
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
α-Halo carbonyls enable meta selective primary, secondary and tertiary C–H alkylations by ruthenium catalysis†
Andrew J.
Paterson
ab,
Callum J.
Heron
ab,
Claire L.
McMullin
a,
Mary F.
Mahon
a,
Neil J.
Press
c and
Christopher G.
Frost
 *a
*a
aDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: c.g.frost@bath.ac.uk; Fax: +44 (0)1225 386231; Tel: +44 (0)1225 386142
bCentre for Sustainable Chemical Technologies, University of Bath, Bath, BA2 7AY, UK
cNovartis Institutes for BioMedical Research, Novartis Campus, Fabrikstrasse, 22, CH-4056, Basel, Switzerland
First published on 26th June 2017
Abstract
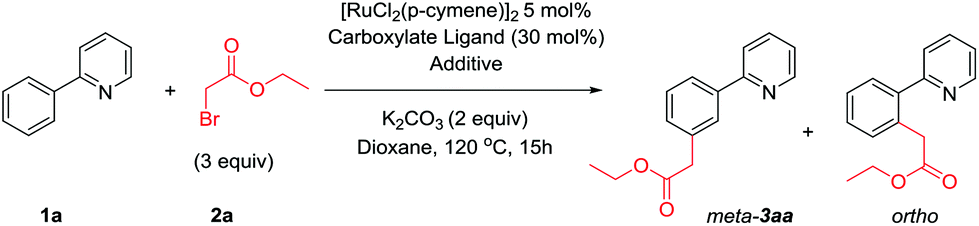
A catalytic meta selective C–H alkylation of arenes is described using a wide range of α-halo carbonyls as coupling partners. Previously unreported primary alkylations with high meta selectivity have been enabled by this methodology whereas using straight chain alkyl halides affords ortho substituted products. Mechanistic analysis reveals an activation pathway whereby cyclometalation with a ruthenium(II) complex activates the substrate molecule and is responsible for the meta selectivity observed. A distinct second activation of the coupling partner allows site selective reaction between both components.
Introduction
The direct functionalization of C–H bonds is an attractive methodology for the atom economic and streamlined synthesis of organic molecules. However, given the relatively low reactivity of C–H bonds and their ubiquity in organic molecules, intrinsic challenges arise when developing new methodologies with adequate reactivity and selectivity. There are however a number of strategies to overcome these challenges and research into this area has received great attention in the last decade.1,2 One of the prevailing methods is the directing group approach. This method utilizes existing functionality within a molecule to coordinate a transition metal catalyst and position it in the vicinity of a C–H bond. This can lead to the formation of a metallacycle which is sufficiently reactive to undergo subsequent functionalization. In the field of aromatic sp2 C–H functionalization, this strategy has enabled a wide range of functionality to be introduced to the ortho position of arenes, utilizing a diverse range of directing groups and transition metal catalysts.3–9 Key to the success of this strategy is the relatively facile formation and conformational stability of the intermediate 5 or 6 membered metallacycles associated with ortho C–H functionalization. However, generating the corresponding metallacycles for more remote C–H bonds such as the meta C–H bond becomes increasingly challenging as ring size increases and conformational stability decreases. Because of this, a number of alternative strategies have been devised (Scheme 1).10,11 | ||
| Scheme 1 Ruthenium catalyzed alkylation of arenes. | ||
In a conceptually similar approach, a range of extended templates containing coordinating pyridine or nitrile groups have been developed, overcoming issues of conformational stability by forming particularly stable metallacycles.12–24 Transient directing groups can also be introduced using carboxylic acids25–29 or norbornene,30–35 effectively installing a temporary directing group to the ortho position, which can undergo a subsequent ortho C–H functionalization. Utilizing non-covalent interactions between a directing group and a substrate can also lead to meta selective reactions.36,37 In an alternative approach, ruthenium catalysis can be employed for the direct meta sulfonation,38,39 secondary40,41 and tertiary41–43 alkylations, bromination44–46 and nitration47 of arenes. These meta selective transformations owe their selectivity to a mechanism involving a directing group assisted ortho cyclometalation, which activates the position para to the newly formed C–Ru bond. Recent studies have also revealed that addition of the coupling partners to the activated substrate are likely to be radical processes whereby a distinct single electron redox cycle could facilitate the generation of the radical species.39,40,42,43,47
As part of our ongoing research into direct meta selective C–H functionalization reactions, we reported the use of α-halo carbonyls as coupling partners for the installation of quaternary carbon centers.42 A range of these reagents are commercially available with the ability to be further modified and have recently been shown to enable synthetically useful mono- and di-fluoromethylations.48,49 We therefore saw their potential as versatile reagents capable of installing other useful functionality to the meta position and enabling previously unprecedented meta selective primary alkylations with ruthenium catalysis, which is currently limited to direct ortho alkylations.50,51 Herein we wish to report the use of α-halo carbonyl reagents to enable useful meta selective primary, secondary and tertiary alkylation reactions. A triaryl phosphine source was necessary to achieve meta selective primary alkylations and mechanistic and computational analysis show that initial cyclometalation with a ruthenium complex is responsible for the meta selectivity observed.
Results and discussion
Optimization
At the outset of our investigation we were interested to achieve meta selective primary alkylations as current ruthenium catalyzed meta selective methodologies are limited to secondary and tertiary alkylations.40,42,43 Given our previous work where reaction with a tertiary α-halo carbonyl could readily furnish meta substituted products,42 we believed that ethyl bromoacetate 2a would be a suitably activated coupling partner to achieve this. We initially began our investigation using conditions known in the field, a ruthenium(II) precatalyst with carboxylate ligands.52–54 However, these resulted in low combined yields of inseparable regioisomers (Table 1, entries 1–4). We previously proposed a dual role of ruthenium in tertiary alkylation reactions; activation of the substrate by cyclometalation, and a single electron redox catalyst to generate an alkyl radical. This proposition was independently supported by the Ackermann group in analogous tertiary alkylation reactions,43 and recently by the Zhao group where ruthenium and ferrocene co-catalysis enabled meta selective benzylations.55 We therefore envisaged a co-catalytic system whereby an additional catalyst could be employed to activate primary alkyl halide coupling partners. A number of copper systems were first employed given their natural abundance and precedence to form alkyl radicals in single electron processes56–59 however no products were observed (entries 6–8). Similarly, photocatalytic ruthenium complexes were also ineffective, providing no benefit over the monocatalytic system (entries 9 and 10). However, the addition of Pd(PPh3)4 significantly improved reactions yields, with 10 mol% loading resulting in near complete selectivity to the meta-substituted product (entry 12). Other palladium sources were less effective however the use of NiCl2(PPh3)2 led to reactions with high meta selectivity (entry 17). During the preparation of this manuscript, work simultaneously published by the Ackermann48 and Wang49 groups demonstrated the use of additional triarylphosphine ligands48 or Pd(PPh3)4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 49 respectively to enable ruthenium catalyzed mono- and di-fluoromethylations. In our system, the addition of free PPh3 also led to reactions with comparable reaction yields and high meta-selectivity suggesting that the previously added metal-phosphine complexes were simply sources of free PPh3 (entry 21). Other phosphine sources gave little additional benefit over PPh3 or were ineffective (entries 23 and 24). Crucially, when no ruthenium complex was employed, no alkylated products were formed (entry 25).
49 respectively to enable ruthenium catalyzed mono- and di-fluoromethylations. In our system, the addition of free PPh3 also led to reactions with comparable reaction yields and high meta-selectivity suggesting that the previously added metal-phosphine complexes were simply sources of free PPh3 (entry 21). Other phosphine sources gave little additional benefit over PPh3 or were ineffective (entries 23 and 24). Crucially, when no ruthenium complex was employed, no alkylated products were formed (entry 25).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Additive | Yielda (%) |
m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :o :o
|
|
a Combined yield for both regioisomers.
b
meta:ortho ratio calculated by 1H NMR.
c Irradiated with blue LEDs.
|
||||
| 1 | No ligand | — | 21 | 2.5:1 |
| 2 | KOAc | — | 24 | 2:1 |
| 3 | MesCOOH | — | 26 | 2.3:1 |
| 4 | AdCOOH | — | 15 | 2:1 |
| 6 | MesCOOH | CuCl (20 mol%), 1,10-Phen (20 mol%) | 0 | — |
| 7 | MesCOOH | CuCl (20 mol%) PMETA (1 eq.) | 0 | — |
| 8 | MesCOOH | Cu2O (10 mol%) 1,10-Phen (12 mol%) | 0 | — |
| 9c | MesCOOH | Ru(bpy)3Cl2 | 21 | 1:1 |
| 10c | MesCOOH | Ru(Phen)3Cl2 | 19 | 0.6:1 |
| 11 | MesCOOH | Pd(PPh3)4 (5 mol%) | 47 | 3.3:1 |
| 12 | MesCOOH | Pd(PPh 3 ) 4 (10 mol%) | 58 |
20:1 |
| 13 | MesCOOH | Pd2(dba)3 (5 mol%) | 17 | 2.5:1 |
| 14 | MesCOOH | Pd(OAc)2 (10 mol%) | 17 | 1.5:1 |
| 15 | MesCOOH | PdCl2(PPh3)2 (10 mol%) | 46 | 10:1 |
| 16 | MesCOOH | Pd(PPh3)4 (15 mol%) | 55 | 19:1 |
| 17 | MesCOOH | NiCl2(PPh3)2 | 38 | 9:1 |
| 18 | MesCOOH | NiCl2(PCy3)2 | 5 | 1:1 |
| 19 | MesCOOH | NiCl2(DME) | 0 | — |
| 20 | MesCOOH | PPh3 (10 mol%) | 51 | 19:1 |
| 21 | MesCOOH | PPh 3 (20 mol%) | 57 |
20:1 |
| 22 | MesCOOH | PPh3 (30 mol%) | 55 |
19:1 |
| 23 | MesCOOH | PCy3 (20 mol%) | 0 | — |
| 24 | MesCOOH | P(4-fluorophenyl)3 | 38 | 15:1 |
| 25 | MesCOOH | PPh3 (no Ru) | 0 | — |
Scope and limitations
With optimized conditions in hand, we aimed to explore the substrate scope with respect to the directing group (Scheme 2). In all cases, near complete selectivity (>20:1) to the meta substituted product was observed. Substitution on the pyridine ring was generally well tolerated although significantly increasing or decreasing electron density had a negative effect on reaction yields. Pyrazole and a range of substituted pyrimidines were also effective directing groups affording the meta alkylated products in good yields. Substitution at the 3 or the 6 position of the pyridine ring completely shut down reactivity, likely due to hindering the ability of the substrate to form a planar cyclometalated complex. Meanwhile conformationally locked benzoquinoline afforded exclusively alkylated product 6aa. X-Ray analysis could unequivocally confirm this regioselectivity (Fig. 1) and supports the proposition that substitution occurs at the position para to the C–Ru bond formed following cyclometalation.
 | ||
| Scheme 2 Directing group scope on unsubstituted arenes Numbers in bold indicate isolated yield when Pd(PPh3)4 was used. aReaction conversion by 1H NMR when Pd(PPh3)4 (10 mol%) was used. bReaction conversion by 1H NMR when PPh3 (20 mol%) was used. | ||
 | ||
| Fig. 1 X-Ray crystal structure of compound 6aa. Ellipsoids are depicted at 30% probability.60 | ||
Next, substitution on the aryl component was considered (Scheme 3). Whereas unsubstituted substrates afforded nearly exclusively the meta substituted products, some regioisomeric products were formed when the electronic properties of the aromatic ring were altered. Generally, electron donating groups at the para position yielded either exclusively the meta product or high selectivity towards this product in modest yields. In contrast, electron withdrawing groups yielded a higher proportion of regioisomeric by-products. Dimethylated substrate 3u afforded none of the meta alkylated product 3ua, likely due to its inability to form a cyclometalated complex. Similarly, incorporating methyl substituents at the ortho or meta position also afforded no products, despite being effective substrates in other ruthenium catalyzed meta alkylations.40,43
 | ||
| Scheme 3 Scope of substituted arenes. aReaction conversion by 1H NMR when Pd(PPh3)4 (10 mol%) was used. bReaction conversion by 1H NMR when PPh3 (20 mol%) was used. cYields quoted are combined yields of both regioisomers when Pd(PPh3)4 was used. dMinor isomer could be assigned as ortho substituted product. | ||
The privileged reactivity of α-halo carbonyls was highlighted when reaction with straight-chain alkyl bromide 2o resulted in no meta alkylated products (Scheme 4) and led to ortho substituted product 1ao in agreement with other work in the field.50,51 No disubstituted products were isolated. The use of Pd(PPh3)4 led to no alkylated products and could be due to undesirable oxidative addition/β-hydride elimination pathways.
 | ||
| Scheme 4 Reaction with unactivated alkyl halide 2o. | ||
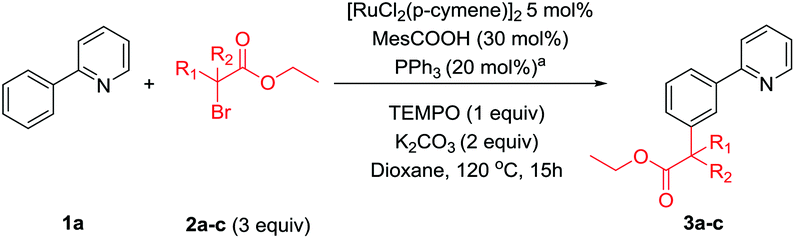
Next, we saw the potential for other α-halo carbonyls to be used to install other useful functionality at the meta position (Scheme 5). When tertiary α-bromo carbonyl reagents were used, the addition of PPh3 did not improve the yield of the corresponding meta substituted products. However, the use of PPh3 with secondary α-bromo carbonyl reagents improved the yield somewhat. Thus, a range of secondary and tertiary alkylated products could be achieved using a [RuCl2(p-cymene)]2 precatalyst with carboxylate (MesCOOH) ligand.40,42,43 In agreement with the reactions carried out in Scheme 2, changing the electronics on pyridine ring generally had a detrimental effect on reaction yields with a 4-Me substituent again proving to be the most effective directing group. We have previously proposed that the key to this type of reactivity when tertiary α-halo carbonyls are employed, is the facile generation of an alkyl radical.42 Captodative stabilization by the electron donating geminal dimethyl substituent along with the electron withdrawing effect of the ester could allow facile homolytic cleavage of the C–Br bond. Thus, coupling partners with solely electron withdrawing groups bound to the α-carbon did not result in alkylated products (3ag, 3ah) whereas the corresponding coupling partners with short alkyl chains introduced could furnish the meta substituted products (3ai). α-Halo ketones could also be effectively coupled (3ae) however α-halo amides were ineffective (3af), again highlighting the importance of captodative stabilization in the coupling partner.
 | ||
| Scheme 5 Scope of α-bromo carbonyl coupling reagents. All numbers indicate isolated yields. aReaction with PPh3 additive (20 mol%). | ||
Next, we investigated the effect of the halide leaving group (Scheme 6). To our surprise, α-chloro carbonyl coupling partners displayed improved performance and this was significant when secondary coupling partners were used, affording the meta substituted products in near quantitative yield. Again, substrates bearing only electron withdrawing groups were ineffective, and support a radical mechanism in these cases. The corresponding α-iodo carbonyl coupling partner was less effective, indicating an order of reactivity of Cl > Br > I, however the reason for this remains unclear.
 | ||
| Scheme 6 Reactions using alkyl halide coupling partners. aReaction with corresponding α-iodo carbonyl. bQuantitative conversion of starting material observed. | ||
Mechanistic considerations
We previously proposed a dual metallic radical based mechanism for meta alkylation reactions involving initial cyclometalation, which activates the position para to the newly installed C–Ru bond for site selective addition.42 Recently, we also showed that substitution happens at the para position of the newly formed C–Ru bond in stoichiometric reactions with cyclometalated complexes in analogous meta sulfonation reactions.39 We were therefore interested to discover the manner in which the alkyl halide coupling partners reacted with the activated arene. In our previous work with meta selective tertiary alkylation reactions, we proposed a second distinct single electron redox cycle that can generate a tertiary alkyl radical, which can add to the cyclometalated complex in a site selective manner.42 To investigate this further, a series of experiments were conducted using radical coupling partner 1,1′-azobis(cyclohexanecarbonitrile) (ABCN, 2n) (Table 2).
Thermal generation of a tertiary radical through loss of nitrogen resulted in no conversion to the meta product when no ruthenium complex was used however when 10 mol% preformed complex [Ru(OMes)2(p-cymene)] was employed, meta alkylated product 1an was formed in a 9% yield showing that ruthenium is essential for the activation of the substrate molecule. Increasing the catalyst loading increased the yield somewhat showing that this is a stoichiometric process. These results support the proposition that cyclometalation activates the position para to the C–Ru bond and external activation of the coupling partner and generation of a tertiary alkyl radical can then result in addition to this complex.
We were then interested to determine whether primary α-halo carbonyl 2a reacted in the same manner as the corresponding secondary and tertiary coupling partners. Reactions with radical scavenger TEMPO were less adversely affected than when the corresponding secondary or tertiary coupling partners (2a and 2b) were used but yielded no meta products when 3 equivalents were used (Table 3). However, this does not conclusively imply a radical mechanism as unlike the secondary or tertiary α-halo carbonyl coupling partners that were shown to be effective in Scheme 4 or the thermally generated tertiary radical formed from ABCN, the radical formed from homolytic cleavage of the C–Br bond in 2a would not benefit from any captodative stabilization.
Furthermore, we have shown theoretically that electrophilic mechanisms are also plausible for cyclometalated ruthenium complexes (Fig. 2 and ESI†). We have applied computational methods to model the electronic properties of the cyclometalated intermediates based on the work of Ritter and co-workers.61 This approach accurately predicts reaction regioselectivity using relative nucleophilic Fukui indices calculated from carbon NBO values and has been applied to 2-phenylpyridine (1a) and other cyclometalated ruthenium complexes.62 The relative Fukui indices in Fig. 2 show that if the organic substrate alone was the active species, then reactivity would most likely occur at C10; para to the pyridine ring due to increased electron density at this position. However, the regioselectivity of the substrate is altered after cyclometalation (Aa), with the most electron rich carbon site for functionalization now indicating addition at C11; the C–H position para to the new Ru–C bond. Cyclometalated complexes containing a phosphine ligand (Ba and Ca) also display similar electronic properties and could serve the additional purpose of blocking the coordination sphere of the ruthenium metal which could otherwise lead to ortho substituted products via an oxidative addition/reductive elimination pathway. The precise role of the phosphine nevertheless remains unclear and could also be involved in the activation of the α-halo carbonyl coupling partner.
 | ||
| Fig. 2 Relative nucleophilicity Fukui indices (fA−) calculated for computed substrates 1a and the corresponding cyclometalated complexes. Calculations were performed at the BP86/6-31G** & SDD(Ru) level of theory. Fukui indices were calculated with NBO total atomic charges from the optimized neutral structure. The most reactive C–H position is highlighted in red. | ||
Based on our most recent mechanistic observations and on previous work conducted by ourselves42 and others40,43 in the field, we propose the following mechanism for meta-selective alkylations with primary, secondary and tertiary α-halo carbonyls (Scheme 7). Reaction of a substrate molecule with a ruthenium carboxylate complex results in a cyclometalated complex activated at the position para to the C–Ru bond. Reaction of this complex with secondary or tertiary radicals externally generated by a single electron Ru(II)/Ru(III)X process then leads to the formation of a cyclometalated arene radical. Single electron oxidation, rearomatization and protodemetalation then leads to the meta substituted products. Primary α-halo carbonyl radicals on the other hand could either undergo either a single electron, or electrophilic addition onto the cyclometalated complex. More detailed mechanistic studies are necessary to accurately determine this process and are currently underway.
 | ||
| Scheme 7 Plausible catalytic cycles. | ||
Conclusions
In summary, we have reported the use of α-halo carbonyls as versatile reagents for the direct meta functionalization of arenes. The procedure is operationally simple and has enabled a range of primary, secondary and tertiary alkylations with the capacity for further synthetic elaborations. A phosphine source was crucial for the installation of primary alkyl groups allowing primary α-halo carbonyls to be coupled selectively to the meta position and was also beneficial for secondary coupling partners. Conversely, straight chain alkyl halides afforded solely ortho substituted products. α-Chloro carbonyls displayed the highest reactivity affording meta substituted products quantitatively in some cases. Experimental and computational mechanistic analysis highlight a dual activation pathway whereby cyclometalation with ruthenium activates the substrate molecule at the position para to the C–Ru bond and is responsible for the meta selectivity observed. Synergistic activation of the α-halo carbonyls then enables site selective alkylation with net meta selectivity.Acknowledgements
We are grateful to the University of Bath, EPSRC DTC in Sustainable Chemical Technologies and Novartis for funding. We acknowledge the valuable assistance of Dr Anneke Lubben (Mass Spectrometry, University of Bath), Dr John Lowe and Dr Catherine Lyall (NMR Spectroscopy, University of Bath). We thank the University of Bath for access to its High Performance Computing Facility.Notes and references
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed.
- D. Y.-K. Chen and S. W. Youn, Chem. – Eur. J., 2012, 18, 9452–9474 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- K. M. Engle, T. Mei, M. Wasa and J. -Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed.
- D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–918 CrossRef CAS PubMed.
- L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC.
- M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS.
- J. Li, S. De Sarkar and L. Ackermann, Top. Organomet. Chem., 2016, 55, 217–258 CrossRef.
- J. Yang, Org. Biomol. Chem., 2015, 13, 1930–41 CAS.
- D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed.
- H. Dai, G. Li, X. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567–7571 CrossRef CAS PubMed.
- L. Wan, N. Dastbaravardeh, G. Li and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 18056–18059 CrossRef CAS PubMed.
- S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778–18781 CrossRef CAS PubMed.
- G. Yang, P. Lindovska, D. Zhu, J. Kim, P. Wang, R. Tang, M. Movassaghi and J. -Q. Yu, J. Am. Chem. Soc., 2014, 136, 10807–10813 CrossRef CAS PubMed.
- M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760–5763 CrossRef CAS PubMed.
- L. Chu, M. Shang, K. Tanaka, Q. Chen, N. Pissarnitski, E. Streckfuss and J.-Q. Yu, ACS Cent. Sci., 2015, 1, 394–399 CrossRef CAS PubMed.
- Y. Deng and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 888–891 CrossRef CAS PubMed.
- M. Bera, A. Maji, S. K. Sahoo and D. Maiti, Angew. Chem., Int. Ed., 2015, 54, 8515–8519 CrossRef CAS PubMed.
- S. Li, H. Ji, L. Cai and G. Li, Chem. Sci., 2015, 6, 5595–5600 RSC.
- S. Li, L. Cai, H. Ji, L. Yang and G. Li, Nat. Commun., 2016, 7, 10443 CrossRef PubMed.
- M. Bera, S. K. Sahoo and D. Maiti, ACS Catal., 2016, 6, 3575–3579 CrossRef CAS.
- T. Patra, R. Watile, S. Agasti, T. Naveen and D. Maiti, Chem. Commun., 2016, 52, 2027–2030 RSC.
- J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429–9432 CrossRef CAS PubMed.
- J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109–4112 CrossRef CAS PubMed.
- J. Luo, S. Preciado and I. Larrosa, Chem. Commun., 2015, 51, 3127–30 RSC.
- J. Luo, S. Preciado, O. Araromi and I. Larrosa, Chem. – Asian J., 2016, 11, 347–350 CrossRef CAS PubMed.
- N. Y. P. Kumar, A. Bechtoldt, K. Raghuvanshi and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 6929–6932 CrossRef CAS PubMed.
- Z. Dong, J. Wang and G. Dong, J. Am. Chem. Soc., 2015, 137, 5887–5890 CrossRef CAS PubMed.
- P.-X. Shen, X.-C. Wang, P. Wang, R.-Y. Zhu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 11574–11577 CrossRef CAS PubMed.
- X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334–338 CrossRef CAS PubMed.
- P. Wang, G.-C. Li, P. Jain, M. E. Farmer, J. He, P.-X. Shen and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14092–14099 CrossRef CAS PubMed.
- P. Wang, M. E. Farmer, X. Huo, P. Jain, P. X. Shen, M. Ishoey, J. E. Bradner, S. R. Wisniewski, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 9269–9276 CrossRef CAS PubMed.
- H. Shi, P. Wang, S. Suzuki, M. E. Farmer and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14876–14879 CrossRef CAS PubMed.
- Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712–717 CrossRef CAS PubMed.
- H. J. Davis, M. T. Mihai and R. J. Phipps, J. Am. Chem. Soc., 2016, 138, 12759–12762 CrossRef CAS PubMed.
- O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef CAS PubMed.
- P. Marcé, A. J. Paterson, M. F. Mahon and C. G. Frost, Catal. Sci. Technol., 2016, 6, 7068–7076 Search PubMed.
- N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–84 CrossRef CAS PubMed.
- J. Li, K. Korvorapun, S. De Sarkar, T. Rogge, D. J. Burns, S. Warratz and L. Ackermann, Nat. Commun., 2017, 8, 15430 CrossRef PubMed.
- A. Paterson, S. St. John-Campbell, M. F. Mahon, N. Press and C. G. Frost, Chem. Commun., 2015, 51, 12807–12810 RSC.
- J. Li, S. Warratz, D. Zell, S. De Sarkar, E. E. Ishikawa and L. Ackermann, J. Am. Chem. Soc., 2015, 137, 13894–13901 CrossRef CAS PubMed.
- C. J. Teskey, A. Y. W. Lui and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54, 11677–11680 CrossRef CAS PubMed.
- Q. Yu, L. Hu, Y. Wang, S. Zheng and J. Huang, Angew. Chem., Int. Ed., 2015, 54, 15284–15288 CrossRef CAS PubMed.
- S. Warratz, D. J. Burns, C. Zhu, K. Korvorapun, T. Rogge, J. Scholz, C. Jooss, D. Gelman and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1557–1560 CrossRef CAS PubMed.
- Z. Fan, J. Ni and A. Zhang, J. Am. Chem. Soc., 2016, 138, 8470–8475 CrossRef CAS PubMed.
- Z. Ruan, S. Zhang, C. Zhu, P. N. Ruth, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2017, 3, 2045–2049 CrossRef PubMed.
- Z. Li, L. Li, Q. Li, K. Jing, H. Xu and G. Wang, Chem. – Eur. J., 2017, 23, 3285–3290 CrossRef CAS PubMed.
- L. Ackermann, P. Novák, R. Vicente and N. Hofmann, Angew. Chem., Int. Ed., 2009, 48, 6045–6048 CrossRef CAS PubMed.
- L. Ackermann, N. Hofmann and R. Vicente, Org. Lett., 2011, 13, 1875–7 CrossRef CAS PubMed.
- L. Ackermann and A. Althammer, Org. Lett., 2008, 10, 2299–2302 CrossRef CAS PubMed.
- E. Ferrer Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, J. Am. Chem. Soc., 2011, 133, 10161–70 CrossRef PubMed.
- L. Ackermann, Chem. Rev., 2011, 111, 1315–45 CrossRef CAS PubMed.
- G. Li, D. Li, J. Zhang, D. Shi and Y. Zhao, ACS Catal., 2017, 7, 4138–4143 CrossRef CAS.
- T. Nishikata, Y. Noda, R. Fujimoto and T. Sakashita, J. Am. Chem. Soc., 2013, 135, 16372–16375 CrossRef CAS PubMed.
- X. Zhang, H. Yi, Z. Liao, G. Zhang, C. Fan, C. Qin, J. Liu and A. Lei, Org. Biomol. Chem., 2014, 12, 6790–6793 CAS.
- G. Caillot, J. Dufour, M.-C. Belhomme, T. Poisson, L. Grimaud, X. Pannecoucke and I. Gillaizeau, Chem. Commun., 2014, 50, 5887–90 RSC.
- R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 8069–8077 CrossRef CAS PubMed.
- Crystal structure determination of 6aa: C17H15NO2 (M = 265.30 g mol−1): monoclinic, space group P21/c, a = 17.0838(3), b = 5.23428(9), c = 14.8641(3) Å, β = 90.8577(17)°, U = 1329.01(4) Å3, Z = 4, T = 150.00(10) K, μ(CuKα) = 0.698 mm−1, Dcalc = 1.326 g cm−3, 12751 reflections measured (5.174° ≤ 2q ≤ 146.686°), 2679 unique (Rint = 0.0339) which were used in all calculations. The final R1 was 0.0369 (I > 2σ(I)) and wR2 was 0.0998 (all data). CCDC 1526788† contains the supplementary crystallographic data for for 6aa.
- G. B. Boursalian, W. S. Ham, A. R. Mazzotti and T. Ritter, Nat. Chem., 2016, 8, 1–6 CrossRef PubMed.
- J. A. Leitch, C. L. Mcmullin, M. F. Mahon, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 2616–2623 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1526788. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob01192j |
| This journal is © The Royal Society of Chemistry 2017 |