
Fluorine-containing bistolanes as light-emitting liquid crystalline molecules†

Abstract
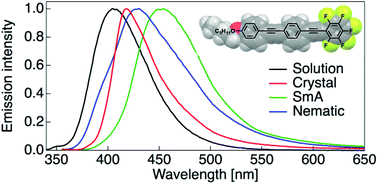
We synthesised a series of dissymmetric bistolane derivatives and evaluated their liquid-crystalline (LC) and photoluminescence properties in detail. In measuring LC behaviours, rational structural design based on the dissymmetric molecular structure and electron-density distribution facilitated the production of the LC phase with a wide temperature range (up to 97 °C). In addition, dissymmetric bistolane derivatives were shown to strongly emit blue-photoluminescence in dilute solution and in crystalline states. It was found that dissymmetric bistolanes possess emissive features in even the LC phase and photoluminescence behaviours such as emission intensity and colour were sensitively switched depending on the molecular aggregate structure caused by applying a thermal stimulus.
 Please wait while we load your content...
Please wait while we load your content...

