
Design, synthesis, and biological activity of second-generation synthetic oleanane triterpenoids†

Abstract
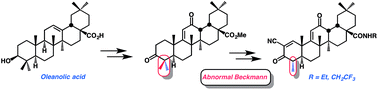
We report the synthesis and biological activity of C-24 demethyl CDDO-Me 2 and the C-28 amide derivatives 3 and 4, which are analogues of the anti-inflammatory synthetic triterpenoid bardoxolone methyl (CDDO-Me) 1. Demethylation of the C-24 methyl group was accomplished via “abnormal Beckmann” rearrangement and subsequent ring A reformation. Amides 3 and 4 were found to be potent inhibitors of the production of the inflammatory mediator NO in vitro.
 Please wait while we load your content...
Please wait while we load your content...

