodes.
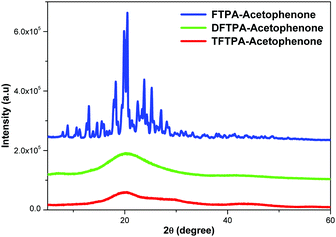 |
| | Fig. 4 Powder X-ray diffraction patterns of yellow phosphors. | |
Photophysical properties
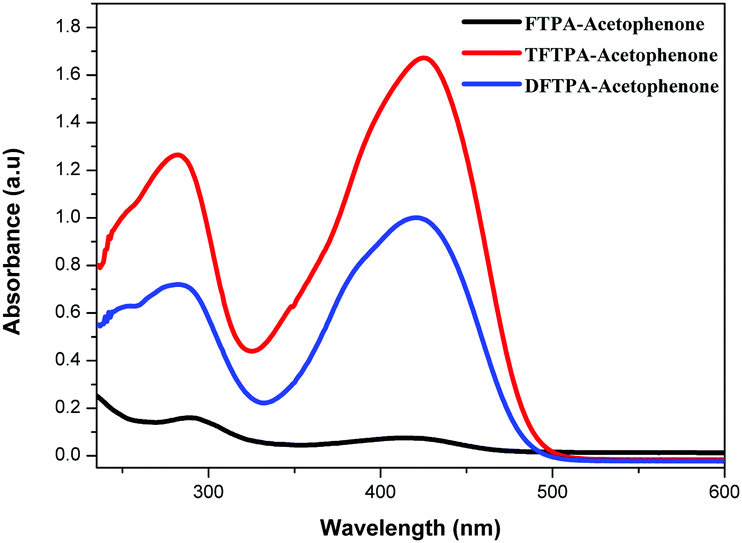
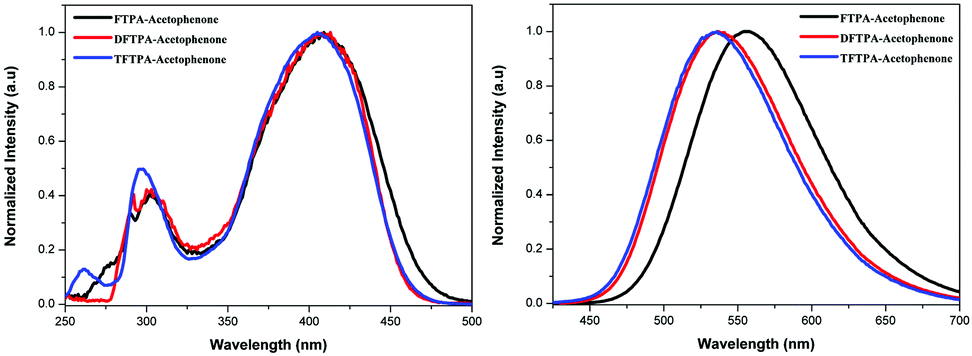
The UV-visible and PL spectra of the synthesized phosphors were measured in dilute CH2Cl2 solutions (Fig. 5 and 6). In the FTPA-acetophenone absorption spectrum, the strong absorption peaks at 292 and 416 nm are ascribed to the π–π* transitions of the conjugated chain.30–32,34 A similar observation has been made in the case of N,N-diphenyl-4-styrylaniline wherein the molecular structure is very similar to that of FTPA-acetophenone.29 The photoluminescence spectra of the FTPA-acetophenone, DFTPA-acetophenone and TFTPA-acetophenone compounds were measured under the monitoring wavelengths of 292 and 425 nm, respectively. The emission spectra of the phosphors are shown in Fig. 6, which indicates that the FTPA, DFTPA and TFTPA-acetophenone phosphors emit at 558, 535 and 532 nm, respectively.
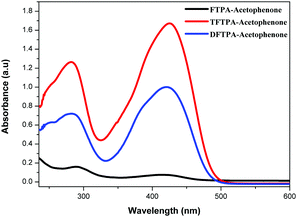 |
| | Fig. 5 Absorption spectra of the TPA substituted acetophenones in CH2Cl2 solvent. | |
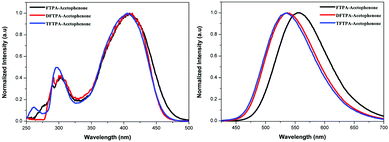 |
| | Fig. 6 Excitation (left) and emission (right) spectra of the TPA substituted acetophenones in CH2Cl2 (10−4 M). | |
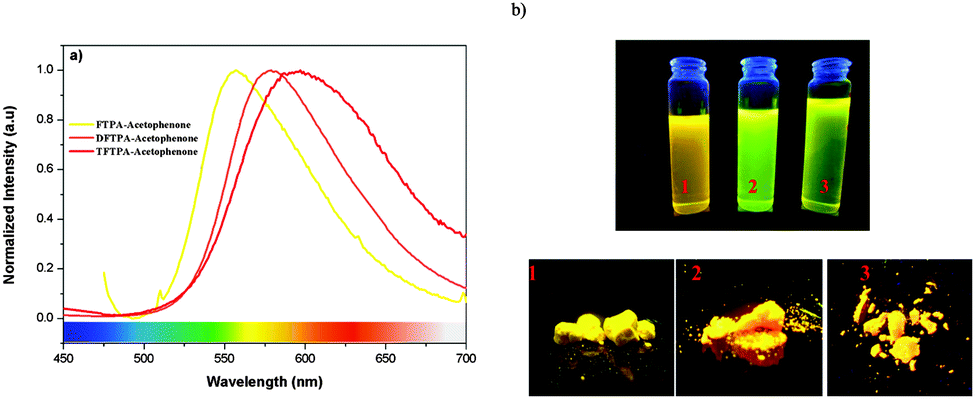
The photoluminescence quantum yields (PLQY) of the currently synthesized phosphors are tabulated in Table 1. The relative comparison of the PL intensity of the yellow phosphors has been made by keeping the FTPA-acetophenone emission intensity as one, and the calculated relative emission intensity has been tabulated for DFTPA and TFTPA-acetophenone in Table 1. The relative intensity of TFTPA-acetophenone is 1.58 times higher than that of FTPA-acetophenone. Solid state photoluminescence studies revealed that the FTPA, DFTPA and TFTPA-acetophenone phosphors emit at 556, 576 and 594 nm, respectively (Fig. 7). The observed red shift in the solid state emission can be attributed to the enhanced aggregation induced emission (AIE).33,35 The solvatochromism of the synthesized phosphors is mentioned in the ESI.†
Table 1 Photophysical and CIE chromaticity values of the yellow phosphors
| Yellow phosphors |
λ
abs
(nm) |
λ
em (nm) |
PL intensity comparison |
PLQY (Φ) |
CIE coordinates (x, y) |
| Solution |
Solid |
|
In CH2Cl2 solution.
Solid.
|
| FTPA-acetophenone |
292, 416 |
558a/556b |
1 |
0.44 |
0.4236, 0.5367 |
0.4562, 0.5002 |
| DFTPA-acetophenone |
284, 420 |
528a/578b |
1.05 |
0.25 |
0.2853, 0.5029 |
0.5202, 0.4660 |
| TFTPA-acetophenone |
282, 423 |
522a/594b |
1.58 |
0.22 |
0.2877, 0.4668 |
0.5504, 0.4393 |
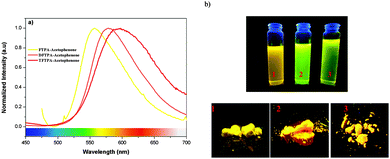 |
| | Fig. 7 (a) Solid state emission spectra of the TPA substituted acetophenones. (b) Digital photographs of the yellow phosphors in solution and the solid state under illumination by a UV lamp. | |
Incorporation of heteroatoms into an organic luminophore can lead to intramolecular charge transfer (ICT), which can induce electronic perturbation, such as polarization. This can considerably alter the photophysical properties of the organic phosphor, particularly the color tunability. The AIE of organic phosphors is emitted in the long wavelength region due to the polarization effect triggered by the D–A interaction (triphenylamine and keto moieties). As shown in Fig. 7, the powders of the yellow phosphors emit a yellow light, while in dilute solution the compounds have a yellowish green color. Similar observations (AIE in the solid state) have been made in the case of dipyrrolylquinoxaline difluoroborate36 and benzothiazole–enamide37 complexes.
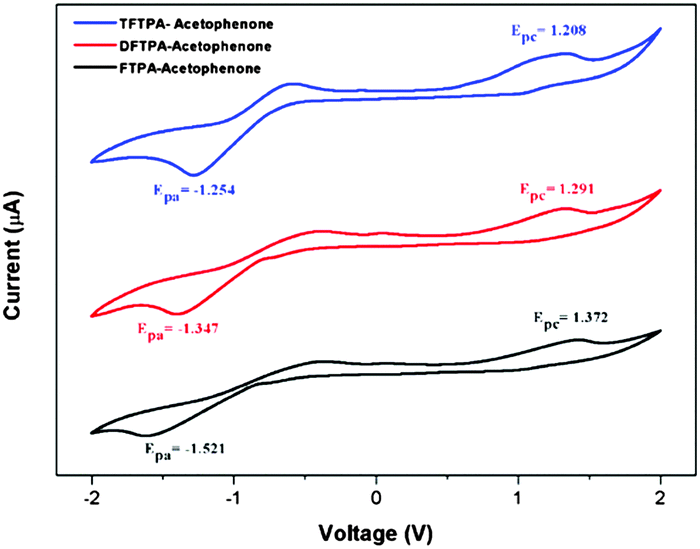
Cyclic voltammetry studies
The HOMO and LUMO energy levels of all the phosphor materials can be calculated from the onset oxidation potential (Eox(onset)) and the onset reduction potential (Ered(onset)).
Fig. 8 shows that all the phosphor materials exhibit two reversible oxidation process in the anodic sweep. No reduction signal was detected for any of the phosphors during the cathodic scan. The HOMO levels determined from Eonset for FTPA-acetophenone (−5.7721 eV) was notably higher than that of DFTPA-acetophenone (−5.6915 eV) and TFTPA-acetophenone (−5.6085 eV). These data show that the incorporation of one to three acceptor groups on the TPA moiety has a great impact on the HOMO and LUMO levels. More specifically, the HOMO and LUMO level values gradually decrease and increase, respectively (Table 2). The overall energy gap of the phosphor materials decreases in a uniform manner. The HOMO levels of DFTPA and TFTPA-acetophenone decrease to ∼0.1 eV lower than FTPA-acetophenone, reflecting the fact that the outer acetophenone units are of electron-withdrawing nature through an inductive or π-polarization effect.
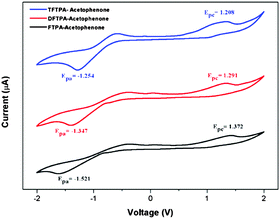 |
| | Fig. 8 Cyclic voltammetry curves of the three yellow phosphors (Epa and Epc are the cathodic and anodic potentials, respectively). | |
Table 2 Electrochemical data and theoretical HOMO–LUMO gaps
| Yellow phosphors |
E
onsetred (V) |
E
onsetox (V) |
HOMO (eV) |
LUMO (eV) |
Energy gap (ΔE, eV) |
| FTPA-acetophenone |
−1.521 |
1.372 |
−5.772 |
−2.879 |
2.89 |
| DFTPA-acetophenone |
−1.347 |
1.291 |
−5.691 |
−3.052 |
2.63 |
| TFTPA-acetophenone |
−1.254 |
1.208 |
−5.608 |
−3.145 |
2.46 |
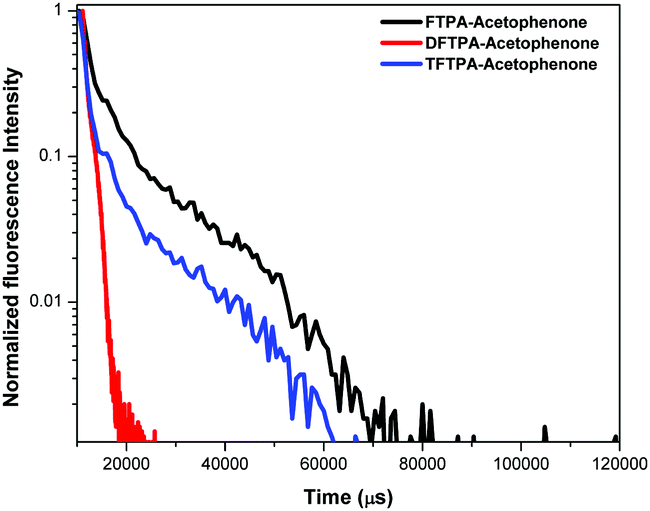
Lifetime measurements
Lifetime decay curve analysis of the phosphors is shown in Fig. 9. The measured data was fitted with the single exponential function given by the equation (t) = I0 + A1exp(−t/τ), where A1 is the scalar quantity obtained from the curve fitting, I0 = 0 is the offset value, t is the time in ms and τ is the decay time value for the exponential component. The luminescence lifetime values (τ) of the FTPA-acetophenone, DFTPA-acetophenone and TFTPA-acetophenone phosphors were found to be 3.91, 9.88 and 2.40 μs, respectively.
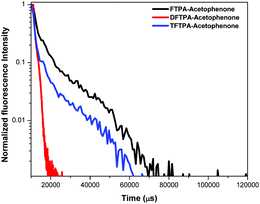 |
| | Fig. 9 Lifetime measurements of the yellow phosphors. | |
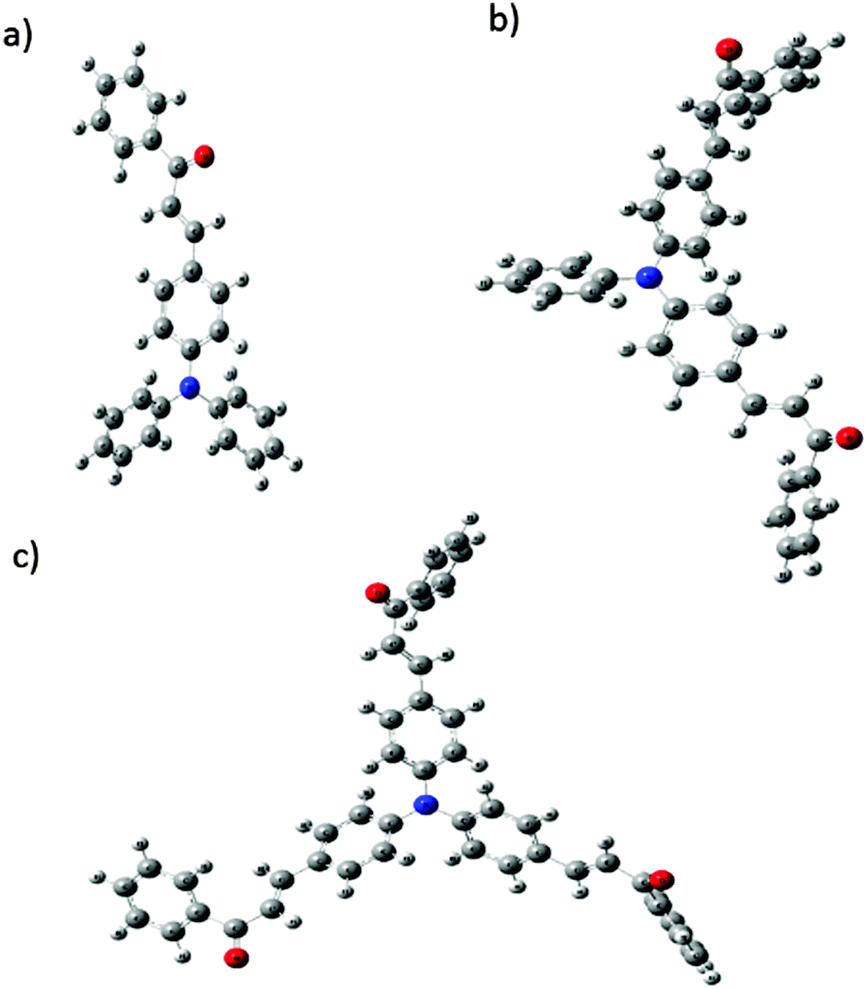
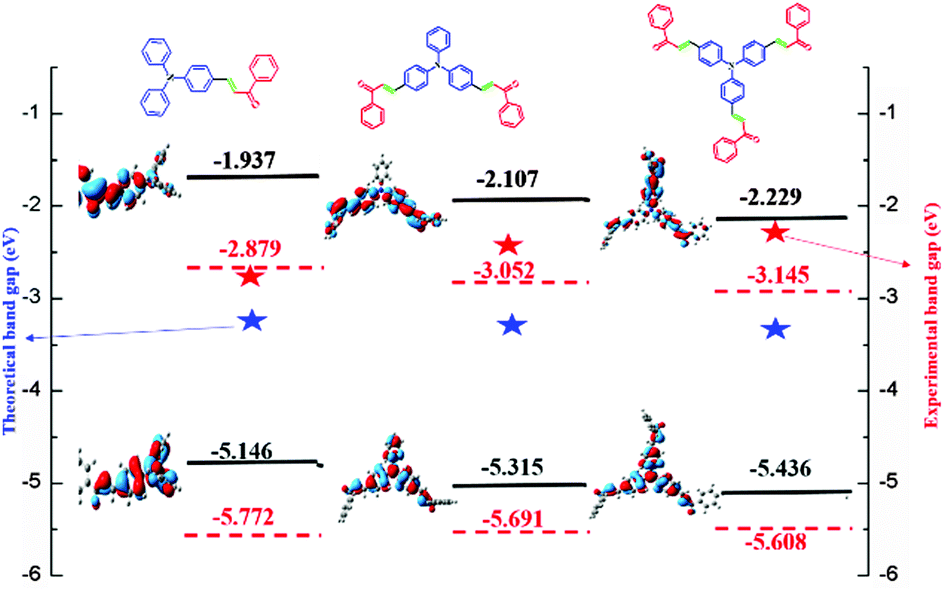
Computational studies of FTPA, DFTPA and TFTPA-acetophenone
Optimized geometries of the FTPA, DFTPA and TFTPA-acetophenone phosphors are given in Fig. 10. Their corresponding frontier orbital energies and gas phase UV-vis spectral wavelengths are tabulated in Table 3. The frontier orbital (LUMO+1 to HOMO−1) diagrams of all the phosphors are presented in the ESI† (Fig. S1). The computed vertical transitions and their oscillator strengths and configurations are mentioned in the ESI† (ST1). The comparison of the HOMO–LUMO energy gaps of the phosphor materials determined theoretically and experimentally are shown in Fig. 11.
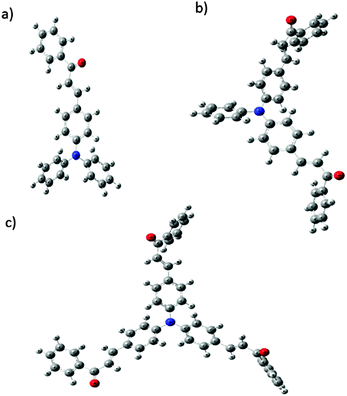 |
| | Fig. 10 Optimized geometries of (a) FTPA, (b) DFTPA and (c) TFTPA-acetophenone. | |
Table 3 Calculated absorption (gas phase), HOMO, LUMO, singlet and triplet energy levels of the phosphors in CH2Cl2 media at the TD-DFT/B3LYP level
| Yellow phosphors |
HOMO (eV) |
LUMO (eV) |
Energy gap (ΔE, eV) |
λ
abs (nm) (gas phase) |
Singlet energy (S1) (eV) |
Triplet energy (T1) (eV) |
| FTPA-acetophenone |
−5.146 |
−1.937 |
3.209 |
293, 423 |
2.716 |
2.047 |
| DFTPA-acetophenone |
−5.315 |
−2.107 |
3.208 |
299, 433 |
2.671 |
2.072 |
| TFTPA-acetophenone |
−5.436 |
−2.229 |
3.207 |
305, 435 |
2.689 |
2.117 |
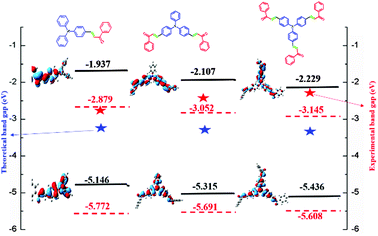 |
| | Fig. 11 HOMO–LUMO energy gap diagram of the yellow phosphors. | |
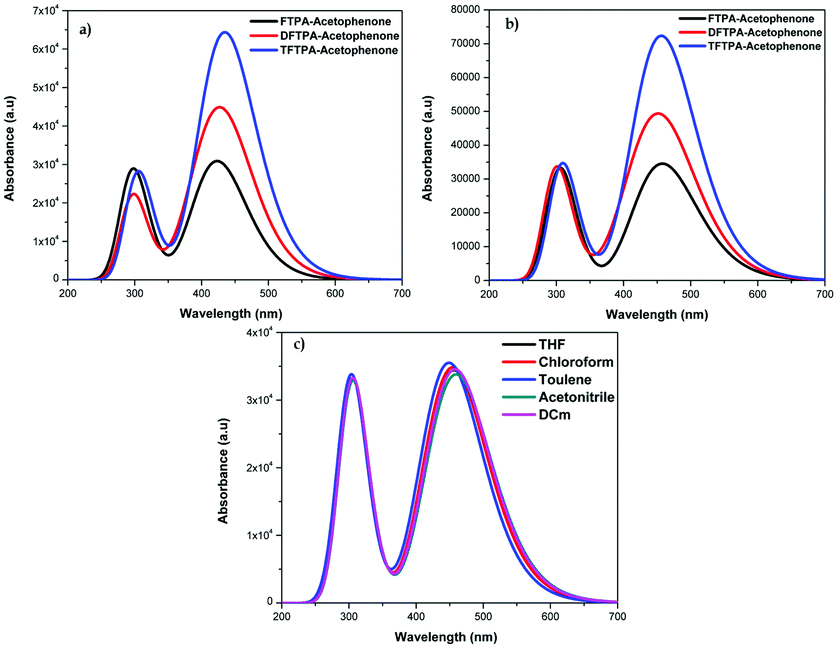
The HOMO–LUMO energy gap of TFTPA-acetophenone is less than that of FTPA-acetophenone, which suggests that there will be a bathochromic shift. This is exactly what happened when we consider the gas phase UV-vis spectral wavelength values from Table 1. However the situation is different when we see the DCM solvent phase UV-vis results. Fig. 12(b) depicts the DCM solvent phase UV-vis spectra for FTPA, DFTPA and TFTPA-acetophenone. Here for the FTPA molecule, two distinct peaks are found at 308 and 457 nm. As we move from FTPA to DFTPA it is expected that there will be a red shift, but a small blue shift occurs in the first peak at 305 nm, and the second peak shifts towards red (459 nm). For TFTPA the first peak appears at 309 nm and the second peak at 456 nm, indicating a red shift and blue shift respectively compared to FTPA and DFTPA-acetophenone. These results match the experimental findings given in Fig. 5. To see the solvent effect on the absorption spectra for these phosphors we took FTPA and calculated the UV-vis spectra with five different solvents: THF, chloroform, DCM, toluene and acetonitrile. The corresponding plot is presented in Fig. 12(c). For all the cases two distinct peaks appear in the UV-vis spectra, for example in the case of toluene solvent the peaks appear at 305 and 448 nm. The peaks are gradually red shifted as the solvent polarity changes. The order is as follows: toluene < chloroform < THF < DCM < acetonitrile.
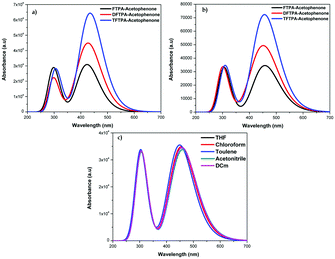 |
| | Fig. 12 UV-vis spectra of the FTPA, DFTPA, TFTPA-acetophenones in (a) the gas phase, (b) the DCM solvent phase, and (c) FTPA-acetophenone in different solvent phases. | |
Singlet (S1) and triplet (T1) energy levels
In order to utilize the phosphor as a host material for red emitting organo-phosphors (iridium or europium-based molecular complexes), it is necessary to know the triplet energy level values. The singlet and triplet energies of the currently synthesized organic phosphors were calculated using the TD-DFT/B3LYP approach. The corresponding energies of the organic phosphors are tabulated in Table 3. The calculated triplet energy level values for the currently synthesized yellow phosphors are in the range of 2.0 to 2.1 eV, which matches well with the known host material Alq3.38
White light emission under fluorescence microscope and CIE color coordinates
In order to evaluate the white light emission and suitability to be used in WLEDs, the organic phosphors were excited under blue photons in a fluorescence microscope and the pictures are shown in Fig. 13. One can clearly observe that all the phosphors emit white light under blue ray excitation, and we believe that the currently synthesized phosphors are suitable for WLEDs.
 |
| | Fig. 13 (a) FTPA, (b) DFTPA and (c) TFTPA-acetophenone phosphors under blue ray excitation. | |
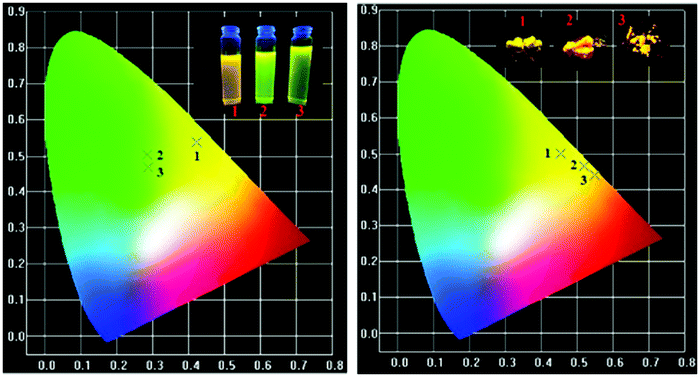
The CIE chromaticity coordinates of the synthesized phosphors and the corresponding emission intensities are tabulated in the ESI† (ST3). The CIE color coordinates of the currently synthesized phosphors show higher color saturation with a yellowish-green and yellow color (Fig. 14).
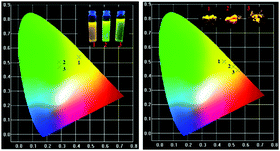 |
| | Fig. 14 CIE color coordinates of the (1) FTPA, (2) DFTPA and (3) TFTPA-acetophenone phosphor materials in solution and the solid state. | |
Conclusion
In summary, we have successfully synthesized a series of triphenylamines with multiple acceptors as yellow organic phosphors and demonstrated that increasing the number of acceptor groups will lead to a red shift in the emission spectrum (yellow to yellowish orange color, due to the π–π interaction and aggregation induced emission). Solvent-dependent photoluminescence studies (solvatochromism) were carried out for all three yellow phosphors and the spectral data were interpreted in terms of the Lippert–Mataga equation. All the phosphors were thermally stable up to 300 °C, which clearly indicates that all the phosphors are useful for the fabrication of white LEDs/OLEDs. These outcomes offer a great platform for the development of practical hybrid lighting devices based on purely organic phosphors.
Acknowledgements
This work is funded by the Department of Science and Technology (DST), Government of India INSPIRE award no. IFA12-CH-48 (VS) and IFA14-CHE-151 (SG).
References
-
(a) C. J. Humphreys, MRS Bull., 2008, 33, 459–470 CrossRef;
(b) R. Haitz and J. Y. Tsao, Phys. Status Solidi A, 2011, 208, 17–29 CrossRef CAS.
- S. Nakamura, T. Mukai and M. Senoh, J. Appl. Phys., 1994, 76, 8189–8191 CrossRef CAS.
- Y. Q. Li, G. de With and H. T. Hentzen, J. Lumin., 2006, 116, 107–116 CrossRef CAS.
-
T. Kanou, Handbook of Phosphors, Ohm, Tokyo, 1987 Search PubMed.
-
(a) Y. Zhu and N. Narendran, Jpn. J. Appl. Phys., 2010, 49, 100203 CrossRef;
(b) Y.-H. Won, H. S. Jang, K. W. Cho, Y. S. Song, D. Y. Jeon and H. K. Kwon, Opt. Lett., 2009, 34, 1–3 CrossRef CAS PubMed.
-
(a) W. Ding, J. Wang, M. Zhang, Q. Zhang and Q. Su, J. Solid State Chem., 2006, 179, 3582–3585 CrossRef CAS;
(b) P. P. V. Weiler, C. Hecht, A. Tücks, A. S. Wochnik, A.-K. Henß, D. Wiechert, C. Scheu, P. J. Schmidt and W. Schnick, Nat. Mater., 2014, 13, 891–896 CrossRef PubMed;
(c) S. Lee, T. Chan and T. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 40–44 CrossRef CAS PubMed;
(d) H. Zhu Chun, C. Lin, W. Luo, S. Shu, Z. Liu, Y. Liu, J. Kong, A. En, Y. Cao, R.-S. Liu and X. Chen, Nat. Commun., 2014, 5, 4312–4328 Search PubMed.
- Y. Li, A. Delsing, G. With and H. Hintzen, Chem. Mater., 2005, 17, 3242–3248 CrossRef CAS.
- H. Kim, J. Jin, Y. Lee, S. Lee and C. Hong, Chem. Phys. Lett., 2006, 431, 341–345 CrossRef CAS.
- P. Schlotter, R. Schmidt and J. Schneider, Appl. Phys. A: Mater. Sci. Process., 1997, 64, 417–418 CrossRef CAS.
- F. Hide, P. Gozodoy, S. P. DenBaars and A. J. Heeger, Appl. Phys. Lett., 1997, 70, 2664–2666 CrossRef CAS.
- C. Zhang and A. J. Heeger, J. Appl. Phys., 1998, 84, 1579–1582 CrossRef CAS.
- R. W. Martin, P. R. Edwards, R. Pecharroman-Gallego, C. Liu, C. J. Deatcher, I. M. Watson and K. P. O'Donnell, J. Phys. D: Appl. Phys., 2002, 35, 604 CrossRef CAS.
- G. Heliotis, G. Itskos, R. Murray, M. D. Dawson, I. M. Watson and D. D. C. Bradley, Adv. Mater., 2006, 18, 334–338 CrossRef CAS.
- G. Heliotis, P. N. Stavrinou, D. D. C. Bradley, E. Gu, C. Griffin, C. W. Jeon and M. D. Dawson, Appl. Phys. Lett., 2005, 87, 103505 CrossRef.
- E. Gu, H. X. Zhang, H. D. Sun, M. D. Dawson, A. R. Mackintosh, A. J. C. Kuehne, R. A. Pethrick, C. Belton and D. D. C. Bradley, Appl. Phys. Lett., 2007, 90, 031116 CrossRef.
- R. Smith, B. Liu, J. Bai and T. Wang, Nano Lett., 2013, 13, 3042–3047 CrossRef CAS PubMed.
- Z. Yue, Y. F. Cheung, H. W. Choi, Z. Zhao, B. Zhong Tang and K. S. Wong, Opt. Mater. Express, 2013, 3, 1906–1911 CrossRef.
- N. Laurand, B. Guilhabert, J. McKendry, A. E. Kelly, B. Rae, D. Massoubre, Z. Gong, E. Gu, R. Henderson and M. D. Dawson, Opt. Mater. Express, 2012, 2, 250–260 CrossRef CAS.
- H. Chun, C.-J. Chiang, A. Monkman and D. O. O'Brien, J. Lightwave Technol., 2013, 31, 3511–3517 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
-
(a) C. C. Haung, H. F. Meng, G. K. Ho, C. H. Chen, C. S. Hsu, J. H. Huang, S. F. Horng, B. X. Chen and L. C. Chen, Appl. Phys. Lett., 2004, 84, 1195 CrossRef;
(b) Q. Wang, J. Ding, D. Ma, Y. Cheng, L. Wang and F. Wang, Adv. Mater., 2009, 21, 2397 CrossRef CAS;
(c) Y.-S. Park, J. W. Kang, D. M. Kang, J. W. Park, Y.-H. Kim, S.-K. Kwon and J.-J. Kim, Adv. Mater., 2008, 20, 1957 CrossRef CAS;
(d) B. W. D'Andrade, R. J. Holmes and S. R. Forrest, Adv. Mater., 2004, 16, 624 CrossRef.
-
(a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed;
(b) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
-
(a) T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2015, 3, 9981 RSC;
(b) T. Jadhav, B. Dhokale, Y. Patil and R. Misra, RSC Adv., 2015, 5, 68187 RSC.
-
(a) M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC;
(b) R. Maragani, P. Gautam, S. M. Mobin and R. Misra, Dalton Trans., 2016, 45, 4802 RSC;
(c) T. Sheshashena Reddy, R. Maragani, B. Dhokale, S. M. Mobin and R. Misra, RSC Adv., 2016, 6, 7746–7754 RSC.
- L. A. Estrada and D. C. Neckers, Org. Lett., 2011, 13, 3304–3307 CrossRef CAS PubMed.
-
(a) R. Misra, R. Maragani, B. Pathak, P. Gautam and S. M. Mobin, RSC Adv., 2015, 5, 71046–71051 RSC;
(b) R. Misra, A. Ramesh Maragani, A. K. R. Patelb and G. D. Sharma, RSC Adv., 2014, 4, 34904 RSC;
(c) R. Misra, R. Maragani, P. Gautam and S. M. Mobin, Tetrahedron Lett., 2014, 55, 7102–7105 CrossRef CAS.
- C.-H. Yang, H.-L. Chen, Y.-Y. Chuang, C.-G. Wu, C.-P. Chen, S.-H. Liao and T.-L. Wang, J. Power Sources, 2009, 188, 627–634 CrossRef CAS.
-
(a) A. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 785–789 CrossRef.
-
(a) R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS;
(b) G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, J. Chem. Phys., 2006, 124, 094107 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
-
M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- L. Zhang, B. Li, B. Lei, Z. Hong and W. Li, J. Lumin., 2008, 128, 67–73 CrossRef CAS.
- R. Boonsin, G. Chadeyron, J.-P. Roblin, D. Boyer and R. Mahiou, J. Mater. Chem. C, 2015, 3, 9580–9587 RSC.
- N. Armroli, L. De Cola, V. Balzami, J. P. Sauvage, C. D. Dietich Buchecker and J. M. Kern, J. Chem. Soc., Faraday Trans., 1992, 88, 553–556 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Q. Liu, X. Wang, H. Yan, Y. Wu, Z. Li, S. Gong, P. Liu and Z. Liu, J. Mater. Chem. C, 2015, 3, 2953–2959 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- T.-Y. Chu, Y.-S. Wu, J.-F. Chen and C. H. Chen, Chem. Phys. Lett., 2005, 404, 121–125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00031b |
|
| This journal is © the Partner Organisations 2017 |