
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The templation effect as a driving force for the self-assembly of hydrogen-bonded peptidic capsules in competitive media†
M.
Grajda
 ,
M. J.
Lewińska
and
A.
Szumna
*
,
M. J.
Lewińska
and
A.
Szumna
*
Institute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-22 Warsaw, Poland. E-mail: agnieszka.szumna@icho.edu.pl
First published on 1st September 2017
Abstract
Peptide-based cavitands (resorcin[4]arenes substituted with histidine and glutamine hydrazides) exist as monomeric species in polar solvents (DMSO and methanol). Upon complexation of fullerenes, the cavitands wrap around the hydrophobic guests forming dimeric capsular shells (as evidenced by DOSY). The self-assembly of the cavitands is based on the formation of beta-sheet-like binding motifs around the hydrophobic core. In a polar environment, these hydrogen bonded structures are kinetically stable and highly ordered as manifested by a 100-fold increase of intensity of circular dichroism bands, as well as a separate set of signals and substantial differences in chemical shifts in NMR spectra. This behavior resembles a protein folding process at the molten globule stage with non-specific hydrophobic interactions creating a protective and favourable local environment for the formation of secondary structures of proteins.
Our work takes inspiration from the native structures of proteins, which are exemplars of excellence in folding of ordered three-dimensional architectures in polar environments. At the initial stages of protein folding a pivotal role is attributed to hydrophobic interactions. Although these interactions are non-directional, they lead to the formation of a hydrophobic core, a molten globule that constitutes a perfect environment for further formation of directional hydrogen bonds by lowering the local dielectric constant.1–3 Thus, hydrogen bonds around a hydrophobic core are stronger and contribute more significantly to the stability of the native state than hydrogen bonds exposed to the aqueous environment.
This bioinspired approach based on a combination of hydrophobic core formation with an ordering power of hydrogen bonds on the peripheries is widely used for the self-assembly of infinite fibrous structures4 or nanoparticles.5 When the number of building blocks decreases, the strength of hydrophobic clustering is reduced and the above-mentioned strategy becomes less efficient. Therefore, the formation of discrete structures made of only a few building blocks is challenging. Gibb and co-workers reported on the self-assembly of discrete capsular dimers,6 tetramers and hexamers7 in water using hydrophobic interactions only.8 The group of Rebek developed a series of hydrocarbon-based templates for ordering of hydrogen bonded multicomponent capsules in non-polar solvents.9 Fullerenes have been used for the templation of self-assembled structures based on 4-pyrimidinone derivatives (isocytosine or 2-ureido-4[1H]-pyrimidinone derivatives),10–13 amino acid functionalized naphthalenediimides,14S-trityl-cysteine derivatives15 or through pyridinium–anion–pyridinium interactions.16 We have also presented resorcinarenes appended to hydrophobic peptides (Phe, Val) that form well-ordered hydrogen bonded dimers and complex fullerenes (Fig. 1).17
 | ||
| Fig. 1 Comparison of self-assembly processes for peptidic capsules observed in a non-polar environment (previous work) and the discussed process in a polar environment (this work). | ||
In all these examples, hydrogen bonded shells are formed in non-polar media (chloroform, toluene, tetrachloroethane, carbon disulfide) and they are not able to survive in polar environments (even DMSO). In this work, we demonstrate that hydrophobic interactions together with β-sheet-like binding motifs are effective (a) in inducing the self-assembly of peptidic cavitands that otherwise do not self-assemble and (b) in ordering hydrogen bonded shells around hydrophobic cores even in competitive, polar environments (Fig. 1).
In our quest towards self-assembly in polar environments we have designed resorcin[4]arene cavitands substituted with hydrophilic peptides, here containing glutamine and histidine residues (3a and 3b, Table 1). The side chains of these amino acids increase solubility in polar solvents and they additionally can take part in stabilizing or competitive (destabilizing) hydrogen bonding interactions due to their dual donor/acceptor character. The final thermodynamic outcome of such a complex system of interactions is highly context-dependent. For example, polyglutamine sequences in the cores of proteins stabilize β-sheets and β-barrels through hydrogen bonding interactions between side chains.18–21 On the other hand glutamine present in the external shells of other proteins, e.g. insulin does not form stabilizing interactions.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Path | Substrates | Equiv. | Conditions | Yielda of: | |
| (3a)2⊃C60 | 3a | |||||
| a Yields were determined by NMR. | ||||||
| 1 | A | 3a + C60 | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
DMSO (24 h, rt) | 8% | 92% |
| 2 | A | 3a + C60 | 2:1 |
DMSO (24 h, 80 °C) | 14% | 86% |
| 3 | A | 3a + C60 | 2:1 |
Ball-mill (2 h) | 18% | 82% |
| 4 | A | 3a + C60 | 2:10 |
DMSO (24 h, rt) | 12% | 88% |
| 5 | A | 3a + C60 | 2:10 |
DMSO (24 h, 80 °C) | 15% | 85% |
| 6 | A | 3a + C60 | 2:10 |
Ball-mill (2 h) | 19% | 81% |
| 7 | B | 1 + 2a + C60 | 2:8:1 |
DMSO (24 h, 80 °C) | 24% | 86% |
| 8 | B | 1 + 2a + C60 | 2:8:1 |
Ball-mill (2 h) | 21% | 79% |
| 9 | B | 1 + 2a + C60 | 2:8:10 |
DMSO (24 h, 80 °C) | 42% | 58% |
| 10 | B | 1 + 2a + C60 | 2:8:10 |
Ball-mill (2 h) | 22% | 78% |
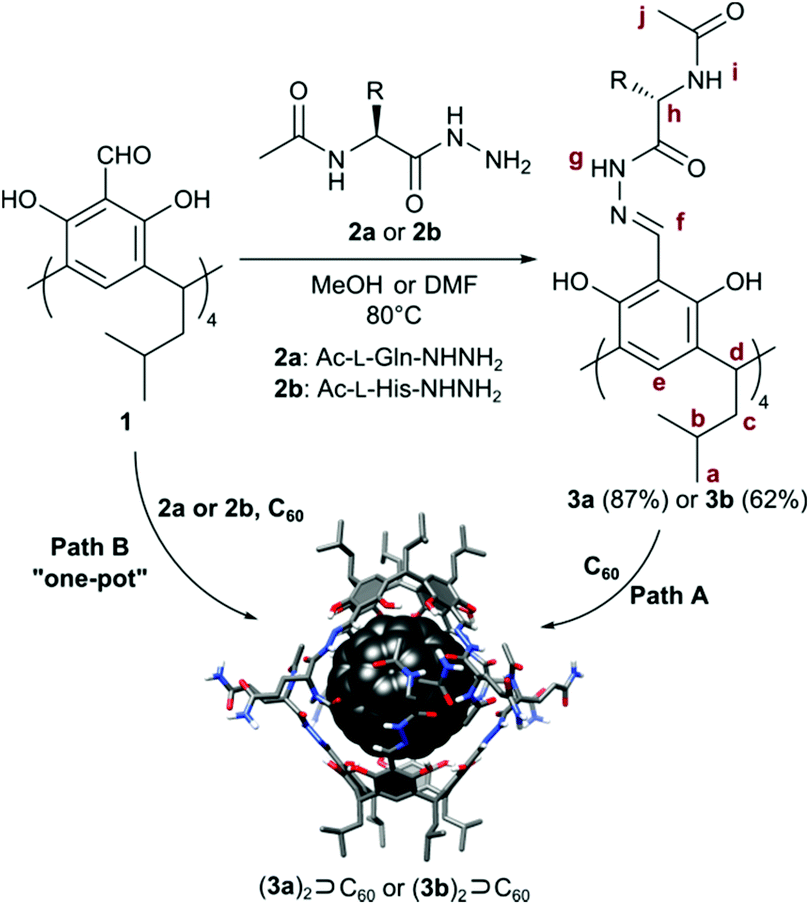
We obtained cavitands 3a and 3b in the reaction of respective amino acid hydrazides 2a or 2b and tetraformylresorcin[4]arene 1 (Table 1). Hydrazones 3a and 3b are soluble in polar solvents: DMSO and methanol but not in water. 1H NMR spectra in DMSO-d6 reveal C4-symmetric structures for both products. Analysis of ROESY NMR spectra indicates the presence of only trivial contacts (originating from covalent proximity). The intramolecular f–j correlation signals that are hallmarks for dimerization via backbone interactions are not present. There are also no indications of specific interactions involving sidechains’ hydrogen bonding groups. These results suggest that hydrazones 3a and 3b do not self-assemble in DMSO and exist as monomeric semi-open cavitands (Fig. 1). This conclusion is also supported by DOSY measurements that reveal a diffusion coefficient of 0.95 × 10−10 m2 s−1 for 3a and 0.95 × 10−10 m2 s−1 for 3b (in DMSO-d6 at 298 K), corresponding to an average diameter of molecule of ∼2.2 nm (predicted value for the cavitand is 2.3 nm, the model is based on the previously reported X-ray structure of the valine derivative).17 This self-assembly behaviour and solubility characteristics of cavitands containing polar side chains are distinctly different from those of the previously reported hydrophobic derivatives that were soluble only in non-polar solvents and formed dimeric capsules in these media.17
In subsequent experiments, we tested the possibility of templation of self-assembly by fullerene C60. For cavitands 3a and 3b a direct complexation experiment is not feasible due to the lack of solubility of fullerene in polar solvents. Therefore alternative ways of reaction activation were employed: (1) thermal activation of samples (heating of 3a + C60 or 3b + C60 in DMSO at 70 °C for 7 days with vigorous stirring, path A); (2) complexation during the chemical reaction of cavitand synthesis (path B); and (3) mechanochemical activation22 in the solid state (Table 1).
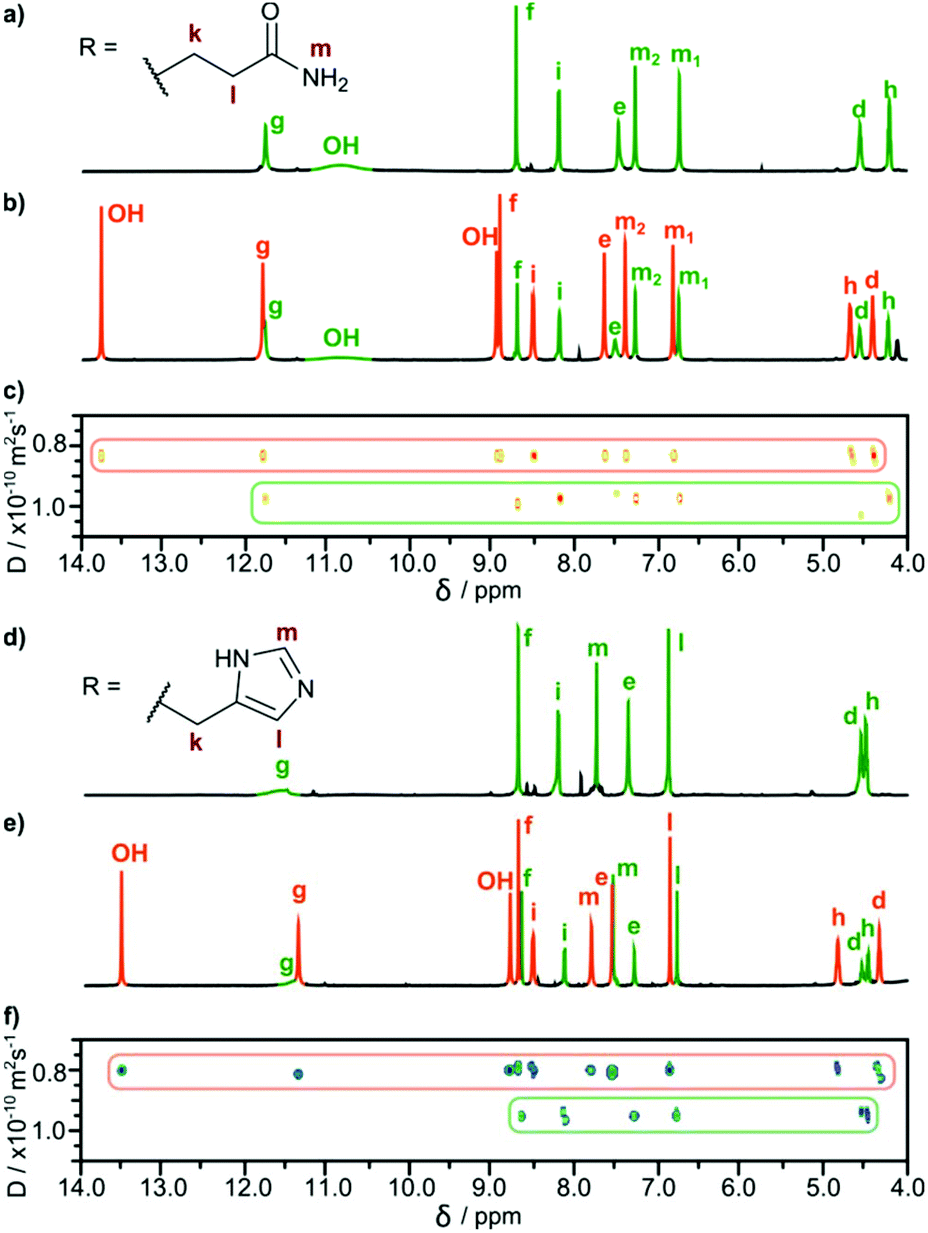
In all cases, the resulting products contained two types of species as revealed by 1H NMR spectra in DMSO-d6 (Fig. 2b and e). One set of signals corresponds to the cavitands (3a or 3b) and the second one is attributed to the dimeric species with encapsulated fullerenes ((3a)2⊃C60 or (3b)2⊃C60). This assignment is based on 13C NMR spectra that reveal the signal of encapsulated fullerene at 141 ppm and on DOSY experiments that clearly indicate a larger size of the newly formed species (Fig. 2c and f). Diffusion coefficients for (3a)2⊃C60 (0.82 × 10−10 m2 s−1) and (3b)2⊃C60 (0.80 × 10−10 m2 s−1) at 298 K in DMSO-d6 correspond to the diameter of 2.7 nm (predicted value for the dimeric capsule is ∼2.7 nm). Additionally, in the ROESY spectra the f–j correlation signals are present for (3a)2⊃C60 and (3b)2⊃C60 indicative of a close contact between hemispheres in a head-to-head arrangement (Fig. 3). This allows us to postulate the capsular structures of complexes (3a)2⊃C60 and (3b)2⊃C60 in DMSO-d6. The corresponding ESI-MS experiments for the products revealed the presence of 3a and (3a)2⊃C60 or 3b and (3b)2⊃C60, without any traces of “empty” capsules, (3a)2 or (3b)2 (ESI†), which proves that the presence of a hydrophobic templating agent is essential for the self-assembly of cavitands 3a and 3b in DMSO.
 | ||
| Fig. 2 1H NMR (600 MHz, DMSO-d6) spectrum of 3a (a) and a mixture of 3a and (3a)2⊃C60 (b); DOSY spectrum of 3a and (3a)2⊃C60 (c); 1H NMR spectrum of 3b (d) and a mixture of 3b and (3b)2⊃C60 (e); DOSY spectrum of 3b and (3b)2⊃C60 (f). | ||
 | ||
| Fig. 3 Dimerization and ordering of the structures in DMSO: ROESY effects indicative of β-sheet-type interactions (a) for (3a)2⊃C60; (b) for (3b)2⊃C60 and (c) predicted binding motif (the 3D structure was obtained by Val → Gln mutation of an X-ray structure of a valine derivative17). | ||
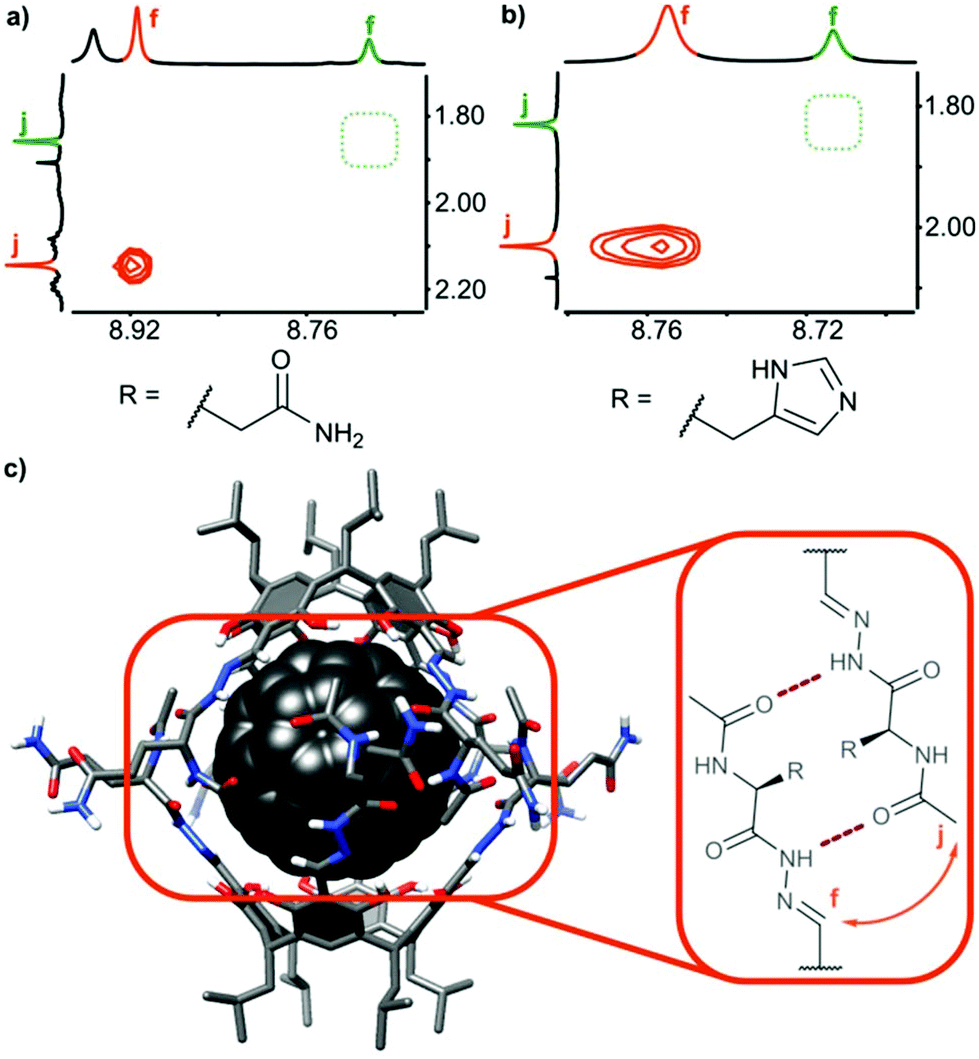
In order to check the stability of hydrogen bonded species in the presence of a competitive solvent that is a hydrogen bond donor and acceptor we performed experiments in methanol. Differences in solubility (Table 2) allowed us to separate a mixture of 3b and (3b)2⊃C60 by selective precipitation of 3b. Spectral data for (3b)2⊃C60 in methanol (CH3OH:CD3OD 9:1) indicate that the complex retains its integrity (Fig. 4). The signal of encapsulated fullerene is present in the 13C NMR spectrum. The diffusion coefficient is in agreement with a diameter of the dimeric complex (2.65 × 10−10 m2 s−1 in methanol at 298 K corresponding to diameter 3.0 nm) and the correlation signal f–j is present in the ROESY spectrum, indicative of a close contact between hemispheres in a head-to-head arrangement. For glutamine derivatives (3a)2⊃C60 and 3a the solubilities in methanol are also different, but we were not able to separate the mixture, due to co-crystallization of the species. However, from a supersaturated methanolic solution we obtained enriched crystals containing up to 60% of a (3a)2⊃C60 (ESI†). After dissolution of the crystals in DMSO intact complex (3a)2⊃C60 was present indicating that also glutamine-based self-assembled species are stable in an environment of methanol.
 | ||
| Fig. 4 Spectra of (3b)2⊃C60: (a) 1H NMR spectrum (600 MHz, CH3OH:CD3OD, 9:1, v:v), (b) 1H NMR spectrum with solvent suppression, (c) DOSY spectrum, (d) comparison of the ECD spectra of 3b (green) and (3b)2⊃C60 (orange) in MeOH (intensities of ECD spectra were scaled using normalization factors from respective UV spectra), (e) ECD spectra. | ||
| Solvent | 3a | (3a)2⊃C60 | 3b | (3b)2⊃C60 |
|---|---|---|---|---|
| Sol. – well-soluble; insol. – insoluble. | ||||
| MeOH | Sol. | Insol. | Insol. | Sol. |
| DMSO | Sol. | Sol. | Sol. | Sol. |
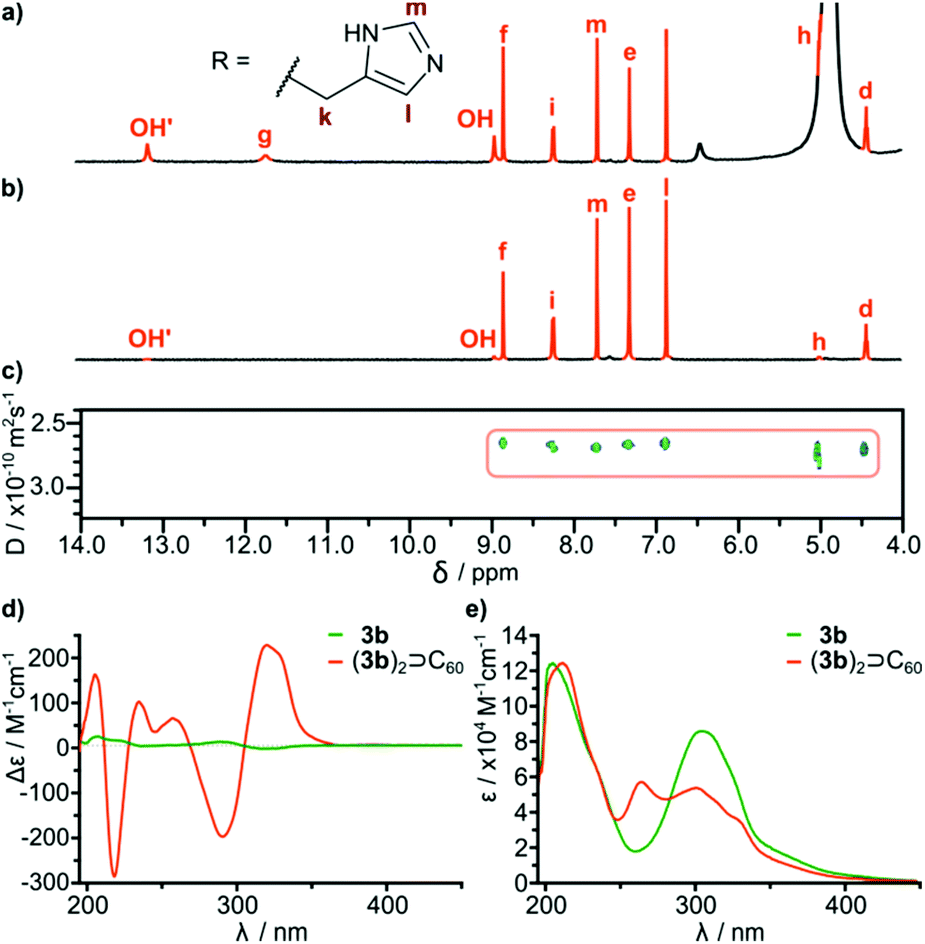
It is important to note that fullerene-templated self-assembly results in the formation of a well-defined hydrogen bonding system with a β-sheet-like binding motif and an ordered inherently chiral conformation. In the 1H NMR spectrum, all signals for hydrogens capable of hydrogen bonding are considerably downfield shifted in (3a)2⊃C60 as compared with cavitand 3a. For example one of the OH protons is shifted from 10.8 ppm to 13.7 ppm, and this proton is postulated to form an intramolecular hydrogen bond. Amide proton i is also upfield shifted in agreement with the formation of an intermolecular hydrogen bond connecting the hemispheres. The formation of a β-sheet-like binding motif is also supported by the increase of three-bond coupling constants 3J(NH-CHα) from 7.2 Hz for 3a to 8.5 Hz for (3a)2⊃C60 and from 8.2 Hz for 3b to 8.9 Hz (DMSO) or 9.6 Hz (MeOH) for (3b)2⊃C60. These coupling constants are directly related to the backbone dihedral φ angles as described by the Karplus relationship for peptides. Higher values are hallmarks of extended β-strand conformations (φ ≈ −139°) over a helix (φ ≈ −57°) or a random coil.23,24 An ordered conformation is also reflected in circular dichroism spectra that exhibit almost a 100-fold enhancement in the intensity of the lowest energy bands for (3b)2⊃C60 as compared with 3b (Fig. 4d). These bands are the most sensitive to conformational inherent chirality and symmetry of these molecules, as they come from the HOMO–LUMO transition involving an inherently chiral chromophore.25 These results indicate that the capsular dimers indeed retain a hydrogen-bonded ordered structure in competitive medium. It is also important to note that there are no traces of dynamic exchange between species at the NMR timescale, indicating that the complexes are also kinetically persistent in this solvent, which is quite surprising considering that semi-open cavitands are also present in the solution.
In summary, we have demonstrated that wrapping of hydrogen-bonded shells around a hydrophobic template is effective in a highly disintegrative environment. DMSO is a strong competitor as a hydrogen bond acceptor and due to its hydrophobic character, while methanol is a strong hydrogen bond donor and acceptor. Therefore, both environments are competitive for aggregates based on hydrogen bonding and hydrophobic interactions. In fact, DMSO or methanol is commonly used for unfolding of natural proteins or disintegration of hydrogen bonded artificial aggregates. Here we demonstrated that the optimal combination of both interactions enables effective self-assembly, although none of the interactions is effective alone. The current system is not water-soluble, but we predict that this strategy can also be extended to aqueous solutions due to even stronger hydrophobic interactions.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Science Center (2013/09/B/ST5/01026) and the Wroclaw Centre for Networking and Supercomputing (grant no. 299). We would like to acknowledge Dr Aleksandra Butkiewicz for CD measurements.Notes and references
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed.
- H. J. Dyson, P. E. Wright and H. A. Scheraga, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13057–13061 CrossRef CAS PubMed.
- K. A. Dill, S. Bromberg, K. Yue, H. S. Chan, K. M. Ftebig, D. P. Yee and P. D. Thomas, Protein Sci., 1995, 4, 561–602 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- I. Leõn, J. Millán, E. J. Cocinero, A. Lesarri and J. A. Fernández, Angew. Chem., Int. Ed., 2013, 52, 7772–7775 CrossRef PubMed.
- C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2004, 126, 11408–11409 CrossRef CAS PubMed.
- H. Gan and B. C. Gibb, Chem. Commun., 2013, 49, 1395–1397 RSC.
- J. H. Jordan and B. C. Gibb, Chem. Soc. Rev., 2015, 44, 547–585 RSC.
- D. Ajami, L. Liu and J. Rebek Jr., Chem. Soc. Rev., 2015, 44, 490–499 RSC.
- E. Huerta, G. A. Metselaar, A. Fragoso, E. Santos, C. Bo and J. de Mendoza, Angew. Chem., Int. Ed., 2007, 46, 202–205 CrossRef CAS PubMed.
- E. Huerta, S. A. Serapian, E. Santos, E. Cequier, C. Bo and J. de Mendoza, Chem. – Eur. J., 2016, 22, 13496–13505 CrossRef CAS PubMed.
- Q. Shi, K. E. Bergquist, R. Huo, J. Li, M. Lund, R. Vácha, A. Sundin, E. Butkus, E. Orentas and K. Wärnmark, J. Am. Chem. Soc., 2013, 135, 15263–15268 CrossRef CAS PubMed.
- D. Račkauskaite, K. E. Bergquist, Q. Shi, A. Sundin, E. Butkus, K. Wärnmark and E. Orentas, J. Am. Chem. Soc., 2015, 137, 10536–10546 Search PubMed.
- G. D. Pantoş, J.-L. Wietor and J. K. M. Sanders, Angew. Chem., Int. Ed., 2007, 46, 2238–2240 CrossRef PubMed.
- G. Markiewicz, A. Jenczak, M. Kołodziejski, J. J. Holstein, J. K. M. Sanders and A. R. Stefankiewicz, Nat. Commun., 2017, 8, 15109 CrossRef PubMed.
- S. J. Park, O.-H. Kwon, K.-S. Lee, K. Yamaguchi, D.-J. Jang and J.-I. Hong, Chem. – Eur. J., 2008, 14, 5353–5359 CrossRef CAS PubMed.
- M. Szymański, M. Wierzbicki, M. Gilski, H. Jędrzejewska, M. Sztylko, P. Cmoch, A. Shkurenko, M. Jaskólski and A. Szumna, Chem. – Eur. J., 2016, 22, 3148–3155 CrossRef PubMed.
- M. F. Perutz, T. Johnson, M. Suzuki and J. T. Finch, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5355–5358 CrossRef CAS.
- M. F. Perutz, R. Staden, L. Moens and I. De Baere, Curr. Biol., 1993, 3, 249–253 CrossRef CAS PubMed.
- M. F. Perutz, Trends Biochem. Sci., 1999, 24, 58–63 CrossRef CAS PubMed.
- M. R. Sawaya, S. Sambashivan, R. Nelson, M. I. Ivanova, S. A. Sievers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. W. Wiltzius, H. T. McFarlane, A. Ø. Madsen, C. Riekel and D. Eisenberg, Nature, 2007, 447, 453–457 CrossRef CAS PubMed.
- M. P. Szymański, H. Jędrzejewska, M. Wierzbicki and A. Szumna, Phys. Chem. Chem. Phys., 2017, 19, 15676–15680 RSC.
- A. C. Wang and A. Bax, J. Am. Chem. Soc., 1996, 118, 2483–2494 CrossRef CAS.
- F. Li, J. H. Lee, A. Grishaev, J. Ying and A. Bax, ChemPhysChem, 2015, 16, 572–578 CrossRef CAS PubMed.
- H. Jędrzejewska, M. Kwit and A. Szumna, Chem. Commun., 2015, 51, 13799–13801 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectra. See DOI: 10.1039/c7ob01925d |
| This journal is © The Royal Society of Chemistry 2017 |