
Design of transition-metal-doped TiO2 as a multipurpose support for fuel cell applications: using a computational high-throughput material screening approach†
Meng-Che
Tsai
 a,
John
Rick
a,
Wei-Nien
Su
b and
Bing-Joe
Hwang
*ac
a,
John
Rick
a,
Wei-Nien
Su
b and
Bing-Joe
Hwang
*ac
aNano Electrochemistry Laboratory, Department of Chemical Engineering, National Taiwan University of Science and Technology, Taipei 106, Taiwan. E-mail: bjh@mail.ntust.edu.tw
bGraduate Institute of Applied Science and Technology, National Taiwan University of Science and Technology, Taipei 106, Taiwan
cNational Synchrotron Radiation Research Center, Hsinchu 30076, Taiwan
First published on 18th August 2017
Abstract
Transition-metal-doped titanium oxide (TiO2, TiMO2) has great potential for resolving carbon corrosion in current applications of proton exchange membrane fuel cells. To quickly search the available materials, a screening methodology is proposed to screen TiMO2 support materials, where M is a doping element selected from the transition metals (TM) to modify the intrinsic properties of anatase-TiO2. A computational strategy was carried out by combining electronic structures and thermodynamic criteria based on high-throughput density functional theory calculations. Several key requirements that strongly influence the catalytic performance occurring on the catalyst systems of platinum (Pt)/TiO2-based supports, e.g., electrical conductivity, oxygen vacancy, Pt adsorption energy and the charge variation of deposited Pt, have been quantitatively assessed. Among all the key requirements, analysis of the electrochemical reaction indicates that a support material with good electrical conductivity is vital. Furthermore, the doping element not only affects the intrinsic properties of anatase-TiO2, but the resultant TiMO2 also changes the characteristics of the deposited Pt atoms. Combining considerations of oxygen vacancy formation energy and Pt adsorption behavior indicated that the strong metal support interactions result in charge variation of the deposited Pt. The key criteria were developed by a set of defined descriptors, then integrated into a guide map to effectively screen promising anatase-TiMO2 support materials. Using this approach, many systems were found which were potentially suitable to enhance the activity and durability of catalysts in several reactions crucial to the energy economy, e.g., the methanol oxidation reaction and the oxygen reduction reaction. The unification of experimental observations and first principle computational material screening offers a route to accelerating the design and development of TiO2-based support materials.
Design, System, ApplicationRecently, it has become apparent that non-carbon support materials are expected to be applied in fuel cell technology in response to serious concerns of carbon corrosion in existing Pt–carbon catalysts. Among the available alternatives, a TiO2-based support is one of the materials with great potential because of its high chemical stability. In this study, a DFT-based material screening approach is performed to search for suitable doping elements for use with TiO2 to develop novel support materials for fuel cell applications, where key descriptors selected from physical and thermodynamic tests are used with emphasis on the desired systems' functionality in a metal catalyst supported on TiO2-based supports. A guide map for the material screening was derived and clearly indicates the characteristics of the potential alternatives. The guide map developed can be used effectively to decide what properties are needed for the combinations of dopant-support materials. The unification of experimental observations and first-principle computational material screening offers a route to accelerating the design and development of TiO2-based support materials. The screening strategy can be viewed as a general, systematic, DFT-based method incorporating both activity and stability criteria in the search for new metal oxide supports. A similar methodology could be further extended to other reactions of interest. |
Introduction
Searching for new energy materials for green technology applications is a challenge. Fuel cell technology is believed to have the potential to become a major source of clean energy1,2 with potentially important applications in transportation. Using methanol as a fuel, fuel cells are environmentally friendly, and can generate electricity from the methanol oxidation reaction (MOR) at the anode and the oxygen reduction reaction (ORR) at the cathode, producing water (H2O) as the byproduct and such a device is known as a direct methanol fuel cell (DMFC). In contrast to DMFCs, a device with hydrogen (H2) as a fuel at the anode is a so-called proton exchange membrane fuel cell. However, both devices have serious cathodic polarization problems in addition to the fact that the commonly used carbon supported platinum (Pt) catalysts have serious issues that need to be overcome. In fuel cells, the degradation of materials is a problem, because of the weak bonding between the catalyst and its carbon support. Additionally, carbon corrosion during cell operation,3 leads to catalyst agglomeration, surface area reduction, and thus, a lower long-term operational efficiency.Recently, non-carbon support materials have been used in fuel cells, and especially metal oxides which are among the alternatives.3 Highly stable titanium dioxide (TiO2)-based supports have shown potential and been reported in the literature.4–7 A high performance fuel cell catalyst support should have excellent electrical conductivity, high corrosion resistivity, and strong bonding between the catalyst particles and the support's surface.3 Although doping by transition metal (TM) can effectively improve its electrical conductivity,4,5,7–17 the low electrical conductivity of TiO2 limits its performance in electrochemical reactions.
Oxygen vacancies (i.e., defects) are ubiquitous on metal oxide surfaces, especially on reducible oxides. The existence of oxygen vacancies can modify the structure of the materials and change the surface's physical and chemical properties, including its reactivity.18 Taking anatase-TiO2 as an example, it is possible to remove surface oxygen by a surface reduction treatment, e.g., a high temperature hydrogen reduction process. However, high temperature treatment causes particle growth and phase transformation from anatase to rutile,19 which results in a drastically decreased surface area, and thus, poor dispersion of the supported catalyst particles. The produced oxygen vacancies on the oxide's surface play an important role on the metal catalyst/defective oxide interface. They may change the properties of the supported metal catalyst and give rise to other reaction sites, i.e., the electron transfer occurs at the catalyst/support interface or a bi-functional effect that accelerates MOR activity.20–22
An investigation of the interfacial properties between Pt and TiO2 (anatase and rutile), using density functional theory (DFT) calculations, concluded that the Pt atoms have strong bonding with the oxygen vacancy sites on the defective surface, that lead to variations in the electronic structure of Pt.23,24 Metal/support interfaces showing ‘strong metal–support interactions’ (SMSI) are of fundamental interest in heterogeneous electrocatalysis as such catalyst/support interactions often exert electronic effects that influence activation energies and reaction rates.12,25,26 Electron transfer is known as the shift in the d-band center (εd) of the surface electrocatalyst, influencing electrocatalytic/intermediate oxide species interactions, and thus, potentially affecting catalytic activity and stability. For example, the higher filling of the Pt 5d orbital may weaken the Pt–CO bonding caused by carbon monoxide (CO) poisoning in the MOR,27 and slightly decrease the O–O bond scission rate in the ORR.
As described previously, the support properties for fuel cell applications play an important role and they can be tuned appropriately to achieve an optimal performance, because the support not only gives good dispersion of the catalyst but also gives some interaction between them. In addition, to propose a methodology with holistic approaches based on the descriptors seems more important and it will enable and facilitate the selection of the suitable support materials. For the necessities of the support, a DFT-based material screening strategy was used which considered performance descriptors for the support materials and assessed the characteristics of the affected Pt nanocatalyst for the MOR and ORR in fuel cell applications. A guide map was developed for the selection of TiO2-based support materials and validated with experimental values found in the literature.
Methodology
The Vienna Ab initio Simulation Package (VASP)28 was used for the periodic DFT calculations that determined the key requirements for the TM-doped TiO2 (TiMO2) as the support materials in fuel cells. The projector-augmented wave (PAW)29,30 generalized gradient approximation (GGA)31 was used to determine the exchange and correlation energies. For the plane wave calculations, a cutoff energy of 400 eV was applied and structural optimization was carried out until the total energy converged to 10−4 eV.In order to obtain the precise electronic structure of the TiO2-based oxides, a screened hybrid functional as proposed by Heyd, Scuseria and Ernzerhof (HSE06)32 was adopted and the details are shown in the literature.33 Structural optimizations of bulk anatase-TiO2 were performed using HSE06 in order to calculate the equilibrium lattice parameters. In each case the atomic positions, lattice vector and cell angle were allowed to relax (within the space group symmetry). This approach minimizes the problems of Pulay stress and changes in basis set which can accompany volume changes in plane wave calculations. A cutoff of 400 eV, and k-point meshes of 6 × 6 × 2, were found to be sufficient for bulk anatase-TiO2 and their doping systems where 25 at% of TM in the bulk model, as shown in Fig. S1 [ESI†]. All the calculations were deemed to be converged when the forces on all the atoms were less than 0.01 eV Å−1.
Because of the interactions between the TM d-orbital and the Ti ions, the TM dopants can create additionally occupied states in the band gap region of TiO2.34 In order to quantitatively evaluate electrical conductivity improvements for different TiMO2, the metal-induced electronic states (MIES), contributed by intermediate states in the band gap region, were calculated by analyzing the partial density of states (PDOS) results, and defined as an area of intermediate states. Large intermediate states significantly help electron conduction,35 resulting in the TiMO2 having improved electrical conductivity.
To obtain the other interactions between Pt and the TiO2 support, periodic DFT calculations were performed in this study. First, a model with an anatase-TiO2 bulk structure was constructed, upon which structural relaxation with 6 × 6 × 2 k-point was performed. A relaxed bulk TiO2 was cut and expanded to a (101) surface model. This TiO2 substrate was constructed as a slab model containing three TiO2 (101) planes: models were separated from their images in the direction perpendicular to the surface using a 14 Å vacuum layer (Fig. S2(a), ESI†). The bottom TiO2 layer remained fixed to the bulk coordinates and full atomic relaxations were allowed for the top two TiO2 layers. In addition, a 3 × 3 × 1 k-point mesh was used in a 2 × 3 supercell and all the calculations were performed using spin polarization. An undoped-TiO2 (101) slab model consisted of 36 Ti atoms and 72 oxygen atoms, whereas different anatase-TiMO2 based structures were modeled by randomly replacing one in ten Ti ions with various 3d, 4d and 5d [TMs from period 4, 5, 6, see Fig. S2(b) (ESI†) in which the ratio of Ti to TM was ∼7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (around 30 atomic% of TM)]. The design concept of 30 atomic% TM dopants used information from a previous experimental study for the doped-TiO2-supported Pt catalysts, e.g., Pt/Ti0.7Ru0.3O2 and Pt/Ti0.7W0.3O2. Therefore, in this study only the effect of the TM dopants for the TiO2 is considered but the optimization by different amount of TM dopants is excluded to simplify the scope of the research.
:3 (around 30 atomic% of TM)]. The design concept of 30 atomic% TM dopants used information from a previous experimental study for the doped-TiO2-supported Pt catalysts, e.g., Pt/Ti0.7Ru0.3O2 and Pt/Ti0.7W0.3O2. Therefore, in this study only the effect of the TM dopants for the TiO2 is considered but the optimization by different amount of TM dopants is excluded to simplify the scope of the research.
The formation energy of an oxygen vacancy (EOvac) was defined with respect to the energy of an oxygen molecule in the triplet state and calculated according to eqn (1):
 | (1) |
Here, the removed oxygen initially relocates to a neighboring Ti–Ti site for the undoped-TiO2 as shown in Fig. S3(a) (ESI†), whereas Fig. S3(b) (ESI†) indicates that the EOvac calculation for anatase-TiMO2 reveals two defects: Ti–Ti and Ti–M.
To investigate the adsorption behavior of Pt deposited on undoped-TiO2, the single Pt atom was put onto different sites, as shown in Fig. S4 (ESI†). The adsorption energy calculation of the single Pt is defined as follows:
| E1Pt = Eslab+1Pt − (Eslab + Eunsupported Pt) | (2) |
In order to obtain the electron transfer between Pt and undoped-TiO2, Bader charge analysis36 was carried out using theoretical computation. The calculated charge variation of the deposited Pt with respect to unsupported Pt (5d96s,1 10 valence electrons of Pt atom) on the various sites is defined as:
| ΔδPt = δunsupported Pt − δsupported Pt | (3) |
Therefore, a positive ΔδPt indicates the electron transfer from the deposited Pt to the support, whereas a negative ΔδPt means that the electron donation was from the support to the Pt atom.
Results and discussion
Electronic structure of anatase-TiO2 and anatase-TiMO2
To investigate the electronic properties of undoped/doped-TiO2, the PDOS were calculated to determine the variations of the electronic structures. Fig. S5 (ESI†) shows that in anatase-TiO2, the Ti3d and O2p electrons contribute mainly to the conduction band and the valence band, respectively, thus forming the large band gap (around 3.2 eV). As shown in the gap region, there is no additional electronic state, therefore, the electron conducting barrier is extremely high inside the material, which is reflected in its poor electronic conductivity. In the electronic structural analysis, the calculated PDOS results, containing the total density of states and the electrons of Ti3d, Mnd and O2p orbitals, were plotted to investigate the influence of the TM dopants, as shown in Fig. S6–S8 (ESI†) and all the results are summarized in the following discussion.Here we introduced one descriptor to represent the improvement of electrical conductivity, namely MIES, which indicates the additional electronic states created by the TM dopants within the band-gap region.34 The additional states significantly help electron conduction,35 resulting in TiMO2 having an improved electrical conductivity. The MIES of all the TiMO2 can be calculated by integrating the area of the additional states in the PDOS of the TiMO2, and the results of this are shown in Fig. 1. A variety of the TM-modified electronic structures for TiO2 are shown in the ESI.† TM dopants including hafnium (Hf), Pt, scandium (Sc), yttrium (Y), and zirconium (Zr), do not create additional MIES, which means not much enhancement for the conduction of electrons. In PDOS analysis, different TM dopants show their significant contributions in the electronic structure and some examples are presented to discuss the characteristics.
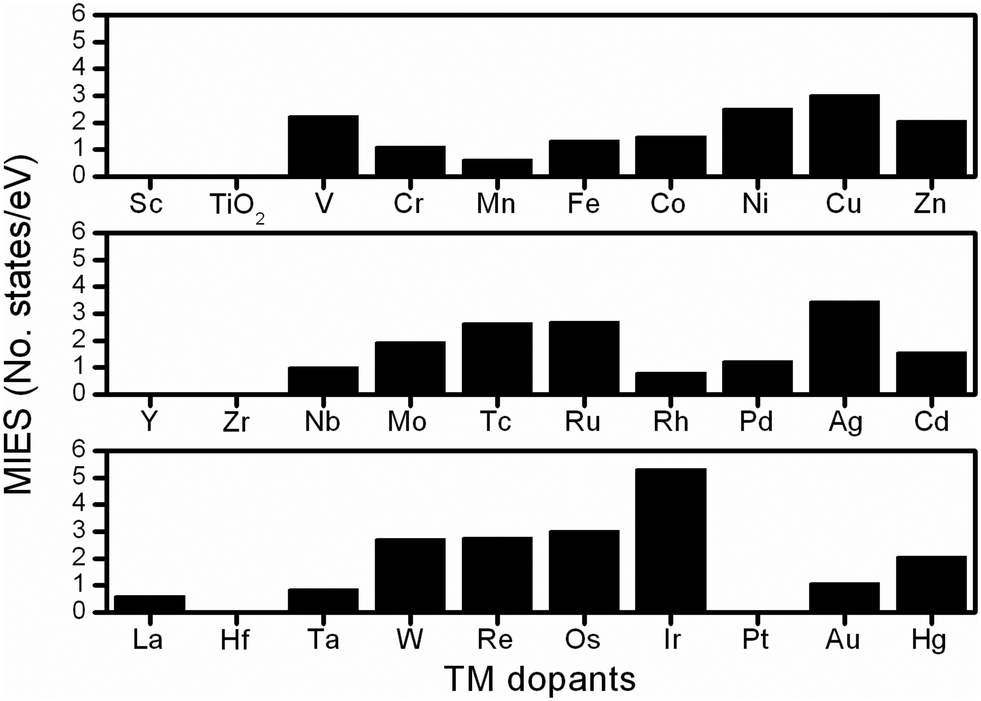 | ||
| Fig. 1 MIES values calculated from the PDOS results of the TiMO2. | ||
For the IIIB group [Sc, Y and lanthanum (La)], their PDOS show that the O 2p orbital was partially localized in the gap region above the Fermi level which indicates the presence of unsaturated oxygen ions in the TiO2 matrix because of the charges of the IIIB elements (3+), and especially in the La-doping the contribution of the O 2p orbital plays a role as an intermediate state. For the IVB group (Zr and Hf), the dopants did not create an additional electronic state in gap region, which indicates no further improvement in the electrical conductivity. For the other TM dopants, their significant MIES in the gap region can also be found in the electronic structures. As seen in the results of PDOS, numerous intermediate states and localized intermediate states (bandwidth <0.15 eV)35 appeared, showing their significant role in modifying the electronic structures. Taking vanadium (V)-doping as an example, a large MIES, mainly consisting of a V-3d orbital, is formed whereas other 3d TMs also exhibit similar behavior. In the 4d TM dopants, niobium (Nb)- and molybdenum (Mo)-doping show a significant feature that the partially occupied Ti 3d electrons were located below their Fermi level which implies that the oxidation state of the Ti ion was decreased whereas the same feature can be found in tantalum (Ta)-doping (5d dopant). In this work, the calculated MIES only provides a reference basis for the modified electronic structure, from which TM dopants can be selected to improve the electrical conductivity of TiO2. A lot of work has been done to prove that many TM dopants can improve the electrical conductivity of TiO2, and the results are summarized in Table S2 (ESI†). Furthermore, the PDOS results show not only the improvement in conductivity but also the interaction between Ti and the TM dopants, and the band alignment for orbital hybridization, which shows useful information for designing the other TiO2-based materials as the co-catalyst.
Formation energy of oxygen vacancy for anatase-TiMO2
For the metal oxide surface, the existence of oxygen vacancies plays a critical role, especially for the interactions between metal catalysts and their oxide supports. In addition, the surface oxygen vacancies also influence variations in the metal catalysts' electronic structures. In this research, for each metal oxide surface, the oxygen, abstracted for vacancy formation, was removed together with its two neighboring Ti atoms (Ti–Ti site) or Ti–M site. Defects originating from the oxygen vacancies were used to investigate the formation energy of vacancies on different anatase-TiMO2 (101) surfaces. For undoped-TiO2 (101), the calculated EOvac was 4.063 eV, which is consistent with previously reported DFT results (4.35 eV).23 It has been shown that the formation energy of oxygen vacancies strongly depends on the nature of the metal oxide. The large EOvac of anatase-TiO2 (101) indicates that the creation of surface oxygen vacancies is very difficult. This result is in agreement with the previous experimental observations, where a high surface area anatase-TiO2 shows a reduction peak around 840 K,19 and indicates that the oxygen vacancy can be produced using thermal treatment at a high temperature leading to a great loss of the surface area.Fig. 2 shows oxygen vacancy formation energies (EOvac) on different anatase-TiMO2 surfaces. Firstly it was observed that the EOvac of TiMIIIBO2 (M = Sc, Y and La) is <0, i.e., negative, indicating that surface oxygen vacancies were spontaneously formed. According to the Bader charge analysis results (Table S1; ESI†), Ti ions of undoped-TiO2 show values typical of Ti4+ (1.1491), whereas the different Ti ions of TiMIIIBO2 take similar δTi/e values, i.e., the charge state of the M ions is 3+, which leads to an excess of oxygen ions. Therefore, when modeling this approach, the amount of stoichiometric oxygen was too high causing the lattice oxygen ions to become unstable during structural relaxation (i.e., structural distortion) so that the oxygen ions were readily removed (lower EOvac) and similar phenomena can be observed with TiMIIBO2 [M = cadmium (Cd), mercury (Hg) or zinc (Zn)]. In addition, some Ti ions of anatase-TiMO2 show that the oxidation state tends to 3+ because the Bader charge value is much larger than 1.1491, i.e., Nb-doped, Ta-doped, tungsten (W)-doped, resulting in higher oxidation states of these dopants.
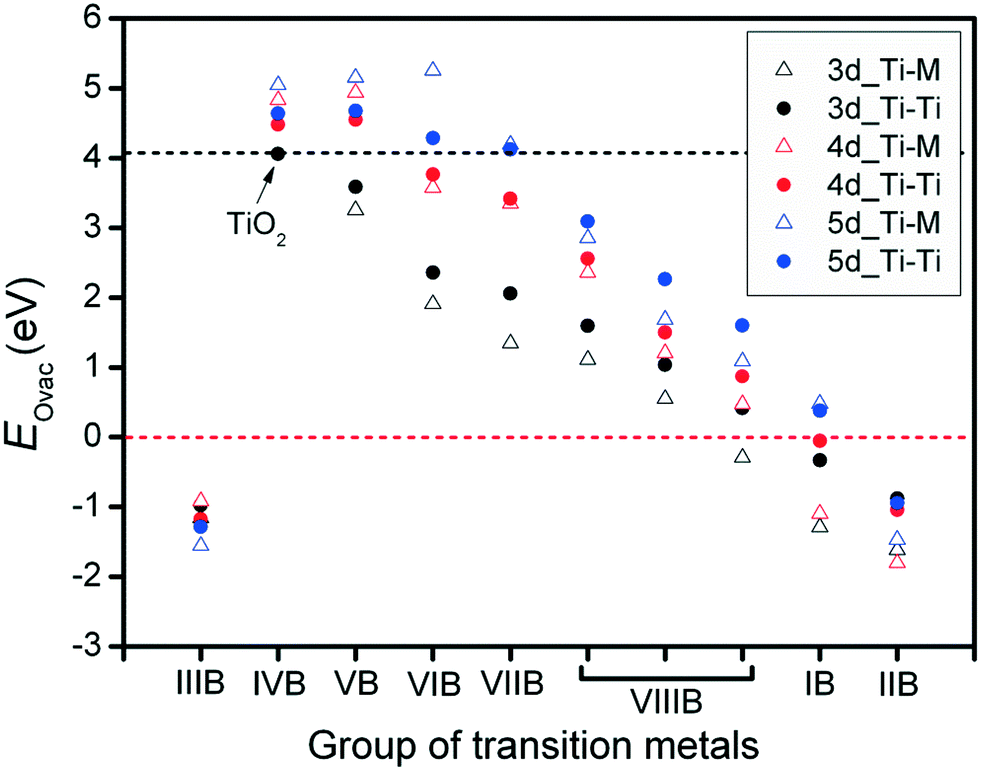 | ||
| Fig. 2 Calculated formation energies of oxygen vacancies for Ti–Ti and Ti–M sites of anatase-TiMO2 (101) surfaces. Black and red dashed lines indicate the values for undoped-TiO2 (101) and the values of EOvac at zero, respectively. | ||
An overview of the EOvac results in Fig. 2 indicates that the variation trend shows a decrease from the IVB group to the IIB group except for IIIB group, even in the TiMIBO2 and TiMIIBO2EOvac values are negative, implying that the d-orbital filling is nearly complete, so that d electrons are not easily shared with oxygen to form a bond, which is attributed to the hybridization between the d-band (TM) and p-band (oxygen). The trend for the EOvac results is similar to the previous study, which shows that the outer electrons can be a descriptor for other thermodynamic properties.37,38 Furthermore, it was found that the EOvac for all the TiM3dO2 is smaller than those of undoped-TiO2, whereas in addition, the surface oxygen ions are preferentially removed from Ti–M sites because of the lower EOvac. For TiM4dO2, TiYO2, TiZrO2 and TiNbO2, it is shown that the surface oxygen ions are preferentially removed from the Ti–Ti sites whereas the rest of the system shows that surface oxygen ions are preferentially removed from the Ti–M sites.
It is worth noting that smaller EOvac is usually expected for most dopants. TiLaO2 and TiHgO2 provide even negative values of EOvac. However, for the TiM5dO2, there are more systems with values of EOvac that are larger than those of undoped-TiO2, i.e., Hf, rhenium (Re), Ta, and W. Furthermore, Ti–M usually provides a lower value of EOvac than Ti–Ti sites. The results clearly indicate that doped systems easily create surface oxygen vacancies in different anatase-TiMO2 (101) variants, and provide useful information to further the understanding of metal catalysts deposited on supports.
Adsorption energy of the single Pt atom on anatase-TiMO2
The interactions between metal catalysts and oxide supports were investigated by calculating the adsorption energies of single Pt atoms (E1Pt) on defect-free, Ti–Ti and Ti–M sites of different anatase-TiMO2 compounds. Comparing the differences of E1Pt enables an analysis of the dispersion, nucleation, and stability of the deposited Pt. For undoped-TiO2 (101), the most stable site for the single Pt atom is the surface oxygen vacancy, situated at the Ti–Ti bridge site, with an adsorption energy of 4.601 eV (see Fig. S4(b), ESI†) for the adsorption configuration, which is in agreement with a previously reported DFT result (4.87 eV).23 Because of the low adsorption energy of the Pt deposited on the defect-free sites of undoped-TiO2 (3.105 eV), the surface oxygen vacancies enhance the interactions between Pt and TiO2, leading to a better dispersion of Pt catalyst on the support, while providing additional nucleation sites. Fig. 3 shows the adsorption energy of a single Pt atom (E1Pt) deposited on different sites. The overall results show a volcano relationship of Pt stability from TiMIIIBO2 to TiMIIBO2, indicating that the deposited Pt atom/support-surface bonding strength increases across the groups in the directions of both IIIB and IIB TM dopants. For TiM3dO2, TiCrO2 provides the weakest E1Pt, for TiM4dO2, TiRuO2 is the weakest E1Pt and for TiM5dO2, TiIrO2 gives the weakest E1Pt. However, in order to create the SMSI, EOvac and E1Pt were combined and used to explore the likelihood of an interface formation. According to the EOvac results, it is known that some TiMO2 supports are more readily reduced than others. For example, the surface of TiTaO2 is not easily reduced, whereas that of TiRuO2 is, because TiMIIIBO2 provides O-vacancy rich surfaces. Therefore, combining the results of EOvac and E1Pt should make it possible to find more systems displaying SMSI.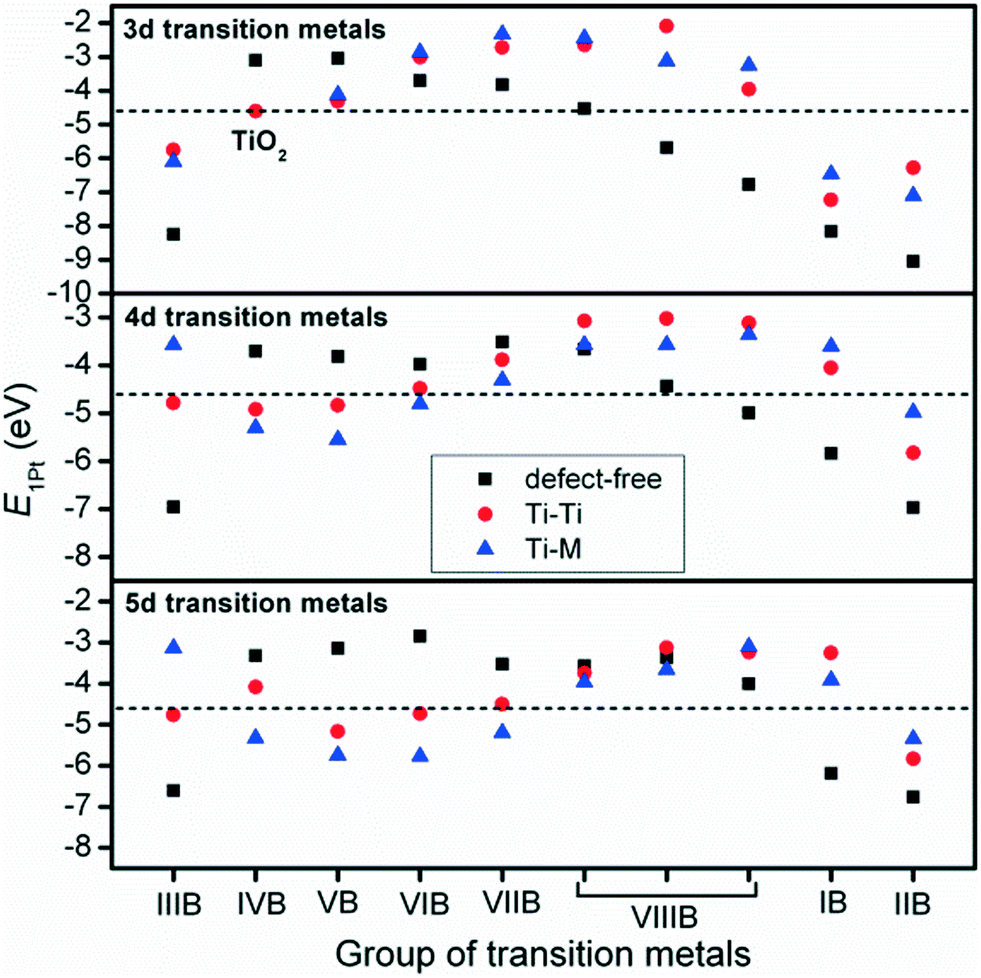 | ||
| Fig. 3 Calculated adsorption energy of a single Pt atom deposited on defect-free (black square), Ti–Ti (red circle) and Ti–M (blue triangle) sites of the anatase-TiMO2 (101) surface, and the black dashed line indicates the E1Pt of the most stable site for Pt/undoped-TiO2. | ||
Consequently, a new descriptor,  , which is defined here as: E1Pt + EOvac, was evaluated, and which indicates that whether Pt atom is thermodynamically favorable on defect site, which can be caused by oxygen spillover. A negative value means that Pt is energetically stable on a defect site. Basically, the trend of
, which is defined here as: E1Pt + EOvac, was evaluated, and which indicates that whether Pt atom is thermodynamically favorable on defect site, which can be caused by oxygen spillover. A negative value means that Pt is energetically stable on a defect site. Basically, the trend of 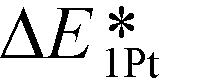 follows the trend of EOvac (Fig. 2). As shown in Fig. S9 (ESI†),
follows the trend of EOvac (Fig. 2). As shown in Fig. S9 (ESI†), 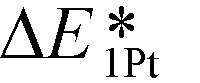 for all the Pt/TiMO2 tested is negative, which implies a strong interaction between Pt and TiMO2 originated from the defect sites, which can determine accordingly the charge state of the deposited Pt (ΔδPt). From the experimental point of view, heat treatment under a H2 environment is recommended to generate the Pt-defect interfaces. In addition, the interaction between the Pt atom and the graphite layer (carbon support) was also calculated, and which shows the Pt adsorption energy is −2.33 eV, which is much weaker than the most stable Pt on all the TiMO2. Therefore, although the Pt adsorptions with some TiMO2 are weaker, they are still stable compared to carbon, and this indicates that TiO2-based materials can effectively enhance the binding strength of Pt to the oxide supports.
for all the Pt/TiMO2 tested is negative, which implies a strong interaction between Pt and TiMO2 originated from the defect sites, which can determine accordingly the charge state of the deposited Pt (ΔδPt). From the experimental point of view, heat treatment under a H2 environment is recommended to generate the Pt-defect interfaces. In addition, the interaction between the Pt atom and the graphite layer (carbon support) was also calculated, and which shows the Pt adsorption energy is −2.33 eV, which is much weaker than the most stable Pt on all the TiMO2. Therefore, although the Pt adsorptions with some TiMO2 are weaker, they are still stable compared to carbon, and this indicates that TiO2-based materials can effectively enhance the binding strength of Pt to the oxide supports.
Charge variation of the deposited Pt atom
To investigate charge variations, single Pt atoms were deposited on defect-free and defect sites, and the Bader charge analysis36 was used to calculate the number of valence electrons for Pt/TiMO2. According to eqn (3), positive values of ΔδPt indicate that the deposited Pt donates electron density to the support and a negative ΔδPt means that the direction of the electron transfer is from the support to Pt. For undoped-TiO2, the single Pt atom deposited on the defect-free site is weakly positively charged (+0.1096 e−). A more important result was that the negative charge density of the Pt atom, on the defect-site, is significant (−0.8757 e−) and which is in agreement with previous DFT results.23 This result provides more evidence that the charge variation of the deposited Pt is able to be controlled by changing the surface condition of TiO2 (101), e.g., a reduction process can be performed to create surface oxygen vacancies to vary the ΔδPt.For TiMO2, variations of ΔδPt are shown in Fig. 4. In the TiM3dO2 series, all Pt deposited on a defect-free site shows a positive charge whereas their positive charges increase as the atomic number increases, except for TiScO2 in which undoped-TiO2 shows the lowest positive charge. In addition, TiScO2, TiNiO2, TiCuO2 and TiZnO2 provide positively charged Pt on defect-free and defect sites, whereas the rest of the TiM3dO2 series provides positively charged Pt only on defect sites. Furthermore, TiM4dO2 and TiM5dO2 show similar trends for the deposited Pt atom and it was even found that some TiMO2 provides negatively charged Pt for both defect-free and defect sites, i.e., TiZrO2, TiHfO2 and TiTaO2.
 | ||
| Fig. 4 Calculated charge variation of a single Pt atom deposited on a defect-free (black square), Ti–Ti (red circle) and Ti–M (blue triangle) sites of anatase-TiMO2 (101), and a black dashed line indicates that ΔδPt is equal to zero. | ||
Although the deposition of a single Pt atom on a metal oxide support is experimentally difficult to achieve, the trend of ΔδPt shown in Fig. 4 predicts the direction of electron transfer between Pt and TiMO2 supports, suggesting that a positively or negatively charged Pt catalyst is needed to enhance the activity for the target reaction. For example, in the anodic reaction of DMFCs, CO poisoning on Pt catalysts is a serious problem because of the strong bonding strength of Pt–CO. Generally, the Pt–CO bonding strength is weakened by higher filling of Pt 5d orbital.27 For the ORR, the Pt catalyst with good activity possesses a slightly higher population of its d-band,5,17,39,40i.e., electronic effect, which implies that electron donation from TiMO2 supports to Pt is beneficial to oxygen reduction. Additionally, the ΔδPt of the Pt deposited on defect sites always shows the direction with a negative change, which means that Pt-defect interfaces are more suitable for the ORR. According to the results, different TiMO2 supports can be used to modify the Pt catalyst's electronic structure.
Guide map for material screening
As mentioned above, good electrical conductivity as well as the possible interaction between catalyst and support are features required for a support material to assist the catalysis. Again, four descriptors, MIES, EOvac, E1Pt, and ΔδPt, were calculated for the TiMO2-based support and Pt, and it will be practically beneficial if these descriptors can be consolidated into a tool allowing quick screening for the potential supports. Considering the TiO2-based material as supports for the fuel cells, some rules were established to extract those key properties, which will benefit the cell's performance. These extracted results are summarized in Fig. 5 and give an indication of the following features: | ||
| Fig. 5 A guide map using four descriptors: MIES, EOvac, E1Pt and ΔδPt, of the TiMO2 as the support for Pt catalysts in fuel cells. Upper (green) and lower (blue) regions represent positively charged and negatively charged Pt, respectively. Improvement of electrical conductivity (MIES) is shown in the different colors of the circles. Solid circles indicate that the surfaces are reducible compared to pure anatase-TiO2, whereas dashed circles represent the opposite case. | ||
♦ MIES values of different TiMO2 in relation to improvements in electrical conductivity, shown by circles of different colors.
♦ EOvac: indicates if TiMO2 possesses a reducible surface, represented by solid (reducible) and dashed (irreducible) circles.
♦ E1Pt: indicates the stability of dispersed Pt catalyst on support. The E1Pt values showing the deposition Pt on defect sites were chosen as the X axis of the figure.
♦ Negative or positive ΔδPt: indicates the electron transfer between Pt and TiMO2, shown on the Y axis. The selected ΔδPt for each doping system corresponds to the selected E1Pt. Here the variations of ΔδPt provide solely the direction of electron transfer between Pt and the TiMO2 oxide support, i.e., a negatively charged Pt or a positively charged Pt.
In general, Fig. 5 is separated into two regions, positively charged and negatively charged Pt, and represents the electron transfer direction effected by the different TiMO2. In the cathodic reaction of fuel cells (ORR), O–O bond scission is thought to be the rate determining step on Pt catalysts, thus generally higher populations of Pt 5d orbitals should mitigate the O–O bond breaking rate and improve the activity. This implies that negatively charged or electron-rich Pt is more favorable. As shown in the literature, the reduced Pt 5d orbital vacancy enhancing the ORR activity has been reported.4,5,7,41 As a result, the electronic effect plays an important role in ORR, whereas the electron transfer direction between deposited Pt and TiMO2 may determine the catalytic performance, and thus, the systems in the region of negatively charged Pt would be recommended as ORR electrochemical catalysts. In addition, a similar requirement for MOR can be found, from a weakening of Pt–CO bonding strength, because of the higher filling of the Pt 5d orbital.27 In fact, the activity of MOR is mainly dominated by a bi-functional effect derived from the catalysts. According to the reaction mechanism of MOR, adsorbed OH produced from H2O dissociation promotes a CO oxidation reaction (CO + OH → COOH), therefore the oxygen vacancies on the TiMO2 surface possibly play a role in providing a site for adsorbed OH. For example, Lobo and Conrad found that adsorption and dissociation of H2O molecules take place on a five-fold coordinated Ru site of the RuO2 (110) surface, however, on perfect RuO2 (110), surface water dissociation is almost negligible.42 The conditions for the dissociative or non-dissociative adsorption of H2O have been investigated on many metal oxide surfaces.43–56 Therefore, those metal cations can perhaps be selected as a dopant to possess the bi-functional effect required for MOR. Additionally, Fig. 5 shows more information about the oxygen vacancy, where the solid circle indicates the EOvac that are lower than TiO2 and are, therefore, those surfaces that can bind with other reaction species to achieve the bi-functional catalysis. These combined results, containing MIES, EOvac, E1Pt and ΔδPt, give the guide that shows how to select the proper TiMO2 for fuel cell reactions, i.e., the systems in the negatively charged Pt region are recommended. However, the results cannot exactly represent the best system. The results only provide a trend whereas choosing the different TM dopants estimates the possible influences caused by these dopants. The focus in this paper is to provide a method or a guide map which enables quick screening of potential supports and dopants, as well as assessing how the doped oxide support affects the properties of the Pt catalyst. Likewise, it is also hoped that the calculations can be helpful when studying experimental findings.
Nowadays, several TM doped-TiO2 supported Pt catalysts have been explored for use as fuel cells, and this information is presented in Table S2 (ESI†). Basically, the systems, shown on the guide map, agree well with most of the experimental results found in the literature, and the guide map provides a good reference for exploring the TM doped-TiO2 supports that show enhanced electrocatalysis such as ORR, and MOR. Yet the evidence is still not clear for the role of the dopant in the Pt/TiMO2. Thus, a more complete and systematic study is necessary. Therefore, by analyzing these key descriptors, the functions of the dopants can be extensively explored and understood. It is believed that the guide map not only helps to understand for the role of dopants but can also be applied to other oxide supports. Based on the summary made using the descriptors, new and potential TiO2-based supports can be designed for different demands.
Conclusions
A high-throughput computational screening approach has been used to develop TiO2-based supports for fuel cell applications. The DFT-based calculations were carried out to investigate the key requirements for anatase-TiMO2 supports in fuel cell applications, which highlighted crucially important factors governing Pt/TiMO2 interfaces. A guide map for the TiMO2 support materials predicted the properties affected by TM dopants, containing MIES, EOvac, E1Pt and ΔδPt data. This study suggests the importance of developing TiMO2 which have improved electrical conductivity and the interaction between Pt and oxide support for enhancing electrochemical reactions such as ORR and MOR. These computationally determined properties can be used to explain experimental phenomena and predict the outcome of future experimental approaches, e.g., measurement of electrical conductivity (or the band gap), defect formation (oxygen vacancy), the electronic property of the Pt catalyst and so on. The screening strategy can be viewed as a general, systematic, DFT-based method incorporating both activity and stability criteria in the search for new metal oxide supports.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Authors are grateful for financial support from the Ministry of Science and Technology (MOST 103-3113-E-011-001-, MOST 104-3113-E-011-001-) and the Top Universities Program from the Ministry of Education. The Authors also wish to thank the National Center for High-performance Computing (NCHC), and the Department of Chemical Engineering, National Taiwan University of Science and Technology (NTUST), for providing computer resources and research facilities.References
- C. Wang, D. van der Vliet, K. L. More, N. J. Zaluzec, S. Peng, S. Sun, H. Daimon, G. Wang, J. Greeley, J. Pearson, A. P. Paulikas, G. Karapetrov, D. Strmcnik, N. M. Markovic and V. R. Stamenkovic, Nano Lett., 2010, 11, 919–926 CrossRef PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- Y.-J. Wang, D. P. Wilkinson and J. Zhang, Chem. Rev., 2011, 111, 7625–7651 CrossRef CAS PubMed.
- V. T. Thanh Ho, K. C. Pillai, H.-L. Chou, C.-J. Pan, J. Rick, W.-N. Su, B.-J. Hwang, J.-F. Lee, H.-S. Sheu and W.-T. Chuang, Energy Environ. Sci., 2011, 4, 4194–4200 Search PubMed.
- V. T. T. Ho, C.-J. Pan, J. Rick, W.-N. Su and B.-J. Hwang, J. Am. Chem. Soc., 2011, 133, 11716–11724 CrossRef CAS PubMed.
- D. Wang, C. V. Subban, H. Wang, E. Rus, F. J. DiSalvo and H. D. Abruña, J. Am. Chem. Soc., 2010, 132, 10218–10220 CrossRef CAS PubMed.
- A. Kumar and V. Ramani, J. Electrochem. Soc., 2013, 160, F1207–F1215 CrossRef CAS.
- K.-W. Park and K.-S. Seol, Electrochem. Commun., 2007, 9, 2256–2260 CrossRef CAS.
- H. Chhina, S. Campbell and O. Kesler, J. Electrochem. Soc., 2009, 156, B1232–B1237 CrossRef CAS.
- S.-Y. Huang, P. Ganesan and B. N. Popov, Appl. Catal., B, 2010, 96, 224–231 CrossRef CAS.
- Y. Liu, J. M. Szeifert, J. M. Feckl, B. Mandlmeier, J. Rathousky, O. Hayden, D. Fattakhova-Rohlfing and T. Bein, ACS Nano, 2010, 4, 5373–5381 CrossRef CAS PubMed.
- C. V. Subban, Q. Zhou, A. Hu, T. E. Moylan, F. T. Wagner and F. J. DiSalvo, J. Am. Chem. Soc., 2010, 132, 17531–17536 CrossRef CAS PubMed.
- Z. Wenfang, L. Qingju, Z. Zhongqi and Z. Ji, J. Phys. D: Appl. Phys., 2010, 43, 035301 CrossRef.
- P. Goswami and J. N. Ganguli, Dalton Trans., 2013, 42, 14480–14490 RSC.
- A. T. Kuvarega, R. W. M. Krause and B. B. Mamba, J. Nanosci. Nanotechnol., 2013, 13, 5017–5027 CrossRef CAS PubMed.
- X. Li, Z. Guo and T. He, Phys. Chem. Chem. Phys., 2013, 15, 20037–20045 RSC.
- A. Kumar and V. Ramani, ACS Catal., 2014, 4, 1516–1525 CrossRef CAS.
- R. Schaub, E. Wahlström, A. Rønnau, E. Lægsgaard, I. Stensgaard and F. Besenbacher, Science, 2003, 299, 377–379 CrossRef CAS PubMed.
- J. Strunk, W. C. Vining and A. T. Bell, J. Phys. Chem. C, 2010, 114, 16937–16945 CAS.
- N. M. Marković, H. A. Gasteiger, P. N. Ross Jr, X. Jiang, I. Villegas and M. J. Weaver, Electrochim. Acta, 1995, 40, 91–98 CrossRef.
- N. P. Lebedeva, M. T. M. Koper, J. M. Feliu and R. A. van Santen, J. Phys. Chem. B, 2002, 106, 12938–12947 CrossRef CAS.
- S. W. Lee, S. Chen, W. Sheng, N. Yabuuchi, Y.-T. Kim, T. Mitani, E. Vescovo and Y. Shao-Horn, J. Am. Chem. Soc., 2009, 131, 15669–15677 CrossRef CAS PubMed.
- Y. Han, C.-J. Liu and Q. Ge, J. Phys. Chem. C, 2007, 111, 16397–16404 CAS.
- H. Iddir, V. Skavysh, S. Öğüt, N. D. Browning and M. M. Disko, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 041403 CrossRef.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- E. Antolini, Appl. Catal., B, 2010, 100, 413–426 CrossRef CAS.
- M. A. Rigsby, W.-P. Zhou, A. Lewera, H. T. Duong, P. S. Bagus, W. Jaegermann, R. Hunger and A. Wieckowski, J. Phys. Chem. C, 2008, 112, 15595–15601 CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, presented in part at the Electronic Structure in Solids '91, Akademie Verlag, Berlin, 1991 Search PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- G. Liu, L. Wang, H. G. Yang, H.-M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
- G. Shao, J. Phys. Chem. C, 2009, 113, 6800–6808 CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford, 1990 Search PubMed.
- F. Calle-Vallejo, N. G. Inoglu, H.-Y. Su, J. I. Martinez, I. C. Man, M. T. M. Koper, J. R. Kitchin and J. Rossmeisl, Chem. Sci., 2013, 4, 1245–1249 RSC.
- F. Calle-Vallejo, O. A. Díaz-Morales, M. J. Kolb and M. T. M. Koper, ACS Catal., 2015, 5, 869–873 CrossRef CAS.
- J.-H. Kim, S. Chang and Y.-T. Kim, Appl. Catal., B, 2014, 158–159, 112–118 CrossRef CAS.
- M.-C. Tsai, T.-T. Nguyen, N. G. Akalework, C.-J. Pan, J. Rick, Y.-F. Liao, W.-N. Su and B.-J. Hwang, ACS Catal., 2016, 6, 6551–6559 CrossRef CAS.
- X. Wang, Y. Orikasa and Y. Uchimoto, ACS Catal., 2016, 6, 4195–4198 CrossRef CAS.
- A. Lobo and H. Conrad, Surf. Sci., 2003, 523, 279–286 CrossRef CAS.
- P. A. Thiel and T. E. Madey, Surf. Sci. Rep., 1987, 7, 211–385 CrossRef CAS.
- V. E. Henrich and P. A. Cox, The Surface Science of Metal Oxides, Cambridge University, Cambridge, 1994 Search PubMed.
- M. A. Henderson, Surf. Sci. Rep., 2002, 46, 1–308 CrossRef CAS.
- M. A. Henderson, S. A. Joyce and J. R. Rustad, Surf. Sci., 1998, 417, 66–81 CrossRef CAS.
- G. Zwicker and K. Jacobi, Surf. Sci., 1983, 131, 179–194 CrossRef CAS.
- V. A. Gercher and D. F. Cox, Surf. Sci., 1995, 322, 177–184 CrossRef CAS.
- M. B. Hugenschmidt, L. Gamble and C. T. Campbell, Surf. Sci., 1994, 302, 329–340 CrossRef CAS.
- M. A. Henderson and S. A. Chambers, Surf. Sci., 2000, 449, 135–150 CrossRef CAS.
- M. A. Henderson, Surf. Sci., 1996, 355, 151–166 CrossRef CAS.
- C. A. Muryn, P. J. Hardman, J. J. Crouch, G. N. Raiker, G. Thornton and D. S. L. Law, Surf. Sci., 1991, 251–252, 747–752 CrossRef CAS.
- D. S. Toledano, P. Metcalf and V. E. Henrich, Surf. Sci., 2001, 472, 21–32 CrossRef CAS.
- S. Warren, W. R. Flavell, A. G. Thomas, J. Hollingworth, P. M. Dunwoody, S. Downes and C. Chen, Surf. Sci., 1999, 436, 1–8 CrossRef CAS.
- T. Kendelewicz, C. S. Doyle, X. Carrier and G. E. Brown, Surf. Rev. Lett., 1999, 06, 1255–1263 CrossRef CAS.
- G. Lu, A. Linsebigler and J. T. Yates, J. Chem. Phys., 1994, 98, 11733–11738 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7me00039a |
| This journal is © The Royal Society of Chemistry 2017 |