
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On-chip label-free protein analysis with downstream electrodes for direct removal of electrolysis products†
Kadi L.
Saar‡
 a,
Yingbo
Zhang‡
a,
Thomas
Müller
ab,
Challa P.
Kumar
a,
Sean
Devenish
b,
Andrew
Lynn
b,
Urszula
Łapińska
a,
Xiaoting
Yang§
d,
Sara
Linse
d and
Tuomas P. J.
Knowles
*ac
a,
Yingbo
Zhang‡
a,
Thomas
Müller
ab,
Challa P.
Kumar
a,
Sean
Devenish
b,
Andrew
Lynn
b,
Urszula
Łapińska
a,
Xiaoting
Yang§
d,
Sara
Linse
d and
Tuomas P. J.
Knowles
*ac
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: tpjk2@cam.ac.uk
bFluidic Analytics Limited, Unit 5 Chesterton Mill, French's Road, Cambridge CB4 3NP, UK
cCavendish Laboratory, Department of Physics, University of Cambridge, J J Thomson Ave, Cambridge CB3 0HE, UK
dDepartment of Biochemistry and Structural Biology, Lund University, SE-22100 Lund, Sweden
First published on 1st December 2017
Abstract
The ability to apply highly controlled electric fields within microfluidic devices is valuable as a basis for preparative and analytical processes. A challenge encountered in the context of such approaches in conductive media, including aqueous buffers, is the generation of electrolysis products at the electrode/liquid interface which can lead to contamination, perturb fluid flows and generally interfere with the measurement process. Here, we address this challenge by designing a single layer microfluidic device architecture where the electric potential is applied outside and downstream of the microfluidic device while the field is propagated back to the chip via the use of a co-flowing highly conductive electrolyte solution that forms a stable interface at the separation region of the device. The co-flowing electrolyte ensures that all the generated electrolysis products, including Joule heat and gaseous products, are flowed away from the chip without coming into contact with the analytes while the single layer fabrication process where all the structures are defined lithographically allows producing the devices in a simple yet highly reproducible manner. We demonstrate that by allowing stable and effective application of electric fields in excess of 100 V cm−1, the described platform provides the basis for rapid separation of heterogeneous mixtures of proteins and protein complexes directly in their native buffers as well as for the simultaneous quantification of their charge states. We illustrate this by probing the interactions in a mixture of an amyloid forming protein, amyloid-β, and a molecular chaperone, Brichos, known to inhibit the process of amyloid formation. The availability of a platform for applying stable electric fields and its compatibility with single-layer soft-lithography processes opens up the possibility of separating and analysing a wide range of molecules on chip, including those with similar electrophoretic mobilities.
Introduction
Microfluidic free-flow electrophoresis (μ-FFE) is a powerful tool for the separation and analysis of charged particles due to its capability to work with small sample volumes, the high separation efficiency that it affords, and its ability to work under steady state flow conditions.1–7 The traditional route to integrating electric fields with microscale channels comprises the incorporation of metal electrodes within the microfluidic chip.8–16 The generation of electrolysis products at the electrode/liquid interface, however, imposes limitations on the stability and sensitivity of devices exploiting these approaches in conductive media. The prime concern in this context is the formation of gas bubbles at the electrode/liquid interface: the gaseous products not only disturb the flow in the microchannels, leading to unstable separation, but they also influence the electric field, making quantitative characterisation challenging. Indeed, as the liquid volumes characteristic of microfluidic devices are on the micro to nanolitre scale, the physical size of bubbles generated within seconds can readily exceed the volumes of microfluidic channels even at comparably low fields of around 20 V cm−1 in conducting buffers which can carry significant current densities.To address this issue, in conventional macroscopic free-flow electrophoresis setups, ion-permeable barriers, such as cellulose nitrate membranes, are introduced between the electrodes and the separation chambers to prevent gaseous electrolysis products from interfering with the separation process.4,17 Several approaches have been presented to similarly reduce the detrimental effects of the formation of the electrolysis products in μ-FFE devices, such as physical separation of the analytical chamber from the electrodes by membranes18–23 or (partial) partitions.3,8,17,24 Additionally, redox electron carriers have been used to suppress the formation and build-up of gas bubbles.25 In spite of alleviating the concerns about the influence of electrolysis products, many of these approaches have limitations, such as intricate fabrication procedures or significant limitations on the applicable electric field and current. In addition, avoiding or displacing gas bubbles does not on its own overcome the issues created by local pH changes due to dissolved electrolysis products or the associated Joule heating. Finally, external electrodes have been used to facilitate device fabrication, alleviating the risk of gases being introduced onto the chip,26–28 but their placement at both inlets and outlets at the same time can still result in electrolysis products and heat flowing through the device.
Due to these difficulties, a number of studies have limited the applied electric fields to small voltages where the problem of gaseous electrolysis products would not be significant.4,10,15,29,30 However, efficient separation for a more general purpose is possible only in the presence of strong electric fields where the deflection of the analyte molecules in the electric field exceeds their diffusion over the same amount of time.2,4,31
Here, we present a strategy for overcoming the specific limitations to the application of high electric fields by placing the electrodes downstream of the microfluidic device to directly and actively transport away the electrolysis products, including heat. Applying the potential off chip allows for large electrode surface areas, thereby decreasing the contact resistance. The electric field can be propagated back into the chip via the use of a conductive electrolyte solution (3M KCl).32 By co-flowing the salt water in a controlled manner with the separation medium, electrical current can flow against the direction of fluid advection on the cathode side, experiencing just a small voltage drop and thus applying a large electric field across the separation channel without the electrolysis products ever coming into contact with the chip itself. Furthermore, the use of hollow metallic tip connectors as external electrodes allows for straightforward integration of this technology with conventional microfluidic devices containing fluidic elements only, while also providing a large active electrode surface area.
We demonstrate the potential of this approach by deflecting charged protein molecules in the electrophoresis area and by further separating both individual proteins and protein complexes from one another in their native environment of aqueous buffers – similar separations are usually performed on solid support media, such as chromatography columns or gel matrices which can interfere with the interactions between the proteins. We also show that the applied electric field can be calibrated which enables the determination of the effective charges of the analytes directly in solution. The fabrication of such devices can be achieved in a single soft-photolithography step and does not require alignment between the fluidic and electronic components. Although demonstrated in the context of μ-FFE, we envision this strategy to be applicable in other circumstances where integration of high electric fields with micron-scale channels is desirable.
Materials and methods
Materials
BSA and human lysozyme were purchased from Sigma Aldrich and used without further purification. The recombinant amyloid-β M1-42, with an N-terminal methionine (hereafter abbreviated as Aβ) was expressed in Escherichia coli from a PetSac plasmid,33 the inclusion bodies were dissolved in 8 M urea, 10 mM tris/HCl, and 1 mM EDTA at pH 8.5 and the Aβ protein was isolated using ion exchange on diethylaminoethyl cellulose resin, followed by two rounds of size exclusion purification in 20 mM sodium phosphate and 0.2 mM EDTA, pH 8.5, using a Superdex 75 HR 26/600 column (GE Healthcare) and one round in 20 mM sodium phosphate and 0.2 mM EDTA, pH 8.0, using a Superdex 75 HR 10/300 column (GE Healthcare). Before each round, the peptide solution was lyophilized and dissolved in 6 M GuHCl and 20 mM sodium phosphate, pH 8.5, as described previously.33,34 The final round of purification served to isolate the pure Aβ monomer and was performed right before the protein was used for the experiment.C-terminal pro-SPC Brichos (hereafter called Brichos) with thioredoxin and His6 tags was expressed in Escherichia coli (the plasmid was a gift from Jenny Presto and Jan Johansson, Karolinska Institute, Sweden) as previously described.35 In short, inclusion bodies were suspended in 2 M urea, 20 mM Tris, 0.5 M NaCl, and 5 mM imidazole, pH 8.0, and the fusion protein was purified on a Ni2+-loaded HiTrap Chelating HP column (GE Healthcare) and cleaved to release the thioredoxin and His6 tag, which was removed using the same chelating column. The Brichos protein was then further purified via ion exchange chromatography using salt gradient elution. The Brichos peak was collected and dialysed against a 20 mM sodium phosphate buffer at pH 7.4. An equimolar amount of amine reactive Alexa488 (Life Technologies) was added from a 5 mM stock dissolved in DMSO to label the Brichos. The solution was incubated at room temperature in the dark for one hour and any unreacted dye was removed by gel filtration in 20 mM sodium phosphate and 0.2 mM EDTA, pH 8.0.
The purified monomeric Aβ was incubated at 37 °C for 60 min with stirring (300 rpm) at a concentration of 36 μM to produce amyloid fibrils, the presence of which was confirmed via a Thioflavin T assay (20 μM Thioflavin T). The fibrils were then incubated at a final concentration of 24 μM with 0.45 μM Alexa-488 labelled Brichos at 23 °C for 48 hours to analyse the binding of the chaperone to the fibrils.
Fabrication of microfluidic devices
The microfluidic devices were fabricated in poly(dimethylsiloxane) (PDMS; Dow Corning) through a single, standard soft-photolithography step of 25 μm height using an SU-8 3025 photoresist on a polished silicon wafer (MicroChemicals GmbH).36,37 The channels were sealed with a quartz slide (Alfa Aesar, 76.2 × 25.4 × 1.0 mm) after both the PDMS and the quartz surface had been activated with oxygen plasma (electronic Diener Femto, 40% power for 15 seconds). The quartz-PDMS devices were then exposed to an additional plasma oxidation step (80% power for 500 seconds) to form silanol groups on the PDMS surface which rendered the channel surfaces more hydrophilic38 to prevent the protein samples from adhering to the PDMS walls of the device.Optical detection
Lysozyme and BSA were used as unlabelled molecules and their movement in the electric field was visualised using an inverted deep-UV fluorescence microscope. The sample was illuminated using a 30 mW 280 nm LED (Thorlabs) exploiting the intrinsic fluorescence of the aromatic residues of the proteins in the deep-UV wavelength range. Briefly, the light was passed through an aspherical lens with a focal length of 20 mm to get a nearly collimated beam and after this onto a dichroic filter cube (280/20–25 nm excitation, 357/44–25 nm emission, 310 nm dichroic beamsplitter). The reflected light from the dichroic mirror was focused onto the sample by an infinity corrected UV objective lens (Thorlabs LMU-10X-UVB; numerical aperture of NA = 0.25), and the emitted light collected through the same objective was passed through the emission filter and focused onto an EMCCD camera (Rolera EM-C2). All the used optics were made out of fused silica to enable high transmission in the UV wavelength region.The labelled Brichos samples were imaged with a fluorescence inverted microscope (Axio Observer D1, Zeiss) equipped with a relevant filter cube (Chroma 49003) and a camera (Evolve 512 EM-CCD, Photometrics).
Results and discussion
Device design
To implement downstream electrodes for free-flow electrophoresis, we designed microfluidic devices containing three wide parallel channels that were connected by narrow (20 μm) perpendicular channels (Fig. 1A). The middle of the three channels (2 mm in width) included the analyte stream which was flanked by the co-flowing buffer (identical to the buffer that the analyte molecules were dissolved in) from both sides. The two side channels (1.2 mm in width) contained the highly conductive electrolyte solution (3M KCl) that transmitted the electric field upstream to the electrophoresis area. While providing electrical conductivity, the connecting channels were designed to have a large hydrodynamic resistance to minimise mass transfer of the electrolyte solution from the side channels to the central electrophoresis channel. They were further designed to have a meandering shape such that the overall area of the device and hence the variations in the photoresist thickness and microfluidic channel heights would be minimised. The flow out from the electrophoresis channel was split towards two separate outlets. This prevents the positive and negative liquid electrodes from coming into close contact with one another which would reduce the electric field across the separation chamber.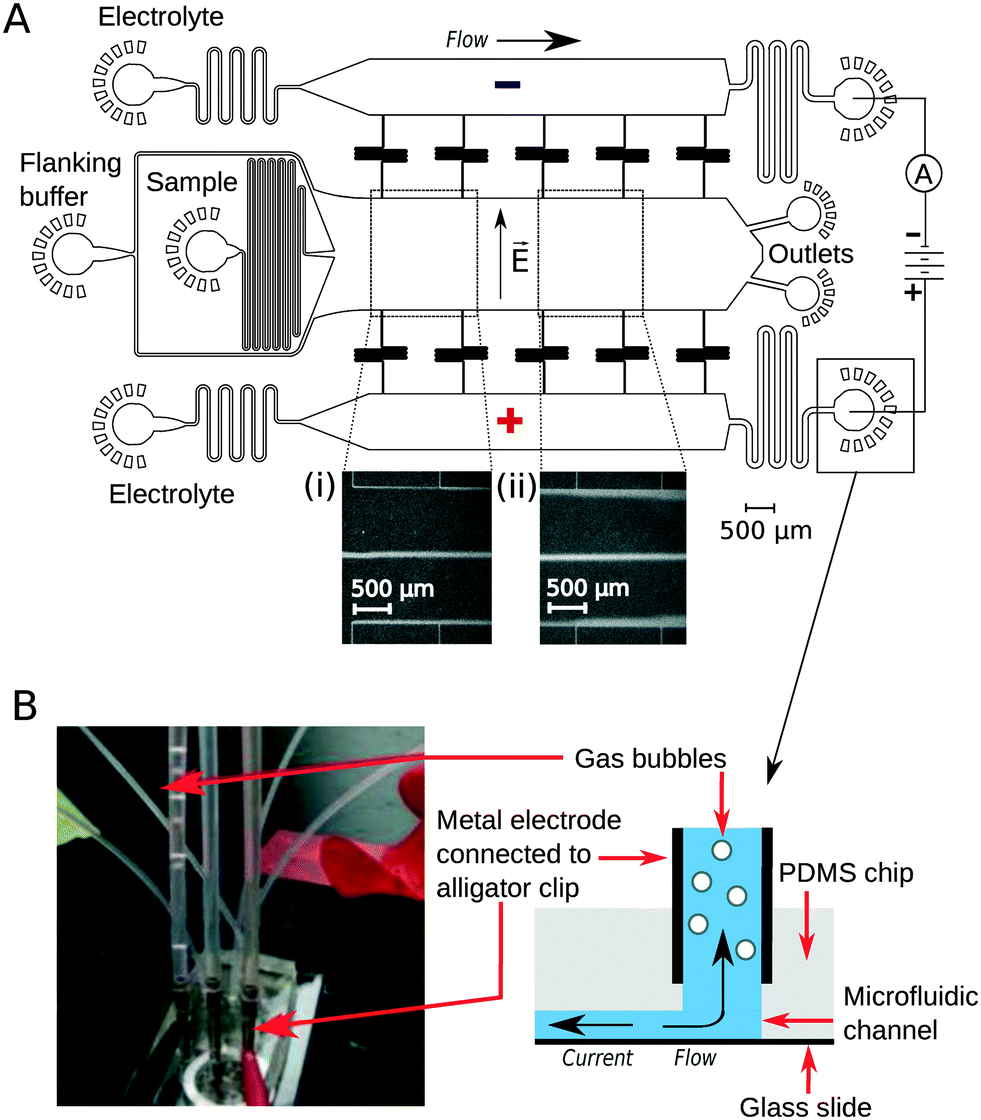 | ||
| Fig. 1 Schematic of the device used in this study. (A) The electrophoresis chamber is connected to a co-flowing electrolyte solution (3M KCl) via narrow perpendicular channels that control the mass transfer of the electrolyte to the electrophoresis chamber forming a thin sheet of electrolyte at the edges of the chamber (inserts (i), (ii); visualised via the addition of trace amounts of bovine serum albumin). The electric field is applied from metal clips at the outlets of the electrolyte channels and it propagates to the electrophoresis channel along with the flowing electrolyte solution. (B) The co-flowing electrolyte solution transports the electrolysis products away from the chip while simultaneously propagating the electric field in the direction opposite to the flow back into the device. | ||
Device operation
The flow of the solutions to the microfluidic channels was controlled by syringe pumps (Cetoni neMESYS modules) to 280 μL h−1 in the main channel and 330 μL h−1 in each of the electrolyte containing channels. This allowed the generation of a thin sheet of the electrolyte solution along the edges of the main channel (Fig. 1A inserts, visualised by adding bovine serum albumin (1 mg mL−1) into the electrolyte solution). The flow rate of the auxiliary fluid was 19 times higher than that of the sample. Hollow metal dispensing tips (20G, Intertronics) were inserted as external electrodes to the outlets of the electrolyte channels and used to apply the electric field (Fig. 1B). Upon application of voltage, the highly conductive KCl solution transmitted the current and electric field to the electrophoresis channel. Gas bubbles nucleated only at the interface of the metal clips and the electrolyte solution and were carried away from the device with the electrolyte flow such that they could not affect the electric or the flow field in the microfluidic device. The fluid was collected in polyethene tubing (Smiths Medical; 800/100/280) connected to 90° bent metal tips. The hydrodynamic “resistors” at the electrolyte outlet (Fig. 1A) were designed to have a hydrodynamic resistance Rh such that the pressure drop on chip would exceed the build-up of hydrostatic pressure in the linear section of the clips (h ≈ 10 mm):| Rh × Q ≫ ρ × g × h = 100 Pa |
To demonstrate the presence of electric field, we injected bovine serum albumin (BSA) molecules via the sample inlet (Fig. 1A) and tracked their movement in the electrophoresis chamber. The protein molecules were dissolved in phosphate buffer (2 mg mL−1 in 10 mM, pH 7) and visualised via their intrinsic fluorescence. This objective was achieved through the use of deep-UV fluorescence microscopy (280 nm excitation; Materials and methods) exploiting the intrinsic fluorescence of the aromatic residues of proteins in this wavelength range. A voltage ramp from 0 V to 54 V with a step size of 3 V was applied using a 500 V bench power supply (Elektro-Automatik EA-PS 9500-06) and the profiles of the protein samples were recorded at the position indicated in Fig. 1A, ii. At this distance, the deflection is small enough to ensure that even at the highest voltage the protein molecules do not interact with the electrolyte that enters the electrophoresis area via the perpendicular connecting channels. The clear deflection of the protein molecules towards the positively charged electrode indicates the presence of electric field (Fig. 2A).
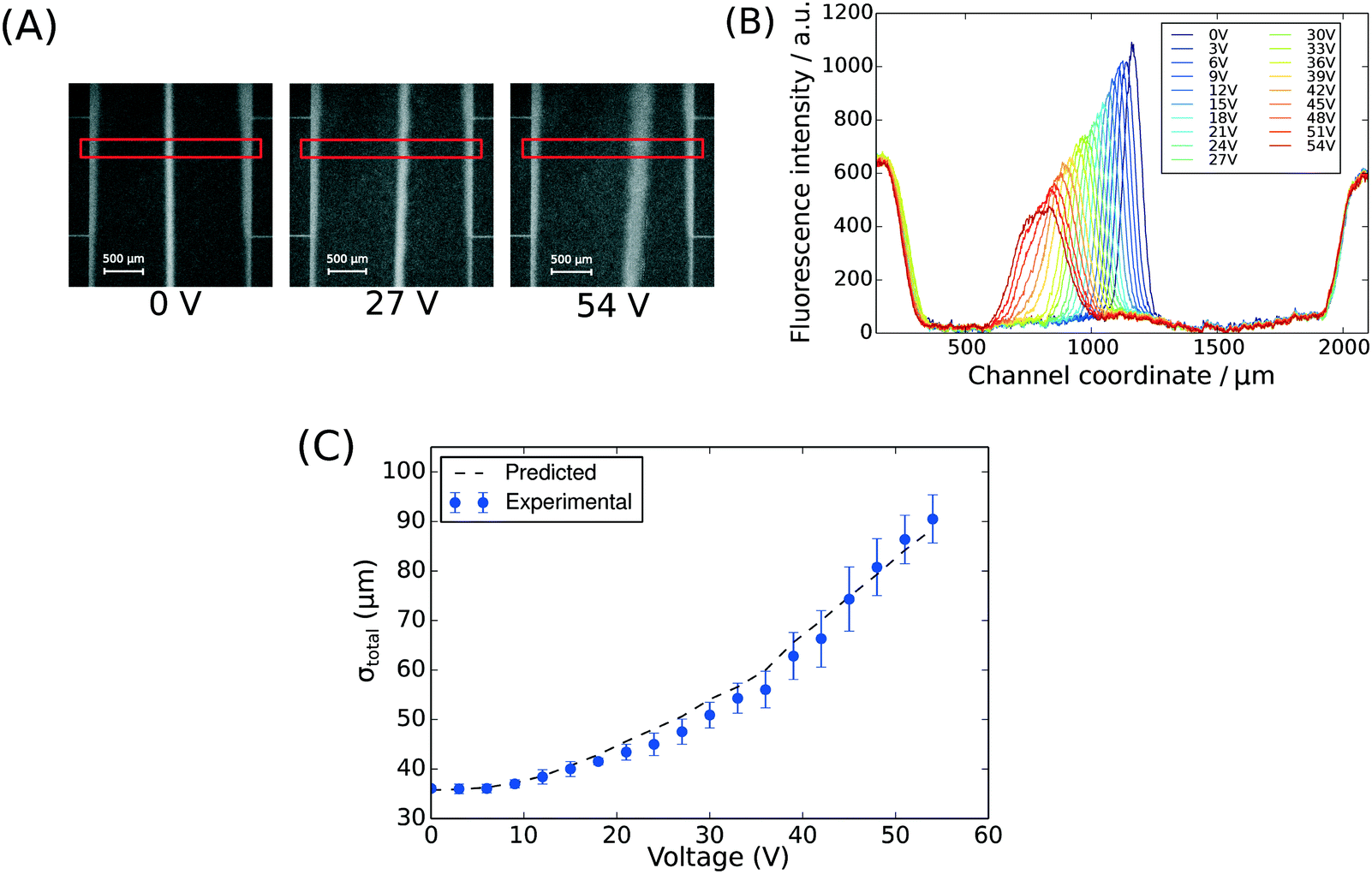 | ||
| Fig. 2 (A) The position of BSA molecules (2 mg mL−1 in 10 mM phosphate buffer, pH 7) was recorded between 0 V and 54 V with a step size of 3 V using deep-UV fluorescence microscopy (280 nm excitation, 340 nm emission) to visualise the protein molecules via their intrinsic fluorescence. The images were taken between the third and the fourth connecting channels counting from upstream (Fig. 1A, ii). The areas annotated by red rectangles (height of 150 pixels) were used to extract the fluorescence profiles. (B) The fluorescence intensity of the BSA sample along the cross-section of the device was extracted at all the voltages to quantify the movement of the molecules in the electric field (average of n = 3 repeats) and to confirm that there was no bulk movement of the fluid in response to the field. (C) The width of the beam of the analyte was observed to increase with its deflection in the electric field as predicted by eqn (1). | ||
To observe the mass transport of the electrolyte into the main separation channel, we supplemented the former solution with a low concentration of BSA (1 mg mL−1; Fig. 1A, insets) and monitored its spatial localisation by UV microscopy. During the application of the electric field, a thin fluorescent layer at the sides of the electrophoresis chamber was observed; however, it stayed at a constant position (Fig. 2B), indicating that there was no bulk movement of the electrolyte fluid into the electrophoresis area due to effects such as electroosmosis. Instead, the interface of the fluorescent sheet was described by the diffusion of the BSA molecules at the interface. Crucially, the diffusion coefficient of the BSA molecules (6 × 10−11 m2 s−1) is less than two orders of magnitude smaller than that of potassium and chloride ions (2 × 10−9 m2 s−1 – the precise value depends on the concentration).39,40 Therefore, within the same residence time, the electrolyte ions diffuse about six times further into the electrophoresis area than the fluorescent BSA marker molecules, i.e. of the order of 100 μm. The average residence time of the fluids within the electrophoresis chamber up to the measurement point is 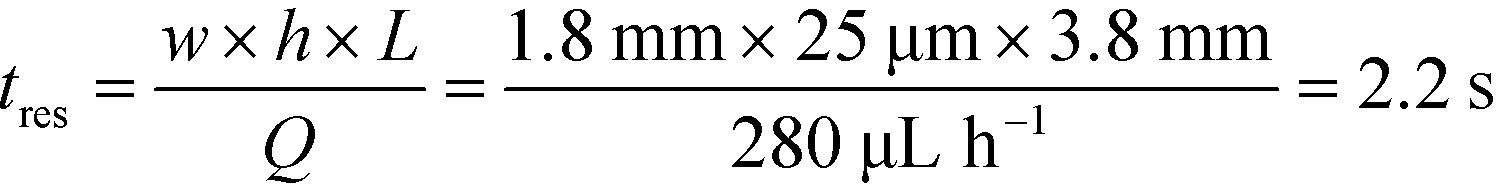 , within this time the electrolyte ions diffuse by around
, within this time the electrolyte ions diffuse by around 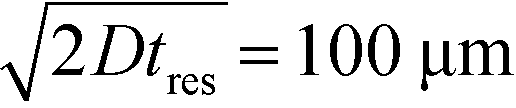 . This number is an approximation as in reality the residence time varies across the cross-section of the device and is higher closer to the edges of the channel than at the centre. With the width of the electrophoresis area being 2 mm, we estimate the electric field across the majority of the width of the electrophoretic chamber to be uniform, which is further confirmed by the linear relationship between the applied electric field and the deflection of the BSA molecules (Fig. 3C).
. This number is an approximation as in reality the residence time varies across the cross-section of the device and is higher closer to the edges of the channel than at the centre. With the width of the electrophoresis area being 2 mm, we estimate the electric field across the majority of the width of the electrophoretic chamber to be uniform, which is further confirmed by the linear relationship between the applied electric field and the deflection of the BSA molecules (Fig. 3C).
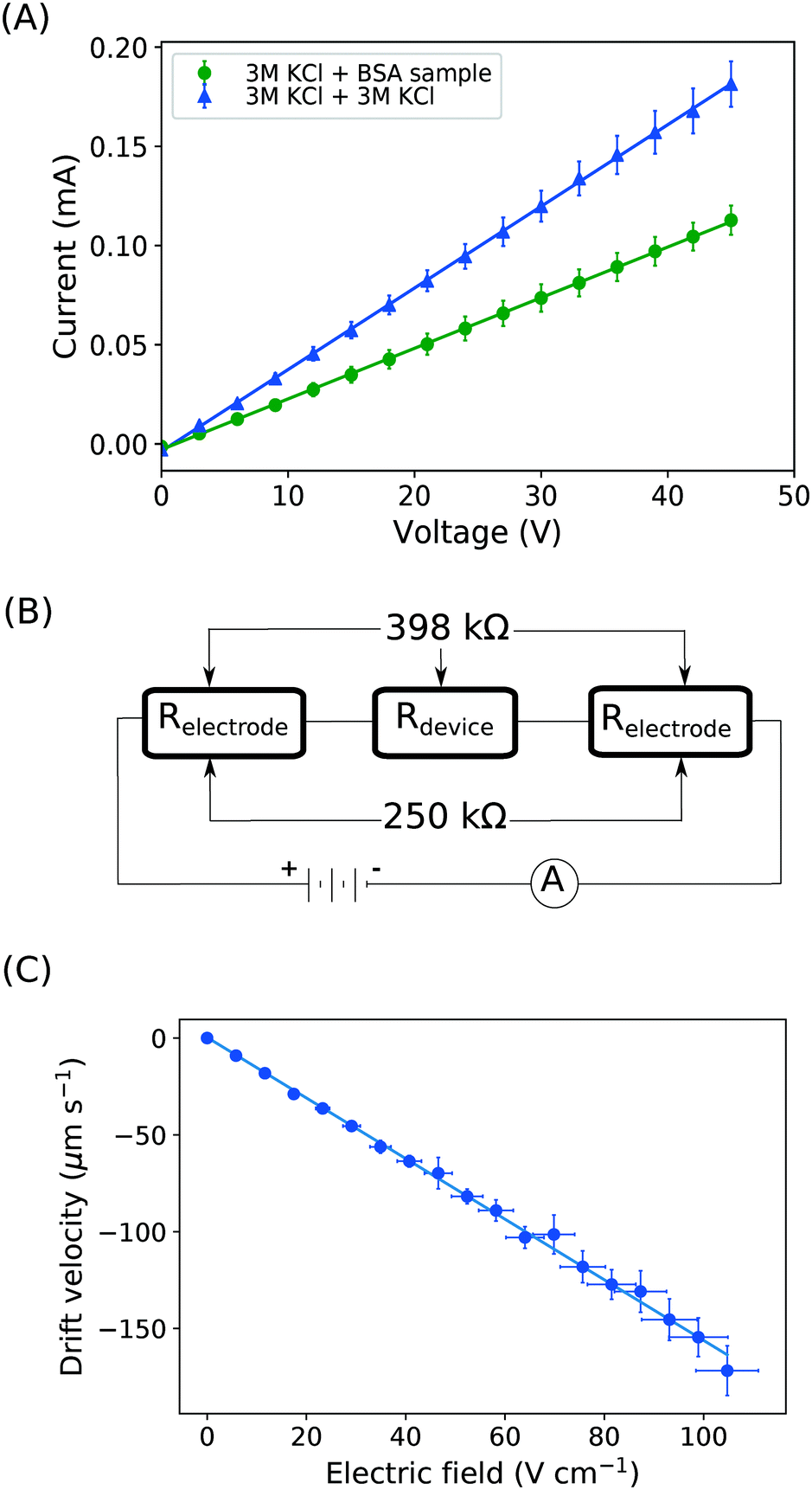 | ||
Fig. 3 (A) Current vs. voltage relationship between the cathode and the anode was recorded first under the conditions where the protein sample and co-flowing buffer were flowed into the separation channel (green circles, average of n = 3 repeats) and then replacing these by a highly conductive electrolyte (3M KCl) solution (blue triangles, average of n = 3 repeats). (B) These data were used to estimate the resistances of the electrodes (250 kΩ) and the overall device (398 kΩ), indicating that the voltage efficiency is around 40%. (C) From the relationship between the drift velocity and the electric field, the electrophoretic mobility of the BSA molecules in pH 7 was estimated to be 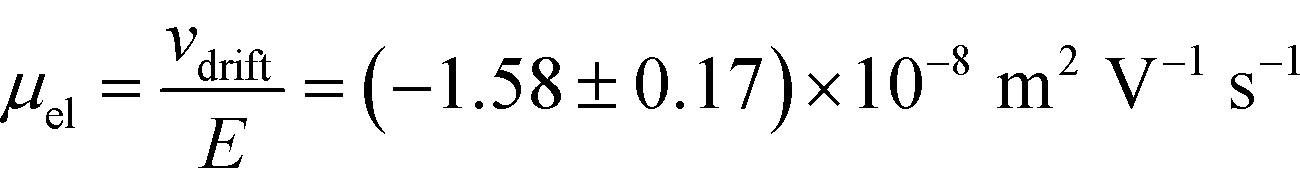 . . | ||
We note that the protein band broadens when the molecules deflect in the electric field (Fig. 2B; average of n = 3 repeats). The observed variance of the analyte band σtotal2 can be described as the sum of the variance contributed by several sources: the finite bandwidth of the sample stream (σinj2), diffusion (σD2), hydrodynamic (σHD2) and electrodynamic (σED2) effects, electrohydrodynamic distortion (σEHD2) and Joule heating (σJH2).41 We estimate that the latter three do not contribute significantly towards the broadening because of the negligible electroosmotic flow (σED2 ∼ 0), no conductivity difference between the sample and the carrier buffer (σEHD2 ∼ 0) and fast heat losses due to the high surface area to volume ratio (σJH2 ∼ 0). The variance of the beam width can thus be approximated as:31
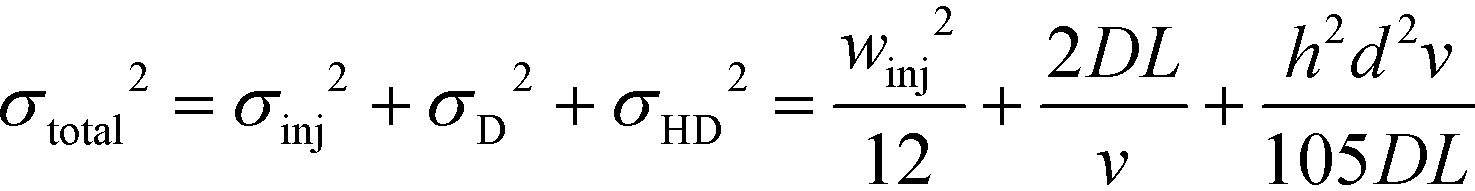 | (1) |
Quantification of electric field and electrophoretic mobilities
With the downstream electrode strategy described in this paper allowing for highly controlled fluid flow, we next demonstrate that it can be used for the characterisation of the effective charges of analytes in solution. To estimate the electrophoretic mobility (μel) of the protein molecules, we set out to quantify the electric field in the electrophoresis chamber. First, the total electrical resistance of the device was determined from the current vs. voltage relationship (Fig. 3A; green circles; average of n = 3 repeats) to be 398 ± 18 kΩ (data recorded simultaneously with the fluorescence profiles of the protein band in Fig. 2B). Next, we noted that if all the channels of the device included an equally conductive solution the electrical resistance of the electrophoretic area would be significantly smaller than that of the connecting channels and the electrolyte channels on the sides. Thus, instead of the sample buffer, a 3 M KCl electrolyte solution was flowed in the electrophoresis chamber (injected via both the inlets of the sample and the flanking buffer – Fig. 1A). This yielded an estimate of 250 ± 11 kΩ for the electrical resistance of the electrodes (Fig. 3A, blue triangles; average of n = 3 repeats). These resistance values agree well with the theoretically estimated electrical resistances of the electrodes and the separation channel (ESI†). From these obtained individual resistances, we concluded the voltage efficiency for our described μ-FFE device to be around 40% (Fig. 3B).Due to the high aspect ratio of the electrophoresis channel 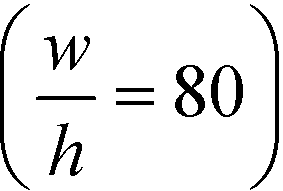 and low Péclet number along its height (Pe = 0.1), the average residence time of the BSA molecules in the section of the channel where the molecules migrate can be estimated as
and low Péclet number along its height (Pe = 0.1), the average residence time of the BSA molecules in the section of the channel where the molecules migrate can be estimated as 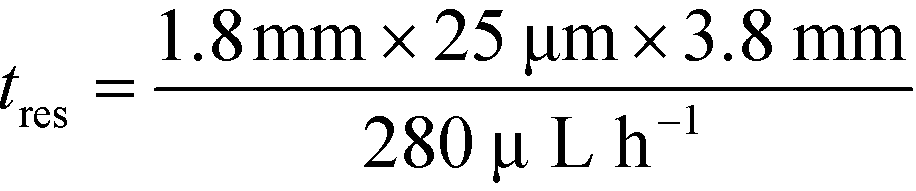 = 2.2 seconds. Using this value, the drift velocities of the BSA molecules at all the individual voltages were estimated and the electrophoretic mobility of the molecules was extracted as
= 2.2 seconds. Using this value, the drift velocities of the BSA molecules at all the individual voltages were estimated and the electrophoretic mobility of the molecules was extracted as 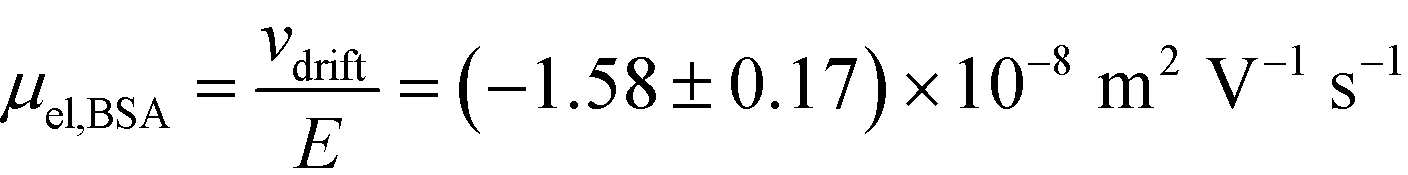 (Fig. 3C). Using literature values for the diffusion coefficient of BSA, the charge of the molecules at pH 7 was evaluated to be around
(Fig. 3C). Using literature values for the diffusion coefficient of BSA, the charge of the molecules at pH 7 was evaluated to be around 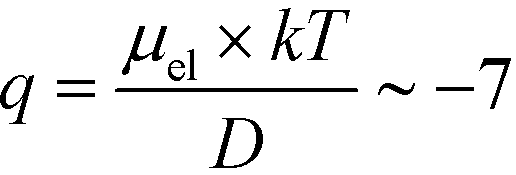 elementary charge units – this estimate is in good agreement with previously obtained values in solutions of similar pH and ionic strength.42,43
elementary charge units – this estimate is in good agreement with previously obtained values in solutions of similar pH and ionic strength.42,43
Separation and analysis of biological mixtures
Finally, the device discussed here presents a combined ability to apply high electric fields and maintain an accurate flow profile through the device even as gaseous electrolysis products accumulate downstream of the microfluidic chip. This opens up the further possibility of determining the effective electrophoretic mobilities of individual protein molecules when they are present in mixtures. We have demonstrated this by applying an electric field to a mixture of BSA (pI 5) and human lysozyme (pI 9) in 10 mM sodium phosphate buffer at pH 7.4. The two molecules were separated under an electric potential (Fig. 4A) and we extracted their individual mobilities as (2.0 ± 0.2) × 10−8 m2 V−1 s−1 for the lysozyme and (−1.7 ± 0.2) × 10−8 m2 V−1 s−1 for the BSA molecules (Fig. 4B).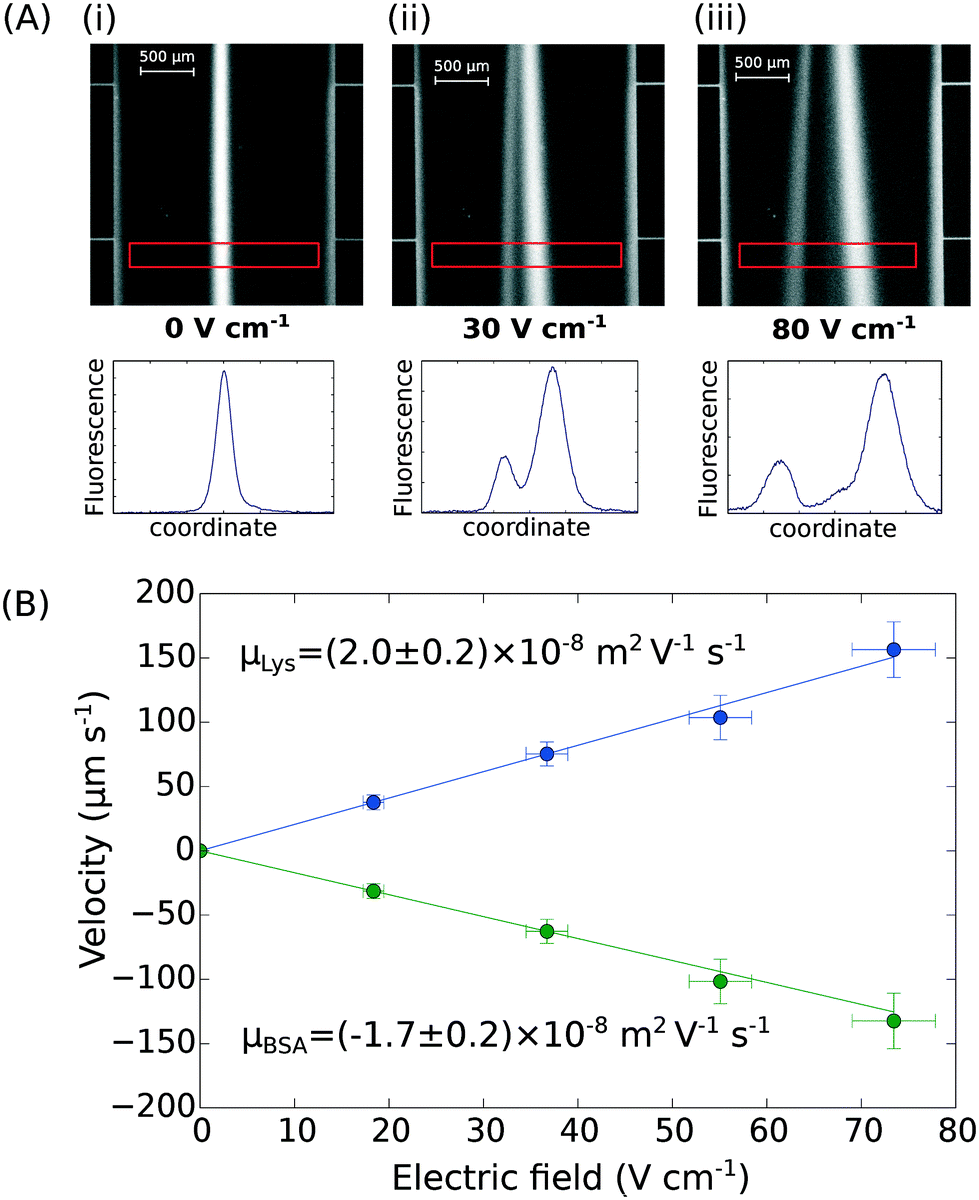 | ||
| Fig. 4 (A) UV fluorescence micrographs and extracted fluorescence profiles (red rectangle) of the separation of bovine serum albumin (2.5 mg mL−1; pI 5) and human lysozyme (4.0 mg mL−1; pI 9) at (i) 0 V cm−1, (ii) 30 V cm−1, and (iii) 80 V cm−1. (B) The profiles were then used for the quantification of the electrophoretic mobilities of the two proteins in the mixture – μLys = (2.0 ± 0.2) × 10−8 m2 V−1 s−1 and μBSA = (−1.7 ± 0.2) × 10−8 m2 V−1 s−1 – calibrating the device as described in Fig. 3A and B. | ||
Given the extensive number of proteins in biological mixtures, such as cell lysate, the separation of such mixtures into all its individual components can be a very challenging goal. Song et al.44 have used free-flow electrophoresis as a preparative technique for mass spectrometry analysis and successfully separated five pI markers (peptides) from one another. They show that further improvements in resolution are possible by placing multiple separation units sequentially and changing the pH closer to the pI of the analytes in each of the next units. The achievable resolution in any of the individual units, however, is limited by the beam broadening effect described in eqn (1).
Indeed, when the flow rate is small, the diffusive broadening term (σD2) starts to dominate eqn (1), whereas at high flow rates the contribution from the hydrodynamic broadening effect (σHD2) becomes dominant – this is because of the increased pressure drop leading to larger differences in the carrier fluid velocity between the fluid layers that are flowing closer to the walls and those at the centre of the channel. The overall broadening is minimal at an intermediate flow rate where the two terms contribute equally. Indeed, ignoring the electroosmotic effect and Joule heating, we can derive an expression for the critical flow rate to channel length ratio (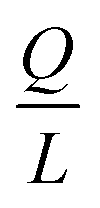 ; ESI†) at which the broadening is smallest and further express this minimal possible broadening as a function of the relative deflection C (fraction of the channel width by which the analyte is deflected):
; ESI†) at which the broadening is smallest and further express this minimal possible broadening as a function of the relative deflection C (fraction of the channel width by which the analyte is deflected):
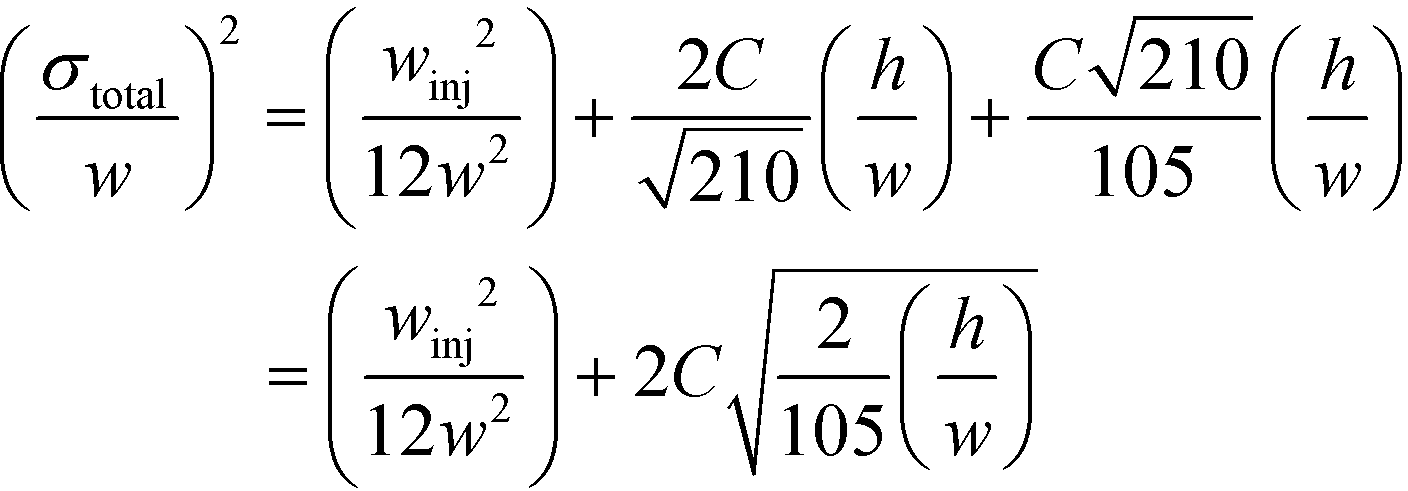 | (2) |
For example, for a relative deflection of C = 45% from the centre of the channel, the minimum possible beam width (defined as 2 × σtotal) is over 8% of the total channel width for the case when the analyte is filling 5% of the channel at the injection and a channel aspect ratio of 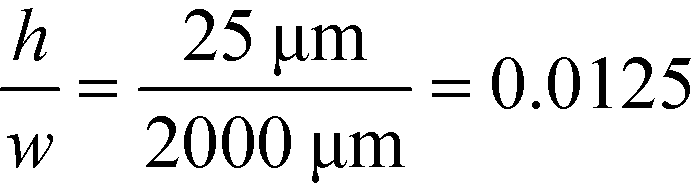 as was used in this work. For a relative injection width
as was used in this work. For a relative injection width 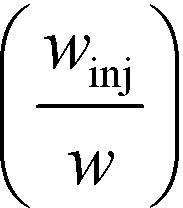 of 1% and a channel aspect ratio of
of 1% and a channel aspect ratio of 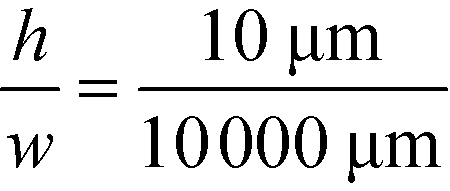 which could be achieved by placing support pillars in the channel, the beam width at a relative deflection of C = 45% could be further reduced to around 2.5%, while the less deflected analytes would remain more confined (e.g. 1.2% at a relative deflection of C = 10% the channel width).
which could be achieved by placing support pillars in the channel, the beam width at a relative deflection of C = 45% could be further reduced to around 2.5%, while the less deflected analytes would remain more confined (e.g. 1.2% at a relative deflection of C = 10% the channel width).
As such, full proteome-level fractionation and selection of specific proteins of interest is likely to be possible only by combining the separation units described here sequentially. Simultaneously, however, with the ability to generate stable electric fields via efficient removal of electrolysis products, the described platform can be used for detecting interactions between proteins and for further separating proteins and protein complexes from one another directly in their native environment. To show the power of this approach, we have examined here the mixture of an aggregate forming protein amyloid-β (Aβ) and a molecular chaperone Brichos.
Aβ is a polypeptide that is prone to self-association and fibril formation; its assembly is believed to play a critical and potentially casual role in the development of Alzheimer's disease.34 The pathway of Aβ aggregation involves a transient heterogeneous mixture including oligomeric species of a range of different association numbers and structural forms. Molecular chaperones are known to play a key role in aiding the folding process of newly synthesised proteins into their native states and to prevent protein aggregation. Specifically, Brichos, which is a protein of approximately 100 amino acids, has been found experimentally to inhibit misfolding and aggregation of Aβ both in vitro and in vivo.35,45
By incubating a mixture of aggregated Aβ (24 μM) and fluorescently labelled Brichos (0.45 μM) and exposing the mixture to the electric field, we detected the formation of a complex between Brichos and Aβ fibrils (Fig. 5A and B) and further fully separated the formed fibril–chaperone complex from the individual chaperone molecules. The occurrence of this interaction is in agreement with an earlier observation of Brichos molecules binding to Aβ fibrils which is believed to inhibit the potential secondary nucleation events that could otherwise occur on the surfaces of the fibrils.46 Non-covalent and reversible interactions between biomolecules, such as proteins, are the basis of an extremely wide range of biophysical and biochemical processes and due to their modulation by solution conditions, these interactions are best studied under native conditions in aqueous environments. As such, we feel that our platform is ideally suited for probing interactions in mixtures as not only is there no interference with a support medium in contrast to more commonly used biophysical separation techniques, such as chromatography or protein gels, it also allows probing these interactions at short timescales not accessible with these more conventional techniques.
 | ||
| Fig. 5 Brichos molecules were labelled with a fluorescent dye (Alexa 488) and mixed with amyloid-β fibrils in order to test their binding to the fibrils. By comparing the behaviour of (A) pure Brichos and (B) Brichos molecules mixed with the fibrils, we concluded the formation of a Brichos–amyloid-β fibril complex and purified this complex from the unbound Brichos molecules. | ||
Conclusions
We have designed and demonstrated a single step lithography approach to fabricate microfluidic devices that can be used to apply high electric fields without the formation of any electrolysis products in the electrophoresis area. This objective was achieved by applying an electric potential to the electrodes situated downstream and outside of the chip and using a flowing electrolyte solution to simultaneously transport the electrolysis products away from the chip and propagate the electric field in the direction opposite to the flow back into the device. We demonstrated the generation of electric fields above 100 V cm−1 and estimated the effective charges of molecules in a mixture by exploiting our ability to apply such fields in a quantitative manner. The availability of a simple strategy to apply strong electric fields within microscale channels in a highly controlled manner opens up the possibility of using microfluidic free-flow electrophoresis in PDMS microfluidics as a preparative technique for the separation of individual proteins or protein complexes while further obtaining quantitative information about their charge states directly in solution during the separation process.Conflicts of interest
Parts of this work have been the subject of a patent application filed by Cambridge Enterprise Limited, a fully owned subsidiary of the University of Cambridge.Acknowledgements
The research leading to these results has received funding from the Engineering and Physical Sciences Research Council (K. L. S.), the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013) through the ERC grants PhysProt (agreement no 337969; T. P. J. K.) and MAMBA (agreement no, 340890; S. L.), the Biotechnology and Biological Sciences Research Council (T. P. J. K.) and the Swiss National Science Foundation (T. M.).References
- N. Pamme, Lab Chip, 2007, 7, 1644–1659 RSC.
- R. T. Turgeon and M. T. Bowser, Anal. Bioanal. Chem., 2010, 394, 187–198 CrossRef PubMed.
- D. Janasek, J. Franzke and A. Manz, Nature, 2006, 442, 374–380 CrossRef CAS PubMed.
- D. Kohlheyer, J. C. T. Eijkel, A. van den Berg and R. B. M. Schasfoort, Electrophoresis, 2008, 29, 977–993 CrossRef CAS PubMed.
- B. R. Fonslow, V. H. Barocas and M. T. Bowser, Anal. Chem., 2006, 78, 5369–5374 CrossRef CAS PubMed.
- J. Wen, E. W. Wilker, M. B. Yaffe and K. F. Jensen, Anal. Chem., 2010, 82, 1253–1260 CrossRef CAS PubMed.
- L.-J. Cheng and H.-C. Chang, Lab Chip, 2014, 979–987 RSC.
- D. E. Raymond, A. Manz and H. M. Wldmer, Anal. Chem., 1994, 66, 2858–2865 CrossRef CAS.
- H. Kobayashi, K. Shimamura, T. Akaida, K. Sakano, N. Tajima, J. Funazaki, H. Suzuki and E. Shinohara, J. Chromatogr. A, 2003, 990, 169–178 CrossRef CAS PubMed.
- H. Lu, S. Gaudet, M. A. Schmidt and K. F. Jensen, Anal. Chem., 2004, 76, 5705–5712 CrossRef CAS PubMed.
- B. R. Fonslow and M. T. Bowser, Anal. Chem., 2005, 77, 5706–5710 CrossRef CAS PubMed.
- V. Kostal, B. R. Fonslow, E. A. Arriaga and M. T. Bowser, Anal. Chem., 2009, 81, 9267–9273 CrossRef CAS PubMed.
- W. Ebina, A. C. Rowat and D. A. Weitz, Biomicrofluidics, 2009, 3, 2605–2609 CrossRef PubMed.
- J.-H. So and M. D. Dickey, Lab Chip, 2011, 11, 905–911 RSC.
- T. W. Herling, T. Müller, L. Rajah, J. N. Skepper, M. Vendruscolo and T. P. J. Knowles, Appl. Phys. Lett., 2013, 102, 184102 CrossRef.
- D. G. Rackus, M. D. M. Dryden, J. Lamanna, A. Zaragoza, B. Lam, S. O. Kelley and A. R. Wheeler, Lab Chip, 2015, 15, 3776–3784 RSC.
- S. Köhler, C. Weilbeer, S. Howitz, H. Becker, V. Beushausen and D. Belder, Lab Chip, 2011, 11, 309–314 RSC.
- M. Mazereeuw, C. M. D. Best, U. R. Tjaden, H. Irth and J. V. D. Greef, Anal. Chem., 2000, 72, 3881–3886 CrossRef CAS PubMed.
- D. Kohlheyer, G. A. J. Besselink, S. Schlautmann and R. B. M. Schasfoort, Lab Chip, 2006, 6, 374–380 RSC.
- J. W. Albrecht and K. F. Jensen, Electrophoresis, 2006, 27(24), 4960–4969 CrossRef CAS PubMed.
- G. Munchow, S. Hardt, J. P. Kutter and K. S. Drese, Lab Chip, 2007, 7, 98–102 RSC.
- J. E. Prest, S. J. Baldock, P. R. Fielden, N. J. Goddard, R. Goodacre, R. O'Connor and B. J. Treves Brown, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2012, 903, 53–59 CrossRef CAS PubMed.
- C. Herzog, G. F. W. Jochem, P. Glaeser and S. Nagl, Microchim. Acta, 2014, 182, 887–892 CrossRef.
- C.-X. Zhang and A. Manz, Anal. Chem., 2003, 75, 5759–5766 CrossRef CAS PubMed.
- D. Kohlheyer, J. C. T. Eijkel, S. Schlautmann, A. Van Den Berg and R. B. M. Schasfoort, Anal. Chem., 2008, 80, 4111–4118 CrossRef CAS PubMed.
- S. Köhler, C. Benz, H. Becker, E. Beckert, V. Beushausend and D. Belder, RSC Adv., 2012, 2, 520–525 RSC.
- S. Jezierski, D. Belder and S. Nagl, Chem. Commun., 2013, 49, 904–906 RSC.
- B. Jung, K. Rose, M. Shusteff, A. Persat and J. Santiago, Liquid and gel electrodes for transverse free flow electrophoresis, US Pat., 8999129, 2015 Search PubMed.
- K. Macounová, C. R. Cabrera and P. Yager, Anal. Chem., 2001, 73, 1627–1633 CrossRef.
- N. W. Frost and M. T. Bowser, Lab Chip, 2010, 10, 1231–1236 RSC.
- B. R. Fonslow and M. T. Bowser, Anal. Chem., 2006, 78, 8236–8244 CrossRef CAS PubMed.
- A. Sciambi and A. R. Abate, Lab Chip, 2014, 14, 2605–2609 RSC.
- D. M. Walsh, E. Thulin, A. M. Minogue, N. Gustavsson, E. Pang, D. B. Teplow and S. Linse, Rev. Geophys., 2009, 276, 1266–1281 CAS.
- E. Hellstrand, B. Boland, D. M. Walsh and S. Linse, ACS Chem. Neurosci., 2009, 1, 13–18 CrossRef PubMed.
- H. Willander, J. Presto, G. Askarieh, H. Biverstål, B. Frohm, S. D. Knight, J. Johansson and S. Linse, J. Biol. Chem., 2012, 287, 31608–31617 CrossRef CAS PubMed.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed.
- D. Qin, Y. Xia and G. M. Whitesides, Nat. Protoc., 2010, 5, 491–502 CrossRef CAS PubMed.
- S. H. Tan, N. T. Nguyen, Y. C. Chua and T. G. Kang, Biomicrofluidics, 2010, 4, 1–8 CrossRef PubMed.
- L. Nuttall, Ann. N. Y. Acad. Sci., 1949, 51, 781–788 CrossRef.
- A. M. Friedman and J. W. Kennedy, J. Am. Chem. Soc., 1955, 77, 4499 CrossRef CAS.
- D. E. Raymond, A. Manz and H. M. Widmer, Anal. Chem., 1996, 68, 2515–2522 CrossRef CAS PubMed.
- U. Böhme and U. Scheler, Chem. Phys. Lett., 2007, 435, 342–345 CrossRef.
- B. Jachimska and A. Pajor, Bioelectrochemistry, 2012, 87, 138–146 CrossRef CAS PubMed.
- Y.-A. Song, M. Chan, C. Celio, S. R. Tannenbaum, J. S. Wishnok and J. Han, Anal. Chem., 2010, 82, 2317–2325 CrossRef CAS PubMed.
- E. Hermansson, S. Schultz, D. Crowther, S. Linse, B. Winblad, G. Westermark, J. Johansson and J. Presto, Dis. Models & Mech., 2014, 7, 659–665 Search PubMed.
- S. I. Cohen, P. Arosio, J. Presto, F. R. Kurudenkandy, H. Biverstål, L. Dolfe, C. Dunning, X. Yang, B. Frohm and M. Vendruscolo, et al., Nat. Struct. Mol. Biol., 2015, 22, 207–213 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7lc00797c |
| ‡ These authors contributed equally. |
| § Current address: Wren Therapeutics Ltd, Cambridge, UK. |
| This journal is © The Royal Society of Chemistry 2018 |